

Abstract
The gep oncogene, defined by the activated mutant of the α-subunit of the G protein G12 (Gα12Q229L or Gα12QL), potently stimulates the proliferation of many different cell types in addition to inducing neoplastic transformation of several fibroblast cell lines. While it has been demonstrated that Gα12QL accelerates G1- to S-phase cell cycle progression, the precise mechanism through which Gα12 communicates to cell cycle machinery is largely unknown. In the present study, we report that the activated—mutational as well as receptor-mediated—Gα12 transmits its proliferative signals to cell cycle machinery by modulating the levels of the S-phase kinase-associated protein 2 (Skp2), an E3 ubiquitin ligase, involved in the regulation of the cyclin-dependent kinase inhibitor (CKI), p27Kip1. Our results show that the expression of Gα12QL leads to an increase in the levels of Skp2 with a correlatable decrease in p27Kip1 levels and subsequent increase in the activities of specific CDKs. By demonstrating that the transient expression of Gα12QL induces an increase in Skp2 levels with resultant downregulation of p27Kip1 in both NIH3T3 and human astrocytoma 1321N1 cells, we establish here that the effect of Gα12 on Skp2/p27Kip1 is cell type independent. In addition, we demonstrate that LPA-stimulated proliferation and changes in Skp2 and p27Kip1 levels in 1321N1 cells could be inhibited by the expression of a dominant-negative mutant of Gα12, thereby pointing to the critical role of Gα12 in LPA-mediated mitogenic signaling. Our findings also indicate that LPA as well as Gα12-mediated upregulation of Skp2 requires a yet to be characterized mechanism involving JNK. Since Skp2 has been identified as an oncogene, and it is overexpressed in many cancers, our results presented here describe for the first time that Skp2 is a novel target in the cell cycle machinery through which Gα12 and its cognate receptors transmit their oncogenic signals.
Keywords: oncogene, G12, LPA, Skp2, JNK
Introduction
GPCR-mediated signaling pathways have recently been identified to play a major role in cancer cell growth and progression.1-5 Many GPCRs have been observed to be overexpressed in various cancer types, and they appear to contribute to tumor cell growth when activated by circulating or locally produced ligands. Confirming this view, a growing list of human cancers including ovarian, breast, prostate, and pancreatic carcinomas has been shown to exhibit aberrant expressions of GPCRs and the ligands that activate them.4-10 In general, ligand-activated GPCRs stimulate diverse physiological responses by catalyzing the guanine nucleotide exchange in the α-subunit of different heterotrimeric G proteins.11 Of the different α-subunits that have been analyzed thus far, the α-subunit of the G protein G12 (Gα12), referred to as the gep oncogene,12 exhibits the most potent mitogenic and oncogenic activities.11-13 Previous studies from our laboratory as well as others have identified different but complementary mechanisms through which Gα12 promotes oncogenic proliferation.11-24 Although these studies have indicated that Gα12 promotes G1/S-phase progression23,24—similar to many oncogenes—the mechanism by which Gα12 communicates to cell cycle machinery is poorly understood. In light of the recent observations that Gα12 and receptors that couple to Gα12 are implicated in the genesis and/or progression of several cancers,11-13 defining the mechanism(s) by which Gα12 accelerates cell cycle progression may prove critically important to identify novel diagnostic, therapeutic, or prognostic targets.
Cell cycle analyses have indicated that mitogenic signaling pathways often converge onto mid to late G1 phase of cell cycle to accelerate cell progression into S phase.25,26 In brief, cell cycle progression is regulated by a coordinated series of phosphorylation events, chiefly mediated by the cyclin-dependent kinase (CDK) family of serine/threonine kinases.25-27 The activities of CDKs are regulated by the stimulatory cyclins and inhibitory cyclin-dependent kinase inhibitors (CKIs).25-28 Once activated, cyclin-CDK complexes drive the cell cycle through its different phases via the phosphorylation of specific downstream targets such as retinoblastoma protein (pRb).29,30 Our studies presented here are focused on identifying the mechanism by which Gα12 communicates to this complex array of interrelated events to accelerate cell cycle progression. We demonstrate here that the expression of the constitutively activated mutant of Gα12 stimulates the expression of cyclin D1 and cyclin A along with an increase in the activities of CDK2 and CDK4 in NIH3T3 cells. We show that the expression of Gα12QL leads to a decrease in the levels of p27-kinase inhibitory protein-1 (p27Kip1), a CKI primarily involved in the inhibition of CDK2 and CDK4.26-28 We establish further that the decrease in p27Kip1 accompanies the upregulation of S-phase kinase associated protein-2 (Skp2), an E3 ubiquitin ligase involved in downregulating p27Kip1 levels.31-34 The ability of transiently expressed Gα12QL to recapitulate similar events in 1321N1 astrocytoma cells indicates that the effect of Gα12 on the levels of Skp1 and p27Kip1 is cell type independent. Our studies presented here also demonstrate that LPA, which has been identified as an oncogenic lipid growth factor in many cancers,4,35-38 stimulates the proliferation of 1321N astrocytoma cells along with an increase in the levels of Skp2 via Gα12. Finally, our studies point to JNK as a novel mediator in LPA/LPAR-Gα12–mediated upregulation of Skp2. Taken together with the findings that Skp2 that has been defined as an oncogene39-42 is overexpressed in many different cancers43-48 similar to the upregulation of LPA-LPAR signaling in many cancers,4,35-38 our data presented here unravel a potential role for Gα12 and its cognate receptors in upregulating the levels of Skp2 in many cancers.
Results
Activated mutant of Gα12 stimulates an increase in cyclin levels
To define Gα12-mediated changes in cell cycle–associated proteins involved in G0-G1-S transition, we examined the profiles of different cyclins in NIH3T3 cells expressing Gα12QL during G1-S progression. After verifying the expression of Gα12QL (Fig. 1A), the levels of G1/S cyclins such as cyclin D1, cyclin E, and cyclin A were monitored by immunoblot analyses using lysates from NIH3T3 cells expressing Gα12QL (Gα12QL-NIH3T3) or the empty vector. Results from this analysis indicated that upon stimulation of cell proliferation by 5% serum, an increased expression of cyclin D1 can be observed in cells expressing Gα12QL by 4 hours following serum stimulation, whereas such an increase can be seen in control cells only from 8 hours onwards (Fig. 1A). In contrast, an increase in cyclin A levels can be seen in Gα12QL-NIH3T3 cells at all the time points compared to the vector control cells (Fig. 1B). In the case of cyclin E, Gα12QL cells showed an increase—albeit small but consistent—in the basal levels compared to the vector controls. Thus, cells expressing Gα12QL primarily showed an increase in the expression profiles of the G1/S-phase cyclins, cyclin D1 and cyclin A. An immunobot analysis of the G1/S phase–associated CDKs, namely, CDK2 and CDK4, showed no changes in their expression levels during these time points (Fig. 1C).
Figure 1.
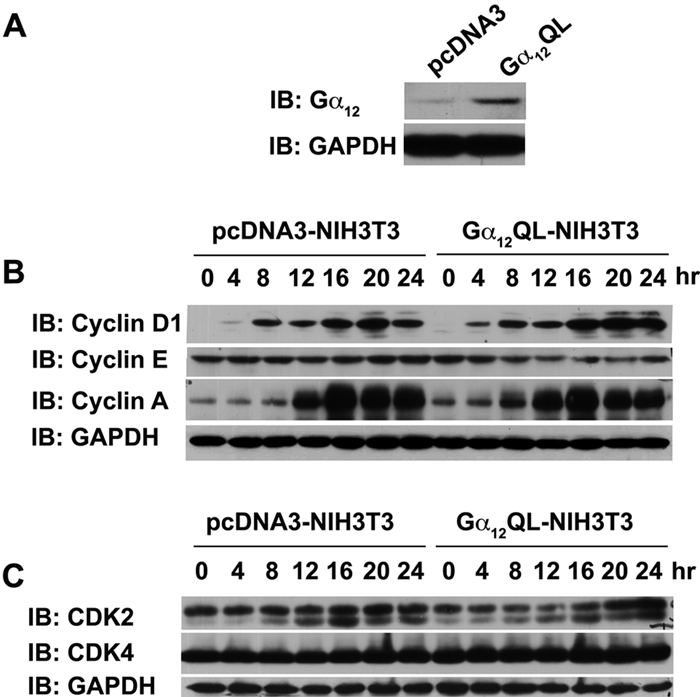
Expression levels of G1/S-phase cyclins and CDKs in Gα12QL-expressing cells. (A) Lysates (50 µg) from pcDNA3 vector control and Gα12QL-transformed NIH3T3 cells (4 × 105) were analyzed for the expression of Gα12QL by immunoblot analysis using antibodies to Gα12. The blot was reprobed with GAPDH to monitor equal loading of proteins. (B) Lysates (50 µg) prepared from vector control and Gα12QL-NIH3T3 cells serum starved for 24 hours followed by stimulation with 5% CS for varying lengths of time were separated on a 10% SDS-PAGE and subjected to immunoblot analysis using antibodies specific to cyclin D1, cyclin A, or cyclin E. The blot was reprobed with antibodies to GAPDH to monitor equal loading of proteins. (C) An identical blot was probed for monitoring the levels of CDK4 and CDK2 using the respective antibodies. The blot was reprobed with antibodies to GAPDH to monitor equal loading of proteins. These analyses were repeated at least 3 times, and the results are from a typical analysis.
Gα12 stimulates an increase in CDK activity
Since increased levels of cyclin D1 and cyclin A are often associated with a corresponding increase in CDK4 and CDK2 activities rather than their levels of expression,25-27 we examined whether the increased levels of cyclins in Gα12QL-expressing cells could be correlated with a corresponding increase in the activities of their catalytic partners CDK2 or CDK4. Lysates prepared from pcDNA3- and Gα12QL-NIH3T3 cells that were stimulated with serum for different lengths of time were subjected to immune complex kinase assays using antibodies specific for CDK2 or CDK4.49 CDK2 immune complex kinase assay was carried out using histone H1 as a substrate,49 whereas CDK4 immune complex kinase assay was carried out using GST-Rb as the substrate.49 As compared to the vector control, cells expressing Gα12QL show a rapid and more potent increase in CDK2 as well as CDK4 activities (Fig. 2A and 2B), while their levels remain unaltered (Fig. 1B). Based on the findings that mitogenic pathways stimulate the rapid phosphorylation of Ser-795 of pRb through the cyclin D1–CDK4 and cyclin A–CDK2 complexes,50,51 the phosphorylation status of Ser795 of pRb has been used as an index of the increased activities of CDK2/4.52 Therefore, we analyzed the phosphorylation of Ser795 of pRb in Gα12QL-expressing cells to further assess and confirm the increase in the activities of CDK2/4. Results from such an immunoblot analysis indicated a rapid increase in the phosphorylated form of pRb in Gα12QL-NIH3T3 cells upon serum stimulation compared to vector control cells (Fig. 2C).
Figure 2.
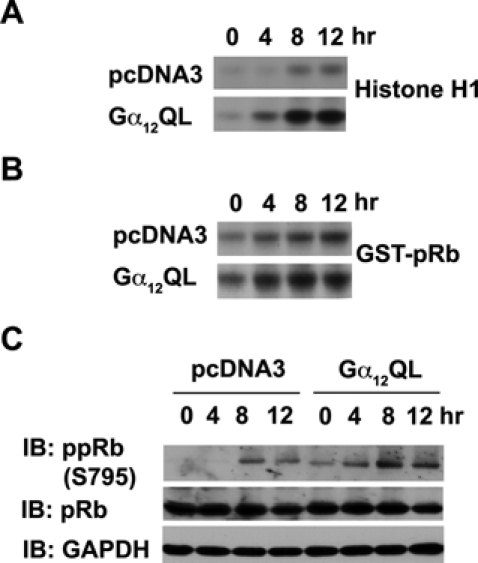
Gα12QL stimulates the activation of CDKs. (A) CDK2 was immunoprecipitated from the lysates (100 µg) prepared from serum-deprived pcDNA3- and Gα12QL-NIH3T3 that were stimulated with 5% calf serum for 0, 4, 8, and 12 hours using antibodies to CDK2. Immune complex kinase assay was carried out using purified histone H1 as a substrate. The phosphorylated histone H1 was separated by SDS-PAGE and visualized by autoradiography. (B) From the lysates (100 µg) as prepared above, CDK4 was immunoprecipitated with antibodies to CDK4, and immune complex kinase assay was carried out using purified recombinant GST-pRb as a substrate. The phosphorylated GST-pRb was separated by SDS-PAGE and visualized by autoradiography. (C) These lysates (50 µg) were separated on a 10% SDS-PAGE, and immunoblot analysis was carried out using antibodies specific to Ser795-phosphorylated pRb. The blot was stripped and reprobed with antibodies to pRb to monitor the expression levels. The blot was further probed with antibodies to GAPDH to monitor equal loading of proteins. The results presented are from a typical set of experiments that were repeated at least 3 times.
Gα12 stimulates promitogenic changes in the levels of p27Kip1 and Skp2
It has been well established that the activities of specific cyclin-CDK complexes are finely and dynamically regulated by distinct CDK inhibitors (CKIs).25-28 Of the different CKIs, p27Kip1 has been observed to play a major role in the regulation of CDK2/4 activities.25-28 Therefore, we monitored the levels of p27Kip1 in response to the expression of Gα12QL. Vector control and Gα12QL-NIH3T3 cells that were made quiescent by 24 hours of serum starvation were stimulated with 5% serum, and the lysates prepared from these cells at different time points were subjected to immunoblot analysis using antibodies to p27Kip1. The results indicated that the cells expressing Gα12QL showed an accelerated decrease in p27Kip1 levels (from 8 hours) compared to vector control cells upon growth stimulation by serum (Fig. 3A). Since the levels of p27Kip1 have been shown to be dynamically regulated via ubiquitin-mediated degradation of p27Kip1 by the E3 ubiquitin ligase Skp2,31-34 we investigated whether the observed decrease in p27Kip1 levels in Gα12QL-NIH3T3 cells is associated with an increase in the expression levels of Skp2. Results from such an analysis indicated an increase in Skp2 levels—during unstimulated as well as serum-stimulated conditions—in Gα12QL cells compared to the vector control cells (Fig. 3B). To establish that the expression of Skp2 is not due to the clonal variation of a specific Gα12QL-expressing clone of NIH3T3 cells, the expression levels of Skp2 and p27 were monitored in 3 different Gα12QL-NIH3T3 clones. Results from such an experiment indicated that an increase in Skp2 levels along with a concomitant decrease in the levels of p27 could be observed in all the Gα12QL-NIH3T3 clones (Fig. 3C), thus establishing that the observed decrease in Skp2 levels is not due to clonal variation. To further confirm that the increased levels of Skp2 are directly in response to Gα12QL, the expression of Skp2 in response to transiently expressed Gα12QL was monitored. Results from this analysis indicated that the transient expression of Gα12QL stimulated the expression of Skp2 with a concomitant decrease in p27 levels (Fig. 3D), thereby confirming that the changes in Skp2-p27Kip1 levels are directly in response to the expression of Gα12QL.
Figure 3.
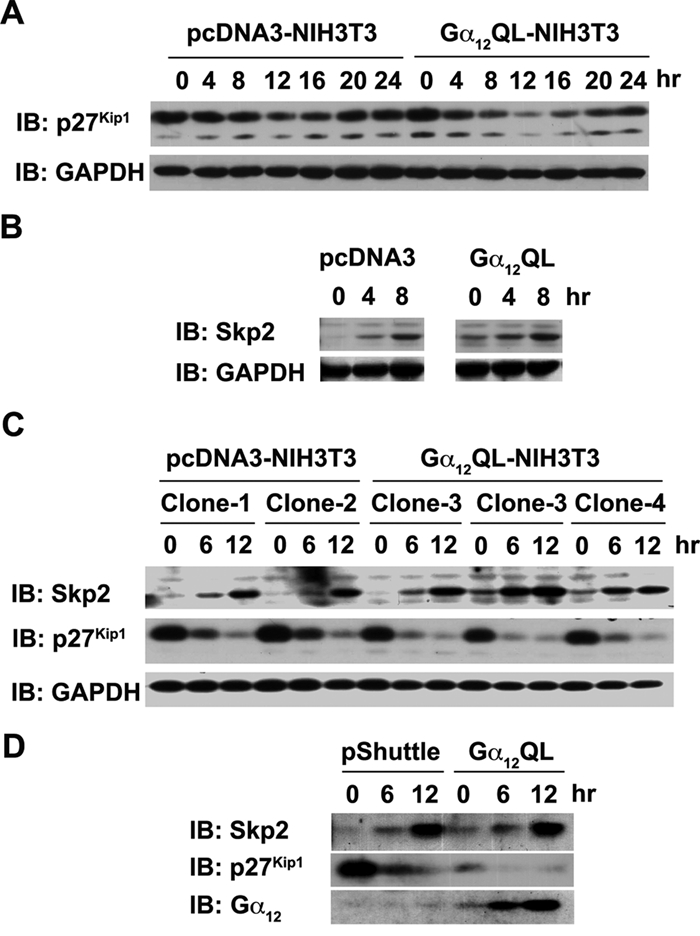
Gα12 modulates the levels of p27Kip1 and Skp2. (A) Lysates (50 µg) from pcDNA3- and Gα12QL-NIH3T3 cells, serum starved for 24 hours and stimulated with 5% CS for the indicated lengths of time, were separated on a 10% SDS-PAGE and subjected to immunoblot analysis using antibodies specific to p27Kip1. The blot was reprobed with GAPDH antibodies to monitor equal loading of proteins. (B) Lysates (50 µg) from pcDNA3-and Gα12QL-NIH3T3 cells that were serum starved for 24 hours and stimulated with 5% CS for 0, 4, and 8 hours were subjected to immunoblot analysis using Skp2-specific antibodies. The blot was probed with antibodies to GAPDH to monitor equal loading of proteins. (C) Lysates (50 µg) were prepared from 2 independent pcDNA3- and 3 Gα12QL-NIH3T3 cell clones that were serum starved for 24 hours and then stimulated with 5% CS for 0, 6, and 12 hours. Immunoblot analysis was carried out using Skp2-, p27Kip1-, or GAPDH-specific antibodies. The experiment was repeated 3 times; results from a typical experiment are presented. (D) NIH3T3 cells that were infected with 600 MOI of Gα12QL expressing adenovirus or empty pShuttle adenovirus were serum starved for 24 hours and then stimulated with 5% CS for the indicated time points (0, 6, and 12 hours). The blot prepared using the lysates (50 µg) from these cells were sequentially probed with antibodies specific to Skp2, p27Kip1, and Gα12.
Regulation of Skp2 by Gα12 is cell type independent
Gα12 has been shown to stimulate mitogenic signaling pathways in many different cell types including the astrocytoma cell line 1321N1.12,22 Therefore, we investigated whether activated Gα12 could induce changes in Skp2 levels in 1321N1 cells. Activated mutant of Gα12 was transiently expressed in 1321N1 cells by infecting them with adenoviral vectors encoding Gα12QL for 24 hours, and the expression levels of Skp2 were monitored using immunoblot analysis. Results indicated that the expression of activated mutant of Gα12 stimulated the proliferation of 1321N1 cells, confirming previous studies (Fig. 4A). When lysates from these cells were subjected to immunoblot analyses to monitor the expressions of Skp2 and p27Kip1, the results indicated that the cells expressing the activated mutant of Gα12 showed an upregulation of Skp2 along with a concomitant decrease in p27Kip1 (Fig. 4B). Together, these findings, for the first time, clearly establish the ability of Gα12QL to increase the expression levels of Skp2 in 2 distinctly different cell types.
Figure 4.
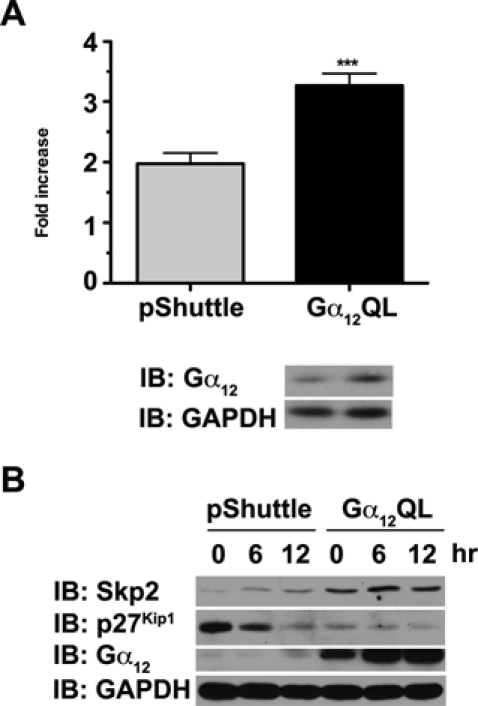
Gα12QL upregulates Skp2 in 1321N1 astrocytoma cells. (A) 1321N1 astrocytoma cells (5 × 103 cells/well) were infected with 600 MOI of either Gα12QL expressing adenovirus or empty pShuttle adenovirus. Proliferation of cells growing under low serum (2% FCS) was determined using an XTT-based cell proliferation kit as described in Materials and Methods. Fold increase was calculated by determining the increase in the absorbance of formazan dye at 492 nm over a period of 24 hours. The expression of Gα12QL was monitored by immunoblot analysis of lysates prepared from an identical set of these cells using Gα12-specific antibodies (lower panel). (B) 1321N1 astrocytoma cells infected with 600 MOI of Gα12QL expressing adenovirus or the empty pShuttle adenovirus were stimulated with 5% FBS for 0, 6, and 12 hours. Lysates (50 µg) from these cells were analyzed for the expression of Skp2, p27Kip1, and Gα12 by immunoblot analysis using respective antibodies. The blot was reprobed with antibodies to GAPDH to ascertain equal loading of proteins.
Gα12 mediates LPA-stimulated cell proliferation and Skp2 upregulation in 1321N astrocytoma cells
The findings that the mutationally activated Gα12 stimulated the upregulation of Skp2 raised an interesting question whether receptor activation of endogenous Gα12 would lead to similar changes in the levels of Skp2 and p27Kip1. This was analyzed with the use of LPA, an oncogenic lipid growth factor, which is known to stimulate cell proliferation via Gα12. After confirming that LPA (60 µM) stimulated the proliferation of 1321N1 cells (Fig. 5A), the expression profiles of Skp2 and p27Kip1 in LPA-stimulated cells were monitored by immunoblot analysis. Results indicated that LPA stimulated a strong increase in the levels of Skp2 along with a drastic decrease in p27Kip1 levels by 24 hours compared to the unstimulated controls (Fig. 5B). To test whether the effects of LPA on Skp2 and p27Kip1 involve Gα12, we analyzed the effect of expressing the dominant-negative mutant of Gα12 on LPA-stimulated changes in Skp2/ p27Kip1 levels. Based on the findings that the C-termini of G protein α-subunits are critical for binding to their cognate receptors,53 an adenoviral vector that encodes the C-terminal 11 amino acids of Gα12 (CT-12) has been successfully used in 1321N1 astrocytoma cells to inhibit Gα12-specific responses.54 Using this adenoviral construct, we analyzed the role of Gα12 in LPA-mediated cell proliferation and associated changes in Skp2-p27Kip1 levels. Results from these analyses indicated that the proliferation of 1321N1 cells stimulated by LPA could be effectively inhibited by the expression of the dominant-negative mutant of Gα12 (Fig. 5C). Similarly, the expression of CT-12 attenuated the LPA-stimulated upregulation of Skp2 along with the associated increase in p27Kip1 levels (Fig. 5D). Taken together, these results clearly establish that the LPA-stimulated increase in Skp2, associated downregulation of p27Kip1, and the resultant cell proliferation in 1321N1 cells require Gα12, the inhibition of which attenuates all of these events.
Figure 5.
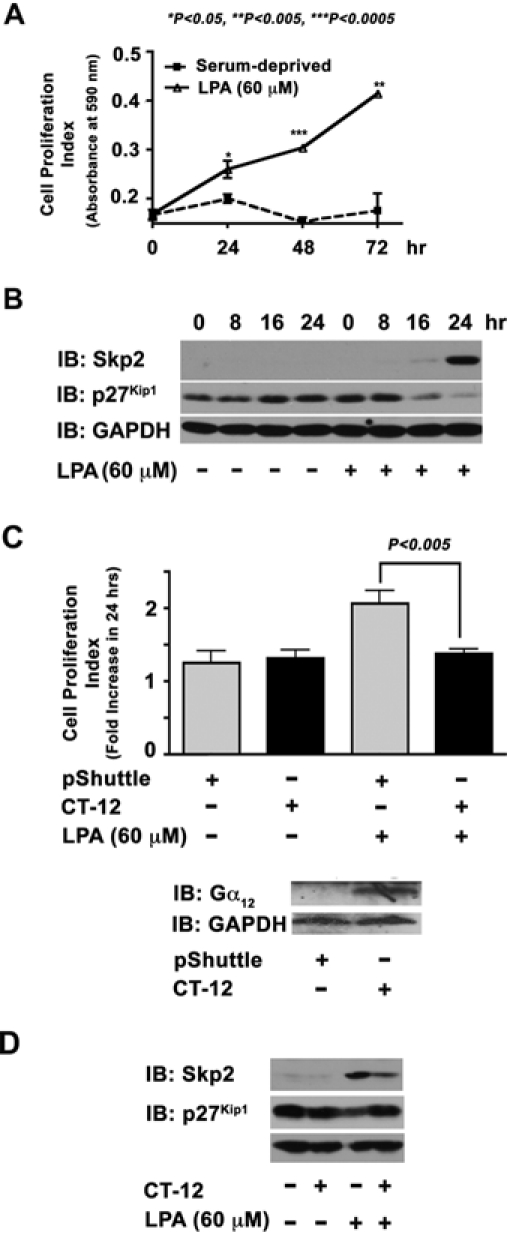
LPA upregulates Skp2 via Gα12. (A) Serum-deprived 1321N1 astrocytoma cells (5 × 103 cells/well) were stimulated with 60 µM of LPA for 24 hours along with the unstimulated control group. The increase in cell numbers at 24, 48, and 72 hours was monitored using crystal violet–based dye binding assay as described in Materials and Methods. A fold increase at 24 hours was calculated to the absorbance at 0 hours. (B) Serum-deprived 1321N1 astrocytoma cells were stimulated with 60 µM of LPA for varying lengths of time (0, 8, 16, and 24 hours) along with unstimulated controls. Lysates (50 µg) from these cells were analyzed for the expression levels of Skp2 and p27Kip1 by immunoblot analysis using respective antibodies. The blot was reprobed with GAPDH antibodies to monitor equal protein loading. (C) Quiescent 1321N1 astrocytoma cells (5 × 103 cells/well) infected with 150 MOI of CT-12 encoding adenovirus or empty pShuttle adenovirus (24 hours) were stimulated with LPA (60 µM) for 24 hours. Cell proliferation was measured using the XTT-based cell proliferation assay as described in Materials and Methods. Fold increase in cell proliferation was calculated by monitoring the increase in the absorbance of the formazan dye at 490 nm over a period of 24 hours. The expression of CT-12 was monitored by immunoblot analysis of lysates prepared from identical set-up experimental samples, using the Gα12 antibody that recognizes the CT-12 (lower panel). (D) Lysates (50 µg) from these cells were subjected to immunoblot analysis using Skp2, p27Kip1, or GAPDH. The results are from a representative set of experiments (n = 3).
Gα12 regulation of Skp2 is mediated by JNK
Being a critical player in G0-G1-S–phase transition, Skp2 is regulated by multiple signaling pathways involving ERKs,55 AKT,56 and FAK.57 However, it is significant to note here that Gα12-mediated mitogenic signaling critically involves the potent activation of JNKs.12,14-16 Therefore, we examined whether LPA-mediated increase in Skp2 levels involves JNK, the dominant signaling pathway utilized by Gα12. As shown in Figure 6A, inhibition of JNKs using SB600125 potently attenuated the increase in Skp2 levels stimulated by LPA in 1321N1 cells. Consistent with this observation, treating NIH3T3 cells expressing Gα12QL with SB600125 resulted in a similar inhibition in the upregulation of Skp2 compared to vehicle-treated Gα12QL-NIH3T3 cells (Fig. 6B). Previous studies have shown that Gα12-mediated activation of JNK involves the small GTPase CDC42.15 Consistent with this observation, inhibition of CDC42 by expressing CDC42 N17, a dominant-negative mutant of CDC42, attenuated Gα12QL-mediated upregulation of Skp2 levels in 1321N1 cells (Fig. 6C). These results pointing out a role for JNK in LPA- and Gα12-mediated upregulation of Skp2 are quite novel and hitherto unreported.
Figure 6.
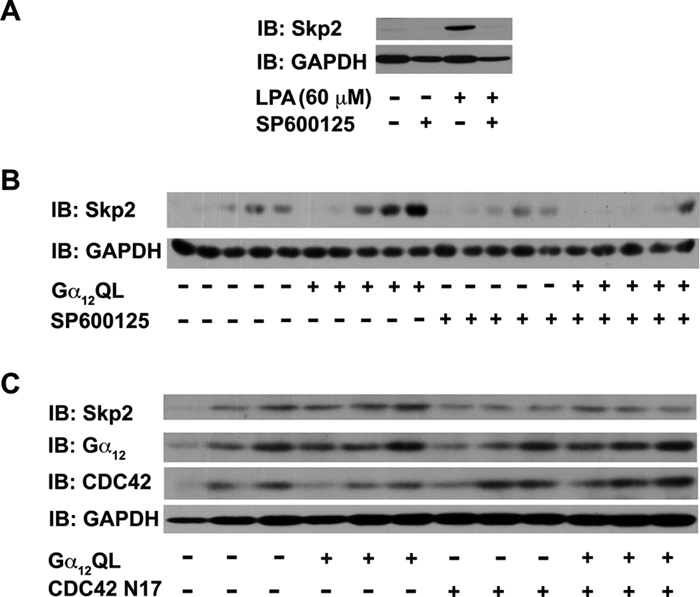
LPA-Gα12–mediated upregulation of Skp2 involves JNK. (A) 1321N1 astrocytoma cells (0.5 × 106 cells/60-mm plate) were treated with SP600125 (50 µM) or DMSO 2 hours prior to stimulation with 60 µM for 24 hours. Lysates (50 µg) from these cells were subjected to immunoblot analysis using antibodies to Skp2. The blot was stripped and reprobed with GAPDH antibodies to monitor equal loading of proteins. (B) Lysates (50 µg) from pcDNA3- and Gα12QL-NIH3T3 cells that were treated with SP600125 (50 µM) or DMSO 2 hours prior to stimulation with 5% CS for 0, 4, 8, 12, and 16 hours and analyzed for Skp2 levels by immunoblot analysis using Skp2 antibodies. The blot was reprobed with GAPDH antibodies to monitor equal loading of proteins. Results are from a typical experiment (n = 3). (C) 1321N1 astrocytoma cells (5 × 103 cells/well) were infected with 600 MOI of Gα12QL-encoding adenoviral vector singly or in combination with CDC42 N17-encoding adenoviral vectors along with appropriate control vectors. After 24 hours, the cells were stimulated with LPA (60 µM) for 24 hours. Lysates (50 µg) from these cells were subjected to immunoblot analysis using antibodies to Skp2, Gα12, CDC42, or GAPDH. The results are from a representative set of experiments (n = 3).
Discussion
Gα12, the α-subunit of G12 that constitutes the gep oncogene, has been shown to be the most potent α-subunit in promoting cell proliferation and neoplastic transformation.11-13,58,59 While studies describing the diversity of signals generated from Gα12 in inducing proliferation have been extensively documented, the mechanism by which mitogenic signaling cues from Gα12 are integrated in to the G1 to S phase of the cell cycle is relatively unknown. The results presented here identify for the first time a multipartite, but interrelated, mechanism(s) through which Gα12 communicates to cell cycle machinery. An overview of these pathways establishes a paradigm in which the multiple targets involved in Gα12-mediated mitogenic signaling converge towards the stimulation and potentiation of CDK activities. The first target of Gα12 signaling appears to be the upregulation of the levels of different G0/G1/S-phase stimulatory cyclins. The second set of targets involves the stimulation of the respective CDKs via p27Kip1 and Skp2. As can be seen in Figure 1, the expression of Gα12 promotes an increase in the levels of cyclin D1, A, and E. Although the mechanism by which Gα12 upregulates these levels was not investigated in the present study, the ability of Gα12 to stimulate Akt signaling in these cells as previously demonstrated by us21 and the subsequent Akt-mTOR–mediated stimulation of cellular translational apparatus appears to play a major role in this process (Ha and Dhanasekaran, unpublished data). Activation of the cyclin D1–CDK4 complex has been shown to play a critical role in inducing G1- to S-phase progression.26-30 Similarly, cyclin A–CDK2 has been established as a key promoter of the S phase of the cell cycle.25,26 By stimulating an increase in the levels of the respective cyclins and their associated CDKs, Gα12 signaling appears to target 2 complementary critical components of cell cycle machinery to ensure accelerated G1/S-phase progression. This is further substantiated by the observed increase in the phosphorylation of Ser795 of pRb in Gα12QL-expressing cells (Fig. 2D) since an increase in the activities of both cyclin D–CDK4 and cyclin A–CDK2 activity has been shown to contribute to the efficient phosphorylation of pRb, thereby relieving its inhibitory effect on S-phase entry. Thus, in addition to upregulating the levels of cyclins, Gα12 is also involved in stimulating the activities of the respective CDKs. The answer to the question as to how Gα12 stimulates the activities of CDK2/4 appears to lie, at least in part, within the ability of Gα12 to modulate the levels of p27Kip1 through Skp2.
It should be noted here that during G1/S-phase progression, p27Kip1 plays a dominant role in negatively regulating CDK2/4.26-28 In turn, p27Kip1 levels are dynamically regulated by the degradation of p27Kip1 by ubiquitin proteosome machinery involving Skp2.31-34 Our results presented here show that Gα12QL stimulates the upregulation of Skp2 along with a concomitant decrease in p27Kip1 levels (Fig. 3A and 3B). Thus, the increase in CDK activities seen in Gα12QL cells possibly results from the decreased levels of p27Kip1 brought out by the upregulated Skp2. Modulation of Skp2 levels appears to be one of the major mechanisms through which mitogens regulate the activities of CDKs in stimulating cell cycle progression.26-34 In this context, our observation that Gα12 signaling stimulates an increase in Skp2 levels along with the downregulation of p27Kip1 in 2 different cell types (Figs. 3 and 4) in which Gα12 promotes proliferation strongly identifies Skp2 as a major signaling target—in addition to cyclins—by which Gα12 promotes G1- to S-phase progression. It should be noted here that Skp2 is also involved in maintaining the optimal levels of cyclin E during G1/S transition.34 Thus, it is likely that the observed decrease in cyclin E levels upon growth stimulation (Fig. 1B) is in response to the elevated Skp2 levels in Gα12QL cells (Fig. 3). Given the primary role of Skp2 in regulating CKIs and other key players in cell cycle progression, the results presented here that Gα12 upregulates Skp2 are highly significant. In addition, considering the potent mitogenic activity of Gα12, our findings unravel Skp2 as the novel target in the cell cycle machinery through which Gα12 accelerates G1- to S-phase cell cycle progression and subsequent cell proliferation.
Our studies presented here also identify 2 newer correlates on LPA-mediated mitogenic signaling: 1) LPA stimulates proliferation along with promitogenic changes in Skp2/p27Kip1 levels via Gα12, and 2) LPA-Gα12–mediated upregulation of Skp2 involves JNK. While the increase in LPA levels and LPA-LPAR–mediated signaling have been implicated in the growth and progression of many tumors, the mechanisms through which it promotes such oncogenic activities are largely unknown. Our results demonstrating that LPA stimulates proliferation of 1321N1 astrocytoma cells along with the upregulation of Skp2 with the resultant downregulation of p27Kip1 that can promote cell cycle progression (Fig. 5A and 5B), both of which can be inhibited by the dominant-negative mutant of Gα12 (Fig. 5C and 5D), constitute the first demonstration of a cell cycle–based mechanism through which the oncogenic growth factor LPA and Gα12, the gep oncogene, stimulate the mitogenic pathway. Another interesting aspect of LPA-Gα12–mediated upregulation of Skp2 relates to our finding that JNK is involved in this process. The expression of Skp2 is known to be regulated by different kinases including ERK, AKT, and FAK.49,55-57 Until now, JNK is not known as one of the kinases involved in the regulation of Skp2. In this context, our results indicating a role for JNK in LPA- and Gα12-mediated upregulation of Skp2 (Fig. 6) are quite important as they identified a so far unknown JNK-dependent mechanism underlying the regulation of Skp2. Although the mechanism(s) by which JNK mediates an increase in Skp2 levels is not clear at present, further studies should clarify its precise role in Gα12-mediated upregulation of Skp2.
Taken together, our results support a model in which Gα12-mediated increase in the levels of stimulatory cyclins as well as the decrease in the levels of p27Kip1 through the upregulation of Skp2 cumulatively contribute to the overall increase in the activities of cyclin-CDK complexes, thereby accelerating G1/S progression. A simplistic working model integrating LPA-LPAR-Gα12 signaling to cell cycle machinery is presented in Figure 7. In light of the observations that 1) Skp2 levels are increased in glioblastomas46 and astrocytic gliomas,47 2) LPA is locally produced in many different cancers including glioblastomas,4,38 3) LPA-LPAR signaling can promote cell growth and cancer progression through the facilitation of autocrine and paracrine signaling loops,4,7,35-39 and 4) Gα12 transmits signals from LPA-LPAR to intracellular effector molecules through JNK,11,58 our identification of Skp2 as the target protein regulated by Gα12 is highly significant. Although finer details of this working model remain to be filled in, with the identification of Skp2 as a major protein being regulated by LPA-LPAR-Gα12 signaling, further studies should define the role of this signaling nexus as well as other critical players involved in this signaling pathway in the genesis and progression of various cancers.
Figure 7.
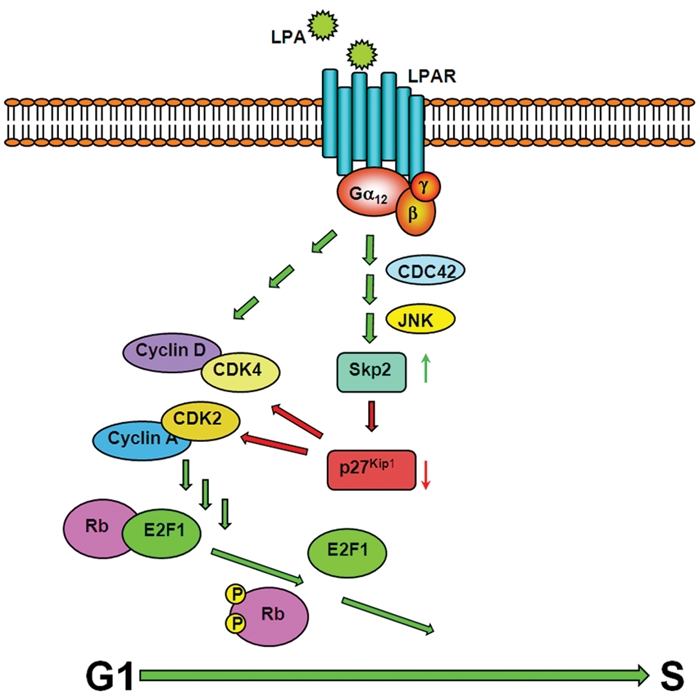
Schematic model for Gα12-mediated cell cycle progression. Receptor or mutationally activated Gα12 accelerates G1-S progression by upregulating the levels of specific cyclins and Skp2, both of which contribute to the increased activities of CDKs. While cyclins stimulate their respective CDKs, CDK activities could be further potentiated by Skp2, which downregulates the levels of p27Kip1, an inhibitory CKI, via the ubiquitin-proteosome degradation system. Gα12-mediated upregulation of Skp2 involves a novel signaling node involving CDC42 and JNK.
Materials and Methods
Cell lines
Parental NIH3T3, 1321N1 astrocytoma cells as well as previously described pcDNA3-NIH3T3 and Gα12Q229L-NIH3T3 cell lines were maintained as described earlier21 in Dulbecco’s modified Eagle’s medium (DMEM) (Cellgro, Mediatech Inc., Manassas, VA) containing 50 U/mL penicillin, 50 µg/mL streptomycin, and 5% calf serum (NIH3T3 cells) or 5% fetal bovine serum (1321N1 cells) at 37°C in a 5% CO2 incubator. The sera were obtained from Life Technologies Inc. (Gaithersburg, MD).
Adenoviral constructs
Construction of adenoviral vector–expressing Gα12QL was carried out using the cDNA insert encoding Gα12QL (1,800 bp) excised from the pcDNA3-Gα12QL vector14 using the restriction nucleases KpnI and XbaI. The cohesive ends were blunted and cloned in the EcoRV site of the pShuttle-IRES-GFP2 vector (Stratagene, La Jolla, CA). The resultant plasmid was linearized using PmeI before it was transformed into Escherichia coli BJ5183-AD-1 cells (pretransformed with pAdEasy-1 plasmid) in which the homologous recombination event with the pAdEasy-1 plasmid containing adenoviral backbone could take place. The recombinant clones were selected by analysis of the PacI-digested DNA from these clones. The positive recombinant DNA was amplified by transforming it on a suitable E. coli strain. The recombinant DNA was cut with PacI and then purified before transfection onto AD293 cells for the virus production. Isolation of the virus was carried out following the standard freeze-thaw procedure. If needed, further amplification of the virus was carried out using AD293 cells. These recombinant adenoviruses were titrated by plaque assay before target cells were infected. Adenoviral vectors expressing the dominant-negative mutant of CDC42 were constructed following identical procedures, using the Bam I–Xho1 insert of CDC42 N17.61 Recombinant adenoviral vector expressing the dominant-negative mutant of Gα12, CT-12,54 was kindly provided by Dr. Yoh Takuwa (Kanazawa University Graduate School of Medicine, Kanazawa, Japan). This virus was amplified by infecting AD293 cells. These recombinant adenoviruses were titrated by plaque assay before using them for infecting the target cells.
Determining the optimal multiplicity of infection (MOI)
Based on the titer obtained from the plaque assay, the volume of virus that corresponds to the particular MOI to be infected was determined. An equal number of NIH3T3 and 1321N1 astrocytoma cells (1 × 106 cells/plate) were plated on 100-mm culture plates and infected with different MOIs (0, 25, 100, 300, and 600) of the adenovirus. Media containing serum were removed from the plates and washed with PBS and then replaced with 2 mL of serum-free medium containing the adenovirus. The plates were kept in the rocker inside the incubator for 2 hours for the cells to get infected. Following this, the plates were replaced with growth medium containing serum and incubated for 24 hours. Lysates were prepared from these cells and analyzed for the expression of proteins encoded by the adenovirus. Based on the expression profile, the optimum MOI to be used for the experiment was determined. The optimal MOI was deduced as the one at which the transgene showed the highest expression with little or no cytotoxic effects. Based on these results, an MOI of 600 was used for both NIH3T3 cells and 1321N1 astrocytoma cells. In experiments in which low serum growth conditions were used, the MOI was reduced to 150.
Cell proliferation assay
The growth profile of 1321N cells in response to LPA was monitored using an assay based on crystal violet staining of live cells following previously published procedures.60 An equal number of 1321N1 cells (5 × 103) grown on 12-well plates were serum starved for 24 hours, after which they were stimulated with 60 µM of LPA for 24, 48, and 72 hours, respectively. At specific time points, cells were fixed using 10% formalin (Fisher Scientific, Pittsburgh, PA) dissolved in PBS for 10 minutes, following which the fixed samples were stained with 0.1% crystal violet (Sigma-Aldrich, St. Louis, MO) for 6 hours. The samples were then washed extensively to remove excess dye and dried overnight. The cell-associated dye was then extracted by incubation with 1 mL acetic acid (Fisher Scientific) for 60 seconds. The optical density of each sample was quantified at 590 nm. Proliferation of 1321N1 astrocytoma cells transiently infected with adenovirus encoding CT-12 or empty pShuttle adenovirus was monitored using the XTT [2,3-bis (2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino) carbonyl]-2H-tetrazolium hydroxide]–based cell proliferation assay kit (Roche Diagnostics, Indianapolis, IN). An equal number of cells (5 × 103 cells/well) grown in 96-well plates were serum starved for 24 hours. These cells were infected with an MOI of 150 of either pShuttle or CT-12 adenovirus in freshly replaced serum starvation medium supplemented with 0.2% fetal bovine serum followed by stimulation with LPA (60 µM) for 24 hours. The absorbance of formazan dye at 490 nm was monitored as an index of actively growing cells using a microplate reader.
Immunoblot analysis
Immunoblot analyses were carried out following the previously published procedures.21 Antibodies to Gα12 (sc-409), cyclin A (sc-596), cyclin D1 (sc-8396), cyclin E (sc-481), CDK4 (sc-601), CDK2 (sc-163), p27Kip1 (sc-528), Skp2 (sc-7164), JNK1 (sc-474), and JNK2 (scv-827) were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Antibodies to phospho-pRb (Ser795; #9301), control Rb (#9302), and phospho-JNK (#9251) were purchased from Cell Signaling Technology Inc. (Danvers, MA). Antibodies to GAPDH (#4300) were purchased from Ambion Inc. (Austin, TX). Peroxidase-conjugated anti-mouse IgG and anti-rabbit IgG were purchased from Amersham Biosciences UK Ltd. (Little Chalfont, Buckinghamshire, UK) and Promega (Madison, WI), respectively.
Immune complex kinase assays
Immune complex kinase assays to monitor the kinase activities of CDK2 and CDK4 were carried out using previously published procedures with appropriate modifications.45 Cells were lysed with kinase lysis buffer (200 µL) containing 20 mM HEPES (pH 7.4), 50 mM β-glycerophosphate, 0.5% Triton X-100, 2 mM MgCl2, 1 mM EGTA, 1 mM dithiothreitol, 2 µg/mL leupeptin, 4 µg/mL aprotinin, 100 µm PMSF, and 1 mM benzamidine. CDK2 or CDK4 in the lysates was immunoprecipitated by incubating 200 µg of protein lysate with the polyclonal antibodies specific to CDK2 (sc-163; 2 µg) or CDK4 (sc-601; 2 µg), respectively, for 1 hour. This was followed by an additional incubation with 20 µL of protein A–Sepharose (Amersham Biosciences Corp., Piscataway, NJ) for 1 hour. The immune complex–bound protein A–Sepharose beads were washed 3 times with lysis buffer. CDK2 immune complex assay was carried out with protein A–Sepharose–bound CDK2 using 5 µg of histone H1 (Roche Diagnostics). CDK4 immune complex assay was carried out with protein A–Sepharose–bound CDK2 using 5 µg of GST-Rb fusion protein (a generous gift from Dr. Fang Liu, Rutgers University, New Brunswick, NJ). The kinase reactions were carried out by resuspending the protein A–Sepharose beads with bound immune complexes containing CDK2 or CDK4 along with appropriate substrates in 40 µL of kinase reaction buffer containing 20 µM [γ-32P] ATP (5,000 cpm/pmol) and incubating at 30°C for 30 minutes. The reaction was stopped by the addition of Laemmli’s sample buffer followed by boiling the sample for 3 minutes. The proteins were resolved by 12% SDS-PAGE, and the dried gels were analyzed by autoradiography.
Acknowledgments
The generous gift of CT-12 adenoviral constructs expressing the dominant-negative mutant of Gα12 by Dr. Y. Takuwa (Kanazawa University Graduate School of Medicine, Kanazawa, Japan) and the critical reading of the paper by Dr. Lakshmi Varadarajalu are gratefully acknowledged.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by grants from the National Institutes of Health [CA123233] and World Class University project funded by Ministry of Education, Science and Technology Development, South Korea [No. R32-2008-000-10098-0].
References
- 1. Li S, Huang S, Peng SB. Overexpression of G protein-coupled receptors in cancer cells: involvement in tumor progression. Int J Oncol. 2005;27:1329-39 [PubMed] [Google Scholar]
- 2. Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7:79-94 [DOI] [PubMed] [Google Scholar]
- 3. Bhola NE, Grandis JR. Crosstalk between G-protein-coupled receptors and epidermal growth factor receptor in cancer. Front Biosci. 2008;13:1857-65 [DOI] [PubMed] [Google Scholar]
- 4. Liu S, Murph M, Panupinthu N, Mills GB. ATX-LPA receptor axis in inflammation and cancer. Cell Cycle. 2009;8:3695-701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arora P, Ricks TK, Trejo J. Protease-activated receptor signalling, endocytic sorting and dysregulation in cancer. J Cell Sci. 2007;120:921-8 [DOI] [PubMed] [Google Scholar]
- 6. Mazzuco TL, Chabre O, Feige JJ, Thomas M. Aberrant GPCR expression is a sufficient genetic event to trigger adrenocortical tumorigenesis. Mol Cell Endocrinol. 2007;265-266:23-8 [DOI] [PubMed] [Google Scholar]
- 7. Mills GB, Eder A, Fang X, et al. Critical role of lysophospholipids in the pathophysiology, diagnosis, and management of ovarian cancer. Cancer Treat Res. 2002;107:259-83 [DOI] [PubMed] [Google Scholar]
- 8. Aly A, Shulkes A, Baldwin GS. Gastrins, cholecystokinins and gastrointestinal cancer. Biochim Biophys Acta. 2004;1704:1-10 [DOI] [PubMed] [Google Scholar]
- 9. Yowell CW, Daaka Y. G protein-coupled receptors provide survival signals in prostate cancer. Clin Prostate Cancer. 2002;1:177-81 [DOI] [PubMed] [Google Scholar]
- 10. Hull MA, Ko SC, Hawcroft G. Prostaglandin EP receptors: targets for treatment and prevention of colorectal cancer? Mol Cancer Ther. 2004;3:1031-9 [PubMed] [Google Scholar]
- 11. Juneja J, Casey PJ. Role of G12 proteins in oncogenesis and metastasis. Br J Pharmacol. 2009;158:32-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Radhika V, Dhanasekaran N. Transforming G proteins. Oncogene. 2001;20:1607-14 [DOI] [PubMed] [Google Scholar]
- 13. Grzelinski M, Pinkenburg O, Büch T, et al. Critical role of Gα12 and Gα13 for human small cell lung cancer cell proliferation in vitro and tumor growth in vivo. Clin Cancer Res. 2010;16:1402-15 [DOI] [PubMed] [Google Scholar]
- 14. Prasad MV, Dermott JM, Heasley LE, Johnson GL, Dhanasekaran N. Activation of Jun kinase/stress-activated protein kinase by GTPase-deficient mutants of Gα12 and Gα13. J Biol Chem. 1995;270:18655-9 [DOI] [PubMed] [Google Scholar]
- 15. Voyno-Yasenetskaya TA, Faure MP, Ahn NG, Bourne HR. Gα12 and Gα13 regulate extracellular signal-regulated kinase and c-Jun kinase pathways by different mechanisms in COS-7 cells. J Biol Chem. 1996;271:21081-7 [DOI] [PubMed] [Google Scholar]
- 16. Collins LR, Minden A, Karin M, Brown JH. Gα12 stimulates c-Jun NH2-terminal kinase through the small G proteins Ras and Rac. J Biol Chem. 1996;271:17349-53 [DOI] [PubMed] [Google Scholar]
- 17. Jiang Y, Ma W, Wan Y, Kozasa T, Hattori S, Huang XY. The G protein Gα12 stimulates Bruton’s tyrosine kinase and a rasGAP through a conserved PH/BM domain. Nature. 1998;395:808-13 [DOI] [PubMed] [Google Scholar]
- 18. Dermott JM, Reddy MR, Onesime D, Reddy EP, Dhanasekaran N. Oncogenic mutant of Gα12 stimulates cell proliferation through cycloxygenase-2 signaling pathway. Oncogene. 1999;18:7185-9 [DOI] [PubMed] [Google Scholar]
- 19. Dermott JM, Ha JH, Lee CH, Dhanasekaran N. Differential regulation of Jun N-terminal kinase and p38MAP kinase by Gα12. Oncogene. 2004;23:226-32 [DOI] [PubMed] [Google Scholar]
- 20. Kumar RN, Shore SK, Dhanasekaran N. Neoplastic transformation by the gep oncogene, Gα12, involves signaling by STAT3. Oncogene. 2006;25:899-906 [DOI] [PubMed] [Google Scholar]
- 21. Kumar RN, Ha JH, Radhakrishnan R, Dhanasekaran DN. Transactivation of platelet-derived growth factor receptor α by the GTPase-deficient activated mutant of Gα12. Mol Cell Biol. 2006;26:50-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aragay AM, Collins LR, Post GR, et al. G12 requirement for thrombin-stimulated gene expression and DNA synthesis in 1321N1 astrocytoma cells. J Biol Chem. 1995;270:20073-7 [DOI] [PubMed] [Google Scholar]
- 23. Kumar RN, Radhakrishnan R, Ha JH, Dhanasekaran N. Proteome analysis of NIH3T3 cells transformed by activated Gα12: regulation of leukemia-associated protein SET. J Proteome Res. 2004;3:1177-83 [DOI] [PubMed] [Google Scholar]
- 24. Mitsui H, Takuwa N, Kurokawa K, Exton JH, Takuwa Y. Dependence of activated Gα12-induced G1 to S phase cell cycle progression on both Ras/mitogen-activated protein kinase and Ras/Rac1/Jun N-terminal kinase cascades in NIH3T3 fibroblasts. J Biol Chem. 1997;272:4904-10 [DOI] [PubMed] [Google Scholar]
- 25. Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79:551-5 [DOI] [PubMed] [Google Scholar]
- 26. Grana X, Reddy EP. Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene. 1995;11:211-9 [PubMed] [Google Scholar]
- 27. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153-66 [DOI] [PubMed] [Google Scholar]
- 28. Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008;14:159-69 [DOI] [PubMed] [Google Scholar]
- 29. Grana X, Garriga J, Mayol X. Role of the retinoblastoma protein family, pRB, p107 and p130 in the negative control of cell growth. Oncogene. 1998;17:3365-83 [DOI] [PubMed] [Google Scholar]
- 30. Adams PD. Regulation of the retinoblastoma tumor suppressor protein by cyclin/cdks. Biochim Biophys Acta. 2001;1471:M123-33 [DOI] [PubMed] [Google Scholar]
- 31. Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol. 1999;9:661-4 [DOI] [PubMed] [Google Scholar]
- 32. Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193-9 [DOI] [PubMed] [Google Scholar]
- 33. Pegano M. Control of DNA synthesis and mitosis by the Skp2-p27-Cdk1/2 axis. Mol Cell. 2004;14:414-6 [DOI] [PubMed] [Google Scholar]
- 34. Nakayama K-I, Hatakeyama S, Nakayama K. Regulation of the cell cycle at the G1-S transition by proteolysis of cyclin E and p27Kip1. Bioche Biophys Res Commun. 2001;282:853-60 [DOI] [PubMed] [Google Scholar]
- 35. Panupinthu N, Lee HY, Mills GB. Lysophosphatidic acid production and action: critical new players in breast cancer initiation and progression. Br J Cancer. 2010;102:941-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Daaka Y. Mitogenic action of LPA in prostate. Biochim Biophys Acta. 2002;1582:265-9 [DOI] [PubMed] [Google Scholar]
- 37. Yamada T, Sato K, Komachi M, et al. Lysophosphatidic acid (LPA) in malignant ascites stimulates motility of human pancreatic cancer cells through LPA1. J Biol Chem. 2004;279:6595-605 [DOI] [PubMed] [Google Scholar]
- 38. Kishi Y, Okudaira S, Tanaka M, et al. Autotaxin is overexpressed in glioblastoma multiforme and contributes to cell motility of glioblastoma by converting lysophosphatidy-lcholine to lysophosphatidic acid. J Biol Chem. 2006;281:17492-500 [DOI] [PubMed] [Google Scholar]
- 39. Latres E, Chiarle R, Schulman BA, et al. Role of the F-box protein Skp2 in lymphomagenesis. Proc Natl Acad Sci U S A. 2001;98:2515-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gstaiger M, Jordan R, Lim M, et al. Skp2 is oncogenic and overexpressed in human cancers. Proc Natl Acad Sci U S A. 2001;98:5043-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hershko DD. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer. 2008;112:1415-24 [DOI] [PubMed] [Google Scholar]
- 42. Signoretti S, Di Marcotullio L, Richardson A, et al. Oncogenic role of the ubiquitin ligase subunit Skp2 in human breast cancer. J Clin Invest. 2002;110:633-41 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43. Yang G, Ayala G, De Marzo A, et al. Elevated Skp2 protein expression in human prostate cancer: association with loss of the cyclin-dependent kinase inhibitor p27 and PTEN and with reduced recurrence-free survival. Clin Cancer Res. 2002;8:3419-26 [PubMed] [Google Scholar]
- 44. Einama T, Kagata Y, Tsuda H, et al. High-level Skp2 expression in pancreatic ductal adenocarcinoma: correlation with the extent of lymph node metastasis, higher histological grade, and poorer patient outcome. Pancreas. 2006;32:376-81 [DOI] [PubMed] [Google Scholar]
- 45. Shigemasa K, Gu L, O’Brien TJ, Ohama K. Skp2 overexpression is a prognostic factor in patients with ovarian adenocarcinoma. Clin Cancer Res. 2003;9:1756-63 [PubMed] [Google Scholar]
- 46. Saigusa K, Hashimoto N, Tsuda H, et al. Overexpressed Skp2 within 5p amplification detected by array-based comparative genomic hybridization is associated with poor prognosis of glioblastomas. Cancer Sci. 2005;96:676-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schiffer D, Cavalla P, Fiano V, Ghimenti C, Piva R. Inverse relationship between p27/Kip.1 and the F-box protein Skp2 in human astrocytic gliomas by immunohistochemistry and Western blot. Neurosci Lett. 2002;328:125-8 [DOI] [PubMed] [Google Scholar]
- 48. Kudo Y, Kitajima S, Sato S, Miyauchi M, Ogawa I, Takata T. High expression of S-phase kinase-interacting protein 2, human F-box protein, correlates with poor prognosis in oral squamous cell carcinomas. Cancer Res. 2001;61:7044-7 [PubMed] [Google Scholar]
- 49. Modiano JF, Domenico J, Szepesi A, Lucas JJ, Gelfand EW. Differential requirements for interleukin-2 distinguish the expression and activity of the cyclin-dependent kinases Cdk4 and Cdk2 in human T cells. J Biol Chem. 1994;269:32972-8 [PubMed] [Google Scholar]
- 50. Connell-Crowley L, Harper JW, Goodrich DW. Cyclin D1/Cdk4 regulates retinoblastoma protein-mediated cell cycle arrest by site-specific phosphorylation. Mol Biol Cell. 1997;8:287-301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J Biol Chem. 1997;272:12738-46 [DOI] [PubMed] [Google Scholar]
- 52. Gao N, Flynn DC, Zhang Z, et al. G cell cycle progression and the expression of G1 cyclins are regulated by PI3K/AKT/mTOR/p70S6K1 signaling in human ovarian cancer cells. Am J Physiol Cell Physiol. 2004;287:C281-91 [DOI] [PubMed] [Google Scholar]
- 53. Gilchrist A, Vanhauwe JF, Li A, Thomas TO, Voyno-Yasenetskaya T, Hamm HE. Gα minigenes expressing C-terminal peptides serve as specific inhibitors of thrombin mediated endothelial activation. J Biol Chem. 2001;276:25672-9 [DOI] [PubMed] [Google Scholar]
- 54. Sugimoto N, Takuwa N, Okamoto H, Sakurada S, Takuwa Y. Inhibitory and stimulatory regulation of Rac and cell motility by the G12/13-Rho and Gi pathways integrated downstream of a single G protein-coupled sphingosine-1-phosphate receptor isoform. Mol Cell Biol. 2003;23:1534-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Villanueva J, Yung Y, Walker JL, Assoian RK. ERK activity and G1 phase progression: identifying dispensable versus essential activities and primary versus secondary targets. Mol Biol Cell. 2007;18:1457-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lin HK, Wang G, Chen Z, et al. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by Akt/PKB. Nat Cell Biol. 2009;11:420-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bryant P, Zheng Q, Pumiglia K. Focal adhesion kinase controls cellular levels of p27/Kip1 and p21/Cip1 through Skp2-dependent and -independent mechanisms. Mol Cell Biol. 2006;26:4201-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Radhika V, Ha JH, Jayaraman M, Tsim ST, Dhanasekaran N. Mitogenic signaling by lysophosphatidic acid (LPA) involves Gα12. Oncogene. 2005;24:4597-603 [DOI] [PubMed] [Google Scholar]
- 59. Goldsmith ZG, Dhanasekaran DN. G protein regulation of MAPK networks. Oncogene. 2007;26:3122-42 [DOI] [PubMed] [Google Scholar]
- 60. Orsulic S, Li Y, Soslow RA, Vitale-Cross LA, Gutkind JS, Varmus HE. Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell. 2002;1:53-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wadsworth SJ, Gebauer G, van Rossum GDV, Dhanasekaran N. Ras-dependent signaling by the GTPase-deficient mutant of Gα12. J Biol Chem. 1997;272:28829-32 [DOI] [PubMed] [Google Scholar]