

Abstract
Microwave-assisted synthesis of 2-oxazolines from carboxylic acids using the open vessel technique is described. This efficient method involves direct condensation of carboxylic acids with excess 2-amino-2-methyl-1-propanol at 170 °C to give the corresponding 2-oxazolines in moderate to excellent yields.
Keywords: Oxazoline, Open vessel mode, Microwave, Carboxylic acid
1. Introduction
Oxazolines have been of great interest1 due to their versatility as protecting groups,2 as chiral auxiliaries in asymmetric synthesis,3 and as ligands for asymmetric catalysis.4 Various methods have been developed for the preparation of 2-oxazolines from carboxylic acids. One early method (see Scheme 1) used conventional heating of equimolar concentrations of carboxylic acids 1 and 2-aminoethanol (2a,R1 = R2 = H) at 200 °C followed by dehydration of N-acyl-2-aminoethanols 3a using phosphorous pentoxide to generate oxazolines 4a;5 thionyl chloride and other reagents have also been used.1 A recent method used 2-chloro-4,6-dimethoxy-1,3,5-triazine and N-methylmorpholine to generate a complex (1 equiv), which upon treatment with carboxylic acids 1 and 3 equiv of 2-amino-2-methyl-1-propanol (2b, R1 = R2 = CH3) at room temperature, gave the corresponding oxazolines 4b.6 More recently, Kangani and Kelley7 reported using bis(2-methoxy-ethyl)aminosulfur trifluoride (Deoxo-Fluor, 2.2 equiv) with carboxylic acids 1 in the one-pot synthesis of amides from secondary amines or 2-oxazolines 4b from 1.8 equiv of 2-amino-2-methyl-1-propanol (2b) at 0 °C. This method reportedly worked better than the triazine complex6 coupling for aliphatic and benzoic acids. We were unable to prepare the corresponding 2-oxazoline of 3,5-dimethoxyphenylacetic acid by this method, however. We now report a general and efficient method which is a variation of the direct synthesis8–10 of 2-oxazolines from carboxylic acids.
Scheme 1.
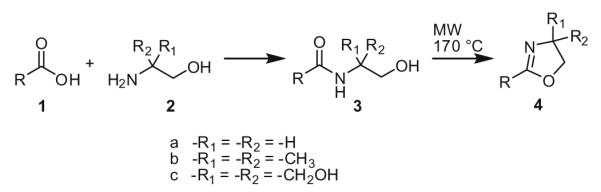
Reaction of carboxylic acids 1 with 2-aminoalcohols 2 to give 2-oxazolines 4.
2. Results and discussion
The use of microwave heating in organic chemistry has become a valuable tool in drug discovery to carry out reactions more efficiently than the use of conventional heating by offering improved reaction yields, decreased reaction times, and solvent-free conditions.11,12 During the course of our work in the characterization of the endocannabinoid metabolome, we recently converted the carboxylic acid moieties of various fatty acids associated with endocannabinoids to 4,4-bis(hydroxymethyl)-2-oxazolines 4c (R1 = R2 = CH2OH) using tris(hydroxymethyl)amino-methane 2c (R1 = R2 = CH2OH) with microwave heating.10 We have now extended this methodology by transforming various carboxylic acids into their corresponding 2-substituted 4,4-di-methyl-2-oxazolines 4b (R1 = R2 = CH3) with microwave heating using 2-amino-2-methyl-1-propanol 2b (see Table 1). Various carboxylic acids and microwave heating conditions were studied for this transformation. Microwave heating in closed vessels led to intermediate amide 3b formation, but not to cyclization. However, by performing the reaction in an open vessel mode, which allows for the dehydration of the amide 3b intermediates, the 2-oxazoline 4b products were prepared in moderate to high yields.
Table 1.
Synthesis of 2-oxazolines 4b from carboxylic acids 1 using open vessel mode microwave heating
| Entry | Acid | Oxazoline product | Reaction conditions | % Yield |
|---|---|---|---|---|
| 1 |

|

|
170 °C, 15 min | 85 |
| 2 |

|

|
170 °C, 15 min | 78 |
| 3 |

|

|
170 °C, 15 min | 79 |
| 4 |

|

|
170 °C, 15 min | 83 |
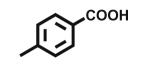
| 5 |
|

|
170 °C, 15 min | 81 |
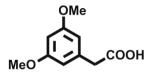
| 6 |
|

|
170 °C, 15 min | 73 |
| 7 |

|

|
170 °C, 15 min | 71 |
| 8 |

|

|
170 °C, 15 min | 80 |
| 9 |

|

|
170 °C, 25 min | 78 |
| 10 |

|

|
170 °C, 25 min | 82 |
| 11 |
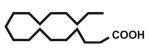
|
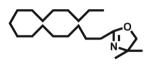
|
170 °C, 40 min | 73 |
| 12 |
|

|
170 °C, 25 min | 78 |
Open vessel mode microwave heating of carboxylic acids in excess 2-amino-2-methyl-1-propanol 2b without solvent at 150 °C for 5 min led to intermediate amides 3b with very little oxazoline 4b formation. The optimized reaction conditions required heating at 170 °C for 15 min, except for entries 9, 10, and 12 which required 25 min and entry 11 that required 40 min for oxazoline formation. Longer heating tends to result in more decomposition byproducts. Yields of 2-phenyl (entry 1, 85%; lit.6 78%; lit.7 97%; lit.9 90%) and aliphatic (entry 11, 73%; lit.6 82%; lit.7 99%; lit.9 84%) 2-oxazolines were comparable to those obtained through the mild methods using coupling and dehydrating reagents or zinc oxide-assisted microwave conditions for these thermally stable carboxylic acids. In addition to aliphatic and aromatic carboxylic acids, entries 3, 4, and 12 demonstrate the application of this open vessel methodology to unsaturated carboxylic acids. The method was also successfully applied to dicarboxylic acids as demonstrated by entries 9 and 10. We were unable to prepare the corresponding 2-oxazoline 4b of 3,5-dimethoxyphenylacetic acid using Deoxo-Fluor, but obtained a 73% isolated yield (entry 6) using open vessel microwave heating with 6 equiv of 2-amino-2-methyl-1-propanol 2b at 170 °C for 15 min. A comparable 68% isolated yield of this oxazoline 4b was obtained from 3,5-dimethoxyphenylacetic acid using 2 equiv of 2-aminoalcohol 2b with longer heating (40 min) conditions, and when using 1 equiv of 2-aminoalcohol 2b , a 60% isolated yield was obtained after 40 min. Thus, using an excess of 2-aminoalcohol 2b, when possible, results in higher conversion to 2-oxazoline, less amide intermediate and other byproduct impurity, and less product decomposition.
3. Conclusions
The reported method proved to be simple and efficient for the conversion of carboxylic acids into 2-oxazolines in good yields. Unlike some of the previously reported methods, our microwave open vessel technique requires only the reactants (solvent-free), relatively short reaction times, and simple chromatographic purifications.
4. Typical procedure
The 3,5-dimethoxyphenyl acetic acid (500 mg, 2.55 mmol) was mixed with 2-methyl-2-aminopropanol 2b (1.36 g, 15.3 mmol, 6 equiv) in a CEM microwave vessel. The resulting mixture was irradiated using the open vessel mode at 170 °C for 15 min. The reaction mixture was quenched with water (4 mL) and extracted with ethyl acetate (3 × 10 mL). The combined organic extracts were dried over magnesium sulfate, and concentrated by rotoeva-poration under vacuum to give the crude product, which was then chromatographed on silica gel using a Biotage SP2 eluting with acetone/hexane (0–30% acetone gradient) to give the corresponding 2-(3,5-dimethoxyphenylmethyl)-2-oxazoline 4b (464 mg, 1.86 mmol, 73% yield, entry 6) as a faint yellow liquid product that was homogeneous by TLC (40:60 acetone/hexane, Rf 0.65).
Supplementary Material
Acknowledgments
This work was supported by Grants DA-7215, DA-3801, DA-152, and DA-9158 from the National Institute on Drug Abuse. We thank Lakshmipathi Pandarinathan for valuable discussions.
Footnotes
Supplementary data Supplementary data (characterizations and 1H NMR spectra of all 2-oxazoline 4b products) associated with this article can be found, in the online version, at doi:10.1016/j.tetlet.2009.07.079.
References and notes
- 1.Gant TG, Meyers AI. Tetrahedron. 1994;50:2297. [Google Scholar]
- 2.Meyers AI, Temple DL, Haidukewych D, Mihelich ED. J. Org. Chem. 1974;39:2787. [Google Scholar]
- 3.Meyers AI. Acc. Chem. Res. 1978;11:375. [Google Scholar]
- 4.Hoarau O, Haddou-Ait H, Castro M, Balavoine GGA. Tetrahedron: Asymmetry. 1997;8:3755. [Google Scholar]
- 5.Wenker H. J. Am. Chem. Soc. 1935;57:1079. [Google Scholar]
- 6.Bandgar BP, Pandit SS. Tetrahedron Lett. 2003;44:2331. [Google Scholar]
- 7.Kangani CO, Kelly DE. Tetrahedron Lett. 2005;46:8917. doi: 10.1016/j.tetlet.2005.10.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang JY, Yu QT, Liu BN, Huang ZH. Biomed. Environ. Mass Spectrom. 1988;15:33. [Google Scholar]
- 9.García-Tellado F, Loupy A, Petit A, Marrero-Terrero AL. Eur. J. Org. Chem. 2003;22:4387. [Google Scholar]
- 10.Williams J, Pandarinathan L, Wood J, Vouros P, Makriyannis A. AAPS J. 2006;8:E655. doi: 10.1208/aapsj080474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wathey B, Tierney J, Lidstrom P, Westmen J. Drug Discovery Today. 2002;7:373. doi: 10.1016/s1359-6446(02)02178-5. [DOI] [PubMed] [Google Scholar]
- 12.Kappe CO. Angew. Chem., Int. Ed. 2004;43:6250. doi: 10.1002/anie.200400655. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.