

Abstract
Introduction
Microdialysis is an important in vivo sampling technique, useful in the assay of extracellular tissue fluid. The technique has both pre-clinical and clinical applications but is most widely used in neuroscience. The in vivo microdialysis technique allows measurement of neurotransmitters such as acetycholine (ACh), the biogenic amines including dopamine (DA), norepinephrine (NE) and serotonin (5-HT), amino acids such as glutamate (Glu) and gamma aminobutyric acid (GABA), as well as the metabolites of the aforementioned neurotransmitters, and neuropeptides in neuronal extracellular fluid in discrete brain regions of laboratory animals such as rodents and non-human primates.
Areas covered
In this review we present a brief overview of the principles and procedures related to in vivo microdialysis and detail the use of this technique in the pre-clinical measurement of drugs designed to be used in the treatment of chemical addiction, neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and as well as psychiatric disorders such as attention-deficit/hyperactivity disorder (ADHD) and schizophrenia. This review offers insight into the tremendous utility and versatility of this technique in pursuing neuropharmacological investigations as well its significant potential in rational drug discovery.
Expert opinion
In vivo microdialysis is an extremely versatile technique, routinely used in the neuropharmacological investigation of drugs used for the treatment of neurological disorders. This technique has been a boon in the elucidation of the neurochemical profile and mechanism of action of several classes of drugs especially their effects on neurotransmitter systems. The exploitation and development of this technique for drug discovery in the near future will enable investigational new drug candidates to be rapidly moved into the clinical trial stages and to market thus providing new successful therapies for neurological diseases that are currently in demand.
1 INTRODUCTION
1.1 Background and historical perspective
Microdialysis is a relatively novel sampling technique which has been extensively used primarily for the characterization and assessment of the neuropharmacodynamic profile of drugs in in vivo rodent as well as non-human primate studies [1]. In recent years, this technique has found extensive application in neurotransmitter research, in particular in the investigation of drug effects on monoamine and amino acid neurotransmitters [2]. Although microdialysis is used as a sampling technique in several organ systems such as blood, eye, liver, muscle etc., it owes its development to attempts at extracellular fluid measurements in the brain; an application for which microdialysis has found most extensive use. The in vivo microdialysis technique essentially began with the push-pull method in the 1960s which examined the possibility of using a semi-permeable membrane to sample free amino acids and other electrolytes in neuronal extracellular fluid. The technique was further improved by the development of the dialysis bag – ‘the dialytrode’ – as a means to collect the dialysis sample. However, the successful use and current popularity of the in vivo microdialysis technique worldwide in quantification of neurotransmitter levels in awake-freely-moving laboratory animals is primarily due to the significant contributions of Ungerstedt and colleagues during the 1970s and 1980s at the Karolinska Institute in Stockholm, Sweden [3,4].
1.2 Principles of microdialysis
In this section we present a summary of the principal concepts involved in the use of this versatile technique. The principle of microdialysis is primarily explained by Fick’s law of diffusion, which results in the passive passage of molecules across a concentration gradient. In this technique, a semipermeable membrane is introduced into the tissue. The membrane is perfused with a liquid that equilibrates with the tissue fluid outside the membrane due to bidirectional diffusion. The microdialysis technique is a complex interplay between the dialysis membrane-containing tube “the probe”, the perfusion liquid, the living tissue and the extracellular fluid containing the molecules of interest. The components of the tissue fluid, depending on the organ system which is perfused, may contain electrolytes, neurotransmitters, neuromodulators etc. which after equilibration are present in the dialysate outflow. Several reviews describe the theory and underlying general principles of in vivo microdialysis in general and brain microdialysis in particular. Vital concepts such as probe recovery, flow rate determination, zero-net flux method, reverse dialysis, data interpretation, spatial and temporal resolution as well as problems such as tissue damage, are discussed in detail in these reviews [1, 4–11].
1.3 General features and procedures of brain in vivo microdialysis
Ungerstedt [4] provides a review of the essential features of brain microdialysis. The most unique feature of the in vivo microdialysis technique is that it allows for a continuous collection of extracellular fluid in live awake animals as opposed to tissue sample obtained after biopsy. The technique also finds use in delivering low molecular weight drugs, as well as drugs which do not cross the blood-brain barrier, to specific brain regions. Drug delivery is accomplished by using the principle of reverse dialysis. The technique also offers an unparalleled advantage as regards analysis of the dialysate fluid. Since the semi-permeable membrane of the dialysis probe allows the transfer of relatively small molecules such as neurotransmitters, the dialysate is free of tissue debris, blood, proteins etc. and thus can be directly analyzed without further purification.
The technique essentially involves surgical implantation of a semi-permeable membrane-containing probe or guide cannula. Perfusion fluid is pumped into the probe via a perfusion pump at an optimum slow rate (generally 1.8 – 2.2 μl/min) and dialysate is collected, post equilibration, via a collection device. Samples are either collected manually, via a fraction collector or injected directly online into an analytical system [see Figure 1]. Chefer and colleagues [12] have described the procedural aspects of brain microdialysis in exacting detail.
Figure 1.
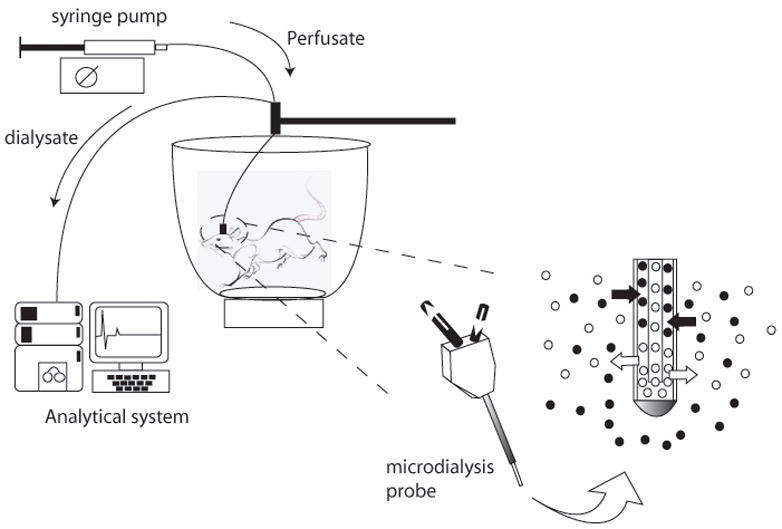
Experimental set up for neurotransmitter sampling using the rodent in vivo microdialysis technique.
1.4 Brain in vivo microdialysis
The in vivo microdialysis technique has been a mainstay of neuropharmacology research and has played a singularly important role in the elucidation of the neurochemical effects of drugs on the synaptic release of monoamine neurotransmitters such as DA, 5-HT and NE; amino acid neurotransmitters such as Glu and GABA as well as ACh in discrete brain regions of laboratory animals such as rodents, both rats and mice, as well as to a limited extent non-human primates [1,5,6,13]. One of the important features responsible for the success of this technique in neurochemical investigation is the development of sophisticated analytical techniques capable of accurate detection of extremely small quantities of the molecules of interest. Developments in analytical chemistry with the advent of techniques such as liquid chromatography, mass spectrometry, electrochemical detection etc have been instrumental in the popular use of the microdialysis methodology in neurochemistry [14–16].
The monoaminergic hypothesis of depression essentially implicates low levels of monoamine neurotransmitters in the pathophysiology of major depressive disorders. This fact has been an important feature in the development of therapeutic agents for the treatment of depression. The overall objective of antidepressant therapy over the past several decades has been to increase monoamine neurotransmitter concentration at the synapse. Antidepressant agents produce elevated monoamine levels either by inhibiting monoamine metabolizing enzymes such as monoamine oxidase or catechol-O-methyltransferase or by monoamine transporter blockade and inhibiting neurotransmitter reuptake. The in vivo microdialysis technique has found tremendous use in the development of second and third generation antidepressants which have revolutionized the pharmacotherapy of major depressive disorders. Microdialysis has been extremely useful in the neurochemical characterization and elucidation of the pharmacological profile of several classes of established as well as novel classes of antidepressants such as selective 5-HT reuptake inhibitors (SSRI), NE reuptake inhibitors (NRI), NE-5-HT reuptake inhibitors (NSRI), 5-HT-NE-DA reuptake inhibitors (SNDRI) etc. Since there exists extensive literature on microdialysis studies investigating the effects of clinically used, established as well as novel antidepressants, this topic will not be discussed in this review. Several excellent reviews highlighting the monoaminerigc theories of depression as well as the role of microdialysis in the development of pharmacotherapeutic agents for the treatment of depression have been cited [17–21].
Here, we review the use of the in vivo microdialysis technique in the elucidation of the neurochemical and pharmacological effects on neurotransmitter systems by drugs used in the treatment of addiction of drugs of abuse such as psychostimulants, neurodegenerative diseases such as AD and PD, and psychiatric disorders such as schizophrenia and ADHD.
2 Neurochemical characterization of amphetamines and the development of novel treatment agents for psychostimulant addiction using in vivo microdialysis
2.1 Psychopharmacology of amphetamines
Amphetamine and its substituted analogs such as methamphetamine (METH), 3,4-methylenedioxymethamphetamine (MDMA), paramethoxyamphetamine (PMA), as well as several others, form a major class of widely abused psychostimulant drugs. Amphetamine abuse is of serious concern and its incidence continues to increase globally and in the United States [22]. The acute effects of these drugs include euphoria, alertness, hyperthermia, hallucinations and hyperlocomotion, while long-term use produces psychosis as well as serious neurotoxic consequences [23]. One of the most significant acute neurochemical effects of this class of drugs is increased neurotransmitter release in multiple brain regions. Amphetamines cause reverse transport of neurotransmitters from the transporters at nerve terminals, promote their release from storage vesicles while inhibiting their reuptake which results in increased synaptic concentrations and enhanced neurotransmission of the biogenic amines DA, NE, 5-HT as well as ACh and Glu in multiple brain regions [24,25]. Amphetamine and various of its substituted derivatives exhibit differential potencies for effects on the various transporters. Amphetamine and METH exhibit considerably greater affinity for the DA transporter (DAT) while MDMA and fenfluramine produce considerable 5-HT release due to their primary effect on the 5-HT transporter (SERT) [26].
2.2 Neurochemical effects of amphetamines: in vivo microdialysis studies
Although in vitro studies using superfused brain slices or synaptosome preparations and in vivo studies employing in vivo voltammetry have contributed to our knowledge of the neuropharmacology of amphetamines, the in vivo microdialysis technique has been of paramount value in characterizing the effects of amphetamines. In this section, we review the neurochemical effects of amphetamine, METH and MDMA on DA and 5-HT release in various brain regions as elucidated using rodent in vivo microdialysis.
2.2.1 DA release
The amphetamine-induced release of DA in the striatum, which is presumed to contribute to its acute hyperlocomotor effect, has been repeatedly demonstrated by investigators. Amphetamine, METH, as well as MDMA, produce a robust increase in extracellular DA concentrations in the striatum. This process has been shown to be both transporter- and impulse-mediated for various amphetamines (e.g. MDMA, amphetamine) [27–30]. Amphetamines have also been shown to produce a robust effect on the dopaminergic mesolimbic and mesocortical pathways, implicated in its addictive properties. Shoblock and co-workers [31] have shown that both amphetamine and METH produced DA efflux in nucleus accumbens (NAc) and the medial prefrontal cortex (PFC) in rats. MDMA has also been shown to elevate limbic, cortical and hippocampal extracellular DA concentrations [32–34]. In addition, activation of 5-HT2A receptors has been shown to contribute to the magnitude of METH- and MDMA-induced DA release in various brain regions in studies employing in vivo microdialysis [35,36].
2.2.2. 5-HT release
Although not as well characterized as the effects of amphetamines on DA release, several reports document the effects of amphetamine and METH on serotonergic neurons and neuronal 5-HT release. Kuczenski et al. [37] reported an increase in extracellular 5-HT in the striatum after amphetamine and METH treatment. However, Gough and colleagues [30] have shown increased 5-HT release in the rat caudate after amphetamine, but not after METH administration. MDMA has been shown to enhance 5-HT release in various brain regions. MDMA administration resulted in a dose-dependent increase in striatal, cortical and hippocampal extracellular 5-HT. Fluoxetine pretreatment attenuated MDMA-induced 5-HT release, providing evidence for a role of SERT in mediating the MDMA-induced neuronal 5-HT efflux [38,39]. Recently, Baumann et al. [40] measured parameters of locomotor activity along with DA and 5-HT microdialysate levels in MDMA-treated rats. Whereas MDMA-induced striatal DA efflux correlated with increased locomotion, the increases in extracellular 5-HT in the striatum and cortex correlated positively with increased stereotypy.
2.3 Agonist therapy for psychostimulant addiction: dual DA/5-HT releasers
In recent years, several pharmacological strategies for the treatment of psychostimulant (e.g., METH and cocaine) abuse have included the development of nonselective inhibitors of DAT and SERT, as well as dual DA/5-HT releasers. [41–44]. Herein, we briefly summarize in vivo microdialysis studies employed in the development of certain novel amphetamine-like compounds with dual DA/5-HT release properties for the treatment of METH and cocaine addiction.
The cocaine- and METH-induced “high” is mediated by elevated extracellular DA concentrations in the mesolimbic reward circuit, which has been proposed as also is the principal cause of addiction. Chronic psychostimulant abuse produces long-term changes in neurochemistry and synaptic plasticity, whereas their withdrawal produces severe mood and behavioral deficits due to impaired 5-HT neuronal function and DA depletion [45]. In view of clinical evidence indicating that negative states associated with drug withdrawal remain an important factor in drug relapse, several strategies have been initiated to target these deficit states. Rothman and colleagues [41,42] discuss the rationale behind the development of dual 5-HT/DA releasers for “agonist therapy” in the treatment of cocaine and METH addiction. Although stimulants such as amphetamine have been used in the treatment cocaine and METH abuse, their use has limited therapeutic value due to inherent abuse potential. It has been hypothesized that increased synaptic 5-HT concentrations counteract the reinforcing and addictive effects of stimulant-induced DA elevations. Notably, the combined administration of phentermine, a DA releaser along with fenfluramine, a potent 5-HT releaser, showed considerable potential in the treatment of cocaine dependence. Thus, one strategy proposed to decrease the abuse liability of candidate medications for stimulant agonist therapy is to add 5-HT releasing properties to these drugs [46].
A series of agents acting on biogenic amine transporters was synthesized and evaluated for their DA and 5-HT release profile in the NAc and PFC utilizing both in vitro synaptosomal preparations and the in vivo microdialysis technique in rodents [41]. The test compounds consisted essentially of substituted amphetamines (e.g. PAL-287, PAL-303, PAL-313, PAL-314 and PAP-353); they produced DA release equivalent to amphetamine-induced DA efflux and exhibited varying potency as 5-HT releasers. Although PAL-287-induced elevations in dialysate DA concentrations that were comparable to amphetamine, its potency for 5-HT release was almost 1,000-fold greater than amphetamine. Despite producing elevated extracellular DA, PAL-287 produced minimal ambulatory activity, in contrast with the hyperlocomotion produced by amphetamine at similarly elevated extracellular DA concentrations. Although the PAL compounds showed potential for self-administration, a sign of positive reinforcement, the effect was inversely proportional to their potency for 5-HT release. PAL-287, which demonstrated pronounced 5-HT release, exhibited minimal self-administration in rhesus monkeys [41]. Negus et al. [47] have shown that the novel dual 5-HT/DA releasing substituted amphetamine compounds, PAL-287, PAL-314 and PAL-353, produced reductions in both cocaine discrimination, as well as cocaine self-administration in rhesus monkeys. Results of these studies highlight the potential usefulness of these agents as targets for agonist therapy for cocaine and METH addiction.
3 Microdialysis studies predict the therapeutic efficacy of drugs used in the treatment of ADHD
ADHD is characterized by symptoms that include hyperactivity, impulsivity, inattentiveness and cognitive impairment. It is thought that the underlying cause of ADHD lies within the PFC and its connection to the striatal areas [48,49]. The lack of inhibitory control centered in the frontal cortex is thought to derive from a dysfunction in dopaminergic and noradrenergic innervations. These innervations are involved in regulating impulsivity and hyperactivity found in ADHD patients [50]. Drugs effective in the treatment of ADHD are thought to act by enhancing the monoaminergic transmission of these pathways through an increase in monoamine levels within the frontal cortex or by direct stimulation of adrenergic and/or dopaminergic receptors. The primary drugs used to treat ADHD include methylphenidate (MPH), atomoxetine (ATX), and guanfacine.
MPH increases both NE and DA levels in the brain through its ability to inhibit monoamine reuptake and cause its release. Although beneficial in the PFC, this excess of monoamines in other brain areas could result in unwanted abuse potential. However, microdialysis studies in male Sprague-Dawley rats demonstrated that there is a preferential increase in DA in the PFC relative to the NAc when low doses of MPH were used [51]. In this study, DA reached maximum levels in both the PFC and NAc 32 minutes after intraperitoneal administration of MPH. At the maximum dose used, 1 mg/kg, MPH produced a 200% and 100% increase in DA over baseline in the PFC and NAc respectively. Similar differences were also observed when MPH was administered orally at 2 mg/kg. An approximate 125% increase above baseline was observed in the PFC relative to a 50% increase above baseline in the NAc [51]. These data indicate the selectivity of MPH in enhancing monoamine levels in the PFC to achieve therapeutic outcomes.
The NE reuptake inhibitor ATX has also been shown to produce increases in both DA and NE in the PFC in the rat as determined by brain microdialysis [52]. However, ATX has also been shown to increase NE levels throughout the brain without increasing DA levels excluding the PFC [53]. The PFC is unique in this regard due to its sole use of the NE transporter for the reuptake of both NE and DA. The inhibition of this transporter effectively increases both monoamines selectively in the PFC [53]. The inability of ATX to increase DA in brain regions other than the PFC has limited its abuse liability. For example, no increase in DA was observed in the NAc after ATX administration in the rat [52].
Recently it has been demonstrated that both ATX and MPH increase histamine in the PFC of the rat [54]. Histamine plays an important role in attention, learning, and memory and has been shown to be activated through dopaminergic mechanisms. The benefit of increasing histamine levels on learning was clearly demonstrated when treating the spontaneously hypertensive rat (SHR) [55,56]. In the study by Liu et al. [55], it was shown that ATX significantly enhanced the performance of the SHR in the Morris water maze. This behavioral test confirms the beneficial effects of ATX on learning and memory through, at least in part, its histamine actions. Rat microdialysis experiments permit investigators to predict the potential efficacy of a drug by measuring drug-induced alterations in monoamine concentrations. In addition, these data also afford researchers an insight into other neurotransmitters that may be affected by drugs. For example, the stimulation of histamine release with DA and/or adrenergic receptor agonists could also be used as a marker of efficacy. Guanfacine, being an α2-adrenoceptor agonist, may also elicit its effects through additional mechanisms. Unfortunately, there are limited microdialysis data on the effects of guanfacine on neurotransmitter release in the brain. As guanfacine-mediated α2A-adrenoceptor agonism occurs post-synaptically, microdialysis would not be useful in the investigation of its clinical efficacy as compared to that of psychostimulants used in the treatment of ADHD [57]. In addition, new therapies that are being proposed may also benefit from analysis by brain microdialysis. The selective DA D4 receptor agonist A-412997 may in part manifest its actions through the release of histamine [58]. Such a release of histamine could help explain its ability to improve cognition [59]. Another example would be the combination of 5-HT1A agonist and NE reuptake inhibitor activity for the treatment of ADHD [60]. Recently, microdialysis studies would help predict novel compounds containing both NE reuptake inhibition and 5-HT1A partial agonist activities [61]. Several of these studies indeed raise the possibility that 5-HT1A partial agonism may contribute to the ability of NE uptake inhibitor to selectively enhance cortical DA transmission [61].
4 Neurochemical characterization of the mechanism of action of typical and atypical antipsychotics using in vivo microdialysis
4.1 Typical and Atypical antipsychotics
The defining characteristic of an atypical antipsychotic drug (APD) is minimal catalepsy in rodents and motor side effects in patients at doses which achieve the presumptive equivalent of an antipsychotic action in rodents, e.g. blockade of conditioned avoidance response, and an antipsychotic effect in patients with schizophrenia who are acutely psychotic. Typical APDs, such as chlorpromazine and haloperidol, achieve their antipsychotic action and cause motor side effects by extensive blockade of striatal D2 receptors. There are several classes of atypical APDs, e.g. those with potent 5-HT2A receptor antagonist properties, e.g. clozapine, risperidone, and aripiprazole, as well as weaker interference with D2 receptor stimulation. In the case of aripiprazole, this weaker blockade with D2 receptor stimulation is achieved by partial agonism at the D2 receptor. The other atypical APDs in clinical use in the US, e.g. asenapine, clozapine, iloperidone, olanzapine, quetiapine, risperidone and ziprasidone, produce DA receptor blockade by direct antagonism of D2 receptors. Because their affinity for 5-HT2A receptors is greater than that for D2 receptors, the interference with D2 receptor stimulation is less than for 5-HT2A receptors. Furthermore, limbic DA release, the likely proximate cause of psychosis, is enhanced during acute psychotic episodes, thus competing with the antipsychotic drugs for the D2 receptor. This competition is likely to enhance the relatively greater blockade of the atypical APDs. Other classes of atypical APDs include the substituted benzamides such as amisulpride and the metabotropic glutamate receptor-2 (mGluR2) agonists, e.g. LY-404039 [62–64]. Because of space limitations, this review will only consider the typical antipsychotic drugs and those atypicals with potent 5-HT2A antagonist properties.
As reviewed elsewhere [65–66], mesolimbic DA release is an important factor in the etiology of positive symptoms (delusions and hallucinations) while cortical DA release is likely to be important to the cognitive dysfunction and negative symptoms characteristic of schizophrenia. Microdialysis in awake-freely-moving or anesthetized rodents and primates has frequently been used to study the action of typical and atypical APDs on cortical and limbic DA efflux. In addition, there have been some studies – utilizing microdialysis – on the effect of typical and atypical APDs on ACh, 5-HT, Glu and GABA efflux. These neurotransmitters play an important role in cognitive function, reality testing and motor function and have an ability to modulate the activity of DA neurons. The current review will highlight some of the key microdialysis studies comparing typical and atypical APDs on the efflux of DA and ACh, in particular.
4.2 Comparison between the regional effects of typical and atypical APDs on neurotransmitter systems: in vivo microdialysis studies
Clozapine and related atypical APDs preferentially increase DA efflux in the medial PFC of rats compared to NAc, the key limbic area as well as the striatum, whereas the reverse is true for typical APDs, i.e. they produce significantly greater increases in DA efflux in the NAc and the striatum compared to the PFC [67,68]. This increase is correlated with their potency as 5-HT2A inverse agonists [69]. The NAc shell is more closely associated with the limbic system, and thus psychosis and reward mechanisms, while the NAc core is associated with the caudate-putamen and thus, with motor function. Clozapine produces equivalent increases in DA efflux in both regions whereas haloperidol preferentially increases DA efflux in the core [70]. The ability of typical APDs to increase cortical and limbic DA efflux can be potentiated and diminished respectivelyby 5-HT2A inverse agonists such as M100907 [71], ACP-103 [72] or SR43469B [73]. Sulpiride, a selective D2/D3 antagonist, without 5-HT2A antagonist properties did not increase cortical DA efflux in rats, but the combination of sulpiride and M100907 was effective in doing so [74]. The ability of the atypical APDs to increase cortical DA efflux can be blocked by the 5-HT1A antagonist WAY100635, suggesting that 5-HT1A agonism is a necessary factor in causing this increase. It is of particular interest that WAY 100635 blocks the cortical DA efflux of atypical APDs, regardless of whether they have intrinsic 5-HT1A partial agonist properties (e.g. clozapine, quetiapine, ziprasidone, and aripiprazole), or lack significant 5-HT1A agonist properties (e.g. olanzapine and risperidone) [69,74]. Similarly Youngren et al. [75] found that clozapine, but not haloperidol, also preferentially increased DA release in the PFC of rhesus monkeys as compared with the striatum. The ability of the atypical APDs to preferentially increase cortical DA efflux would be expected to produce selective stimulation of D1 receptors in the prefrontal cortex which are known to have an important effect on cognition. However, it should be kept in mind that enhanced stimulation of D1 receptors may have a negative effect on cognition, suggesting that there can be adverse effects in at least some patients from atypical APDs. It is of interest in this regard that clozapine has been reported to impair working memory in patients with schizophrenia after six weeks of treatment, but this negative effect is no longer evident at 6 months [76].
Rodent microdialysis studies conducted by several investigators have consistently reported that the atypical APDs clozapine, olanzapine, risperidone and ziprasidone, but not the typical APDs haloperidol, thioridazine and chlorpromazine, produced a robust increase in ACh release in both PFC and the hippocampus [77–79]. The current interest in developing muscarinic M1 or M4 agonists or α7 nicotinic receptor agonists, which also enhance cortical ACh efflux is consistent with this property of the atypical APDs.
N-methyl-D-aspartate (NMDA) antagonists, phencyclidine (PCP), dizocilpine (MK-801) and ketamine induce hyperlocomotion, deficits in cognition and social interaction, as well as disruption of the prepulse inhibition (PPI) of startle. These properties are key elements in the hypoglutamatergic hypothesis of schizophrenia. Microdialysis experiments conducted by several investigators, have shown the effects of APDs on NMDA antagonist-induced behavioral and neurochemical consequences in both rodents and non-human primates. Two recent studies from the Artigas laboratory have shown that the atypical APDs clozapine and olanzapine, but not haloperidol suppressed the NMDA antagonist-induced 5-HT and Glu efflux in the PFC [80,81]. Linn and co-workers [82] reported that clozapine, but not haloperidol, attenuated the PCP-induced disruption of PPI in adult capuchin monkeys. Microdialysis studies have also compared the effect of both typical and atypical APDs on GABA efflux in rodents. Bourdelais and Deutch [83] have reported that administration of clozapine, but not haloperidol, markedly reduced GABA levels in the PFC of rats, with similar results being reported in the rat striatum. The effects of clozapine on GABA levels may play a role in its lack of motor side effects in schizophrenic patients [84].
In conclusion, the studies reviewed indicate that in vivo microdialysis in rodents as well as in non-human primates is an extremely useful method for neurochemical and pharmacological characterization of APDs along with behavioral assays. The importance of the technique is apparent from its role in the elucidation of various receptors in the mechanism of action of known APDs, which consequently remains important in the development of novel APDs.
5 Alterations in striatal Glu in an animal model of PD: correlation between in vivo microdialysis and ultrastructural immunocytochemistry
There is growing interest in the interactions between DA and Glu and it may be the lack of DA in PD that results in dynamic changes in Glu at least within the striatum. The DA terminals originating from the substantia nigra pars compacta (SN-PC) make a symmetrical synaptic contact not only on the dendritic shaft of the medium spiny neuron but also on the neck of the dendritic spine [85]. The asymmetrical synaptic contact on the head of that same spine within the dorsolateral striatum originates from not only the motor cortex but also the thalamus [86] and the nerve terminals contain the neurotransmitter Glu [87]. Therefore, the alterations in Glu levels, which may originate from either the motor cortex or thalamus, may be a target for future pharmacological treatment. Thus, a clearer understanding of glutamate changes in specific brain areas over time could lead to the development of more specific therapies.
5.1 Time–dependent changes in striatal Glu following loss of DA
Using in vivo microdialysis, Lindefors and Ungerstedt [88] were the first to report an increase in the extracellular level of striatal Glu in anesthetized rats 3–4 weeks following a 6-hydroxydopamine (6-OHDA) lesion. Notably, in the same year, Reid et al. [89] reported just the opposite effect. It appears that following the lesion (perhaps after 3–4 weeks), the animals were then tested with the DA agonist apomorphine to evaluate the number of contralateral turns. It is well known that animals continue to be sensitized to the effects of apomorphine weeks after a single injection of the compound [90]. It has been reported that short-term administration of a low dose of apomorphine (0.05 mg/kg/d) has a significant effect on the extracellular levels of Glu within the striatum and the substantia nigra (SN) [90].
Increases in extracellular levels of Glu following a 6-OHDA lesion [88] are in agreement with findings from several other labs who have reported an increase in spontaneous activity of GABAergic striatal neurons after 6-OHDA treatment [91]. It is possible that such an increase in electrical activity may be due to an increase in striatal glutamatergic function. Increased spontaneous neuronal activity within the striatum peaks approximately 4 months following the lesion [91]. This increased neuronal activity starts to decrease at 6 to 8 months and by one year the activity in the lesioned animals was similar to that found in the control group [92].
Time-dependent changes in Glu within the DA-deafferented striatum of rats were reported to occur following a unilateral injection of the neurotoxin 6-OHDA into the medial forebrain bundle [93]. Using both in vivo microdialysis and correlating these findings with alterations in Glu immuno-gold labeling by electron microscopy, changes in striatal glutamatergic function were analyzed at 1 and 3 months following the lesion.
5.2 In vivo microdialysis findings
There was a significant increase in the basal extracellular level of striatal Glu compared to the sham (control) group 1 month following the nearly complete loss of striatal DA [93]. Notably, thes complete opposite was found 3 months following a nigrostriatal lesion, where it was reported that there was a significant decrease in the extracellular levels of striatal Glu [93]. The increase in extracellular striatal Glu 1-month after the near complete loss of striatal DA is consistent with some earlier reports [88,94–99].
Alterations in the extracellular levels of striatal Glu have been observed following partial lesioning of the nigrostriatal pathway with the neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Regardless of whether acute (4 × 20 mg/kg every 2 hours) or subacute (30 mg/kg/d × 7 days) administration of the toxin is used, both paradigms result in bilateral loss of DA terminals in the striatum but either an increase (in the case of acute MPTP) or decrease (in the case of subacute MPTP) in the extracellular level of striatal Glu [100,101]. The increase in extracellular Glu following acute MPTP is similar to what has been reported 1 month following 6-OHDA insult [93]. The decrease in extracellular Glu following subacute administration is similar to what has been reported 3 months after 6-OHDA lesioning [93]. In a subchronic model of PD, using MPTP plus probenecid, an agent that decreases MPTP secretion, [102], it has been reported that nearly 4 months from the onset of administration of the toxin, there was no change in the extracellular level of striatal Glu or in the uptake of Glu into synaptosomes [103]. There also appears to be a lack of correlation between alterations in Glu within the striatum compared to the SN. Using this same MPTP/probenecid model, some of us have recently reported that 4 months following the initiation of drug treatment, there is an increase in the extracellular level of Glu within the SN [103], a finding consistent with the long-standing model of basal ganglia function [104]. At 6 months following a 6-OHDA lesion, there continues to be a significant decrease in the extracellular level of striatal Glu (Meshul et al., unpublished findings), a result similar to what is observed 3 months following such a lesion [93]. In addition, microdialysis of the subthalamic nucleus (STN) in the 6-OHDA lesioned rat model results in an increase in extracellular Glu (Figure 2). The increase in STN glutamate following loss of striatal DA is consistent with the current model of basal ganglia function [104], suggesting that a possible explanation for the increase in activity of the STN is due to increased glutamate release. This would result in activation of the output from the STN to the SN and globus pallidus pars interna, resulting in an eventual decrease in activity of the motor cortex and the classic signs of PD, i.e. bradykinesia and slowness in the initiation of movement.
Figure 2.

Baseline extracellular level of Glu within the STN 3 months after a unilateral lesion of nigrostriatal pathway. Note the significant increase in Glu levels in the 6-OHDA lesioned group compared to the sham group. Values are means ± S.E.M.
* - p < .05 using Student’s t-test.
Some of us have carried out striatal in vivo microdialysis following subchronic administration of MPTP over a 4-week time period in which the mice received a daily dose of 10 mg/kg (Table 1). We find, similar to that reported after subacute administration of MPTP [100], a decrease in the extracellular levels of striatal Glu. Several preliminary in vivo microdialysis studies were carried out to determine the effect of exposure to an enriched environment or forced exercise. Following constant exposure of both young and aged mice to either an enriched environment for 3 weeks or running on a treadmill on a daily basis for 1 hr/day for 3 weeks, we find that an enriched environment leads to an increase in extracellular striatal Glu in both young and aged mice. Infusion of a Glu transporter antagonist results in a varying increase in extracellular Glu (Table 1). Exposure of MPTP-treated mice to either an enriched environment or treadmill exercise results in the partial restoration of DA-labeled neurons in the SN and an increase in locomotor function (Sashkin and Meshul, unpublished findings). Therefore, the increase in the number of DA-labeled neurons following exposure to an enriched environment may be related to the increase in extracellular striatal Glu.
Table 1.
Effect of MPTP, an enriched environment, or treadmill running on striatal extracellular glutamate
| Group | Basal Levels (pmole/ul) | PDC (1 mM infusion for 90 minutes) |
|---|---|---|
| MPTP-Young mice | 0.40 ± .08** | Not determined |
| Vehicle-Young mice | 0.60 ± .05 | Not determined |
| SE-Young Mice | 0.57 ± .01 | 1.01± .01 (76%  ) ) |
| EE-Young Mice | 1.12 ± .16* |
1.59 ± .02* (43% )
|
| SE-Aged Mice | 0.69 ± .03 | 0.88 ± .05 (18% ) |
| EE-Aged Mice | 0.88 ± .05* |
1.3 ± .03* (48% )
|
| Control (no running) | 0.93 ± .04 | 1.13 ± .02 (122% ) |
| Runners | 0.63 ± 0.4** | 0.94 ± .02** (149% ) |
Values are means ± S.E.M in the dialysate sample. PDC: L-trans-pyrrolidine-2,4-dicarboxylic acid (reverses the glutamate transporters and increases Ca++-dependent glutamate release). SE: standard environment; EE: enriched environment.
p < .05 compared to the SE group;
p < .05 compared to the SE or control group.
5.3 Morphological correlates to changes in extracellular Glu
Because of the controversy associated with the origin of the extracellular level of striatal Glu, immuno-electron microscopy was carried out using an antibody against Glu and tagging the primary antibody with a secondary antibody that contains an immuno-gold label. We were the first to report that following subchronic treatment with the DA antagonist haloperidol the changes in asymmetrical synaptic contacts were associated with nerve terminals containing Glu and that there was an increase in the density of such nerve terminal Glu immuno-gold labeling compared to the control group [87]. However, there is a heterogeneous Glu input to the striatum. We have been made aware only recently that nerve terminals making an axospinous contact could still originate from the thalamus [86,105]. The terminals from the cortex and thalamus can be differentiated by their vesicular Glu transporters. The input from the cortex contains the messenger RNA and protein for the vesicular Glu transporter-1 (VGLUT-1), while that from the thalamus contains the vesicular Glu transporter-2 (VGLUT-2) [105,106].
It has been reported that 1 month following a 6-OHDA lesion, there was a decrease in the density of nerve terminal Glu immunolabeling within all asymmetrical synapses [93]. Even though striatal DA levels do not recover, Glu synapses continue to show signs of plasticity. Of particular interest was that 3 months following a nigrostriatal lesion, the relative density of nerve terminal Glu immunolabeling showed an increase compared to the non-lesioned group.
At this point, it was not known if the Glu input from the cortex vs. thalamus was differentially affected following a nigrostriatal lesion. It has been reported that a lesion of the intralaminar thalamic nuclei (centromedian and parafasicular) blocks the 6-OHDA-induced increase in striatal enkephalin and glutamic acid decarboxylase mRNA and cytochrome oxidase mRNA in the subthalamic nucleus [107]. It was recently reported that there is an increase in the number of nerve terminals containing VGLUT-1 protein after MPTP administration in the non-human primate [108], suggesting a change in the Glu input from the cortex. For VGLUT-2, there was a shift in the number of terminals making axospinous vs. axodendritic contact. Since VGLUT-1 is associated with nerve terminals originating from the cortex, we recently carried out VGLUT-1 labeling of the striatum 6 months following a nigrostriatal lesion (Figure 3). We find that at this time point there is a significant decrease in the extracellular level of striatal Glu (Meshul et al., unpublished findings). When the density of nerve terminal Glu immunogold labeling was then determined, it was found that in both VGLUT1 positive and negative nerve terminals, there was an increase in the density of striatal Glu immuno-gold labeling compared to the control group (Figure 4). This suggests that both the cortico- and thalamostriatal pathways are equally affected following the nearly complete loss of nigrostriatal DA. As previously reported: following loss of striatal DA, an increase or decrease in extracellular glutamate is associated with a decrease or increase in the density of glutamate immuno-gold labeling within nerve terminals making an asymmetrical (excitatory) contact onto a dendritic spine [90,93,101,109,110]. This inverse relationship in extracellular glutamate and density of glutamate immuno-gold density is suggested to be due to decreased release of glutamate (i.e., as measured by an increase in extracellular glutamate) resulting in an accumulation of glutamate inside nerve terminals (i.e., an increase in glutamate immuno-gold labeling). It has been reported that more than 90% of the glutamate immuno-gold labeling is associated with the synaptic vesicle and not the nerve terminal cytoplasm [93]. Therefore, based on our ultrastructural immuno-gold labeling data, it would appear that glutamate is not being released from the terminal and there is more immuno-gold labeling of the synaptic vesicles.
Figure 3.
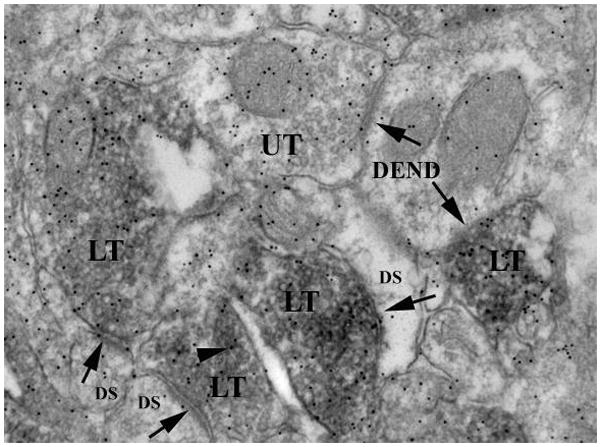
Double labeling for the striatal vesicular Glu transporter-1 (VGLUT-1) and Glu immuno-gold. Electron photomicrograph using the diaminobenzidine technique to localize VGLUT-1 (dark reaction product) and the immunogold technique to localize an antibody against the neurotransmitter, Glu, within the dorsolateral striatum. Within the VGLUT-1 labeled nerve terminal (LT), there is an accumulation of small round synaptic vesicles and 10 nm gold particles indicating the location of the antibody (arrowhead). There is also an unlabeled nerve terminal (UT). The nerve terminals are making an asymmetrical synaptic contact (arrow) with an underlying dendritic spine (DS) or dendrite (DEND).
Figure 4.

Density of striatal nerve terminal Glu immuno-gold labeling (# gold particles/um2) within VGLUT-1 labeled and unlabeled nerve terminals following administration of 6-OHDA. 6-months following the nearly complete loss of striatal DA, the increase in the density of Glu immuno-gold labeling was identical in both VGLUT-1 labeled and unlabeled striatal nerve terminals compared to the control (CTL) group. Values are means ± S.E.M.
* - p < .05 compared to the respective control group using an ANOVA.
Knowing the changes in extracellular glutamate in striatum, SN or STN is of great importance in terms of potential pharmacological treatment. It is possible that treatment with a specific glutamate antagonist may not necessarily be successful since glutamate may be increased in one region but decreased in another and at different times after the loss of DA. In addition, knowing whether the input from the motor cortex (i.e., VGLUT-1) or thalamus (VGLUT-2) is of greater or lesser importance may be crucial for future pharmacological treatment. Agents that can selectively target the VGLUT-1 or VGLUT-2 pathways may prove to be of significant use in combating the changes in glutamate following the loss of nigrostriatal DA.
6 ACh: cognition, memory, Alzheimer’s disease
6.1 Introduction: what’s special about ACh?
ACh with its virtue of being ubiquitous in major brain functions has been a subject of microdialysis studies since the 1980s. Since ACh neither fluoresces nor oxidizes – unlike the catecholamies – it has been necessary to develop an enzymatic assay which facilitates detection of femtomolar (fmol) amounts by HPLC. Recent developments have included mass spectrometry with levels of detection approaching one fmol or less [111].
While sensitive ACh detection has become more accessible, ACh and cholinergic synapses have some unusual neurochemical characteristics that have important consequences for microdialysis studies. First, synthesis of ACh depends on the availability of its precursors glucose and choline, which can become rate-limiting for ACh synthesis when ACh release is strongly stimulated [112]. While choline is routinely measured together with ACh, it should be noted that (a) choline levels are not directly correlated with ACh but are largely controlled by phospholipid synthesis and hydrolysis and (b) the concentration of choline in the extracellular space of the brain is much (100 – 1,000-fold) higher than that of ACh. In fact, the high levels of free choline in the brain complicate the validation of methods to quantify ACh release through choline sensors which reflect ACh release only under strictly controlled conditions [113].
Notably, cholinergic neurons follow an endogenous diurnal rhythm (with higher activity during the night in rodents) and they respond to a wide variety of physiological stimuli (reviewed by Day et al. [114]). Thus, hippocampal and cortical, but not striatal ACh levels typically increase upon sensory stimulation (which includes intraperitoneal injections). Moreover, measuring ACh release during anesthesia should be avoided because most common anesthetics inhibit ACh release. The good news is that ACh in the brain is a purely neuronal product: ACh release is calcium-dependent and is nearly completely (>90%) inhibited by blockers of neuronal impulse flow such as tetrodotoxin (TTX). As a consequence, there are no difficulties of interpretation with ACh compared to e.g. amino acids which have transmitter as well as metabolic pools.
6.2 Microdialysis of ACh: which brain area?
The highest density of cholinergic neurons in the brain and the highest tissue content of ACh in the brain are found in the striatum, resulting in much of the early work with microdialysis focusing on rat striatum [115]. Cholinergic activity in the striatum is controlled by interactions with several other transmitter pathways including cortical fibers that increase ACh levels through NMDA receptors and dopaminergic neurons which act predominantly though D2 receptors to reduce ACh release. PD is a major research area targeting striatal mechanisms. Cholinergic drugs however, are of limited effectiveness in this disease, and most drug development programs focus on glutamatergic or dopaminergic approaches. Cholinergic actions in the NAc are mediated by both muscarinic and nicotinic receptors. Most research in this area however, is basic research aimed at understanding drug addiction and will not be reported here (for reviews, see [114,115]. Similarly, microdialysis studies in the brain stem are important for our understanding of the sleep-wake cycle and anesthesia but limited drug development efforts have been invested in this area.
Most studies in drug development focus on cholinergic projection fibers that originate in the basal forebrain and innervate hippocampal and cortical areas. These cholinergic fibers are involved in higher cognitive functions, making them a prominent target for therapeutic research. AD is a particularly well-studied disease target because cholinergic neurons in the forebrain degenerate in AD, and lack of cholinergic transmission is a major reason for the cognitive (and probably some psychiatric) symptoms of AD. Accordingly, much work has been invested to study cortical and hippocampal cholinergic transmission in vivo by microdialysis [116]. A variety of drugs has been tested for their effects on cortical ACh levels: inhibitory effects were found for example, for opiates, benzodiazepines, and ethanol. Of note, dual-probe microdialysis has been found useful to distinguish local actions of drugs in the hippocampus or cortex from actions in the somatic regions of the neurons in the basal forebrain [117]. Increases of ACh in the hippocampus are often interpreted as a sign of potential pro-cognitive effects [118] although such isolated findings do not provide sufficient evidence in the absence of supporting behavioral experiments.
Various approaches have been used to combine microdialysis with behavioral studies [119]. Microdialysis studies in cortical areas clearly demonstrated that ACh plays a major role in attention [113]. Attention and motivation are reflected in cortical ACh levels and these can be used to test the effects of exogenous drugs. Hippocampal ACh also responds to novelty. As indicated above, sensory stimulation such as injections or stroking of the animal lead to changes of hippocampal ACh release. In studies using learning and memory paradigms such as object recognition, ACh levels increased when new stimuli were given to the animal. However, reconnaissance of known objects (when memory was retrieved) did not cause significant changes of ACh [120]. These studies do not support the concept that cognitive processes are directly related to ACh levels in the brain. Rather, levels of ACh reflect the attention and vigilance of the animal during the testing procedure. Clearly, physiological stimuli such as exploration of a novel environment can be used to enhance ACh release and also to test the effect of drugs on this physiologically induced cholinergic stimulation. The cholinergic link can be strengthened by using cholinergically impaired animals in the testing procedure [121]. Drug development studies now routinely employ parallel measurements of microdialysis and behavioral studies to characterize pro-cognitive actions of cholinergic modulators (see below).
6.3 Cholinesterases, microdialysis and drug development
ACh is the only classical neurotransmitter inactivated by enzymatic hydrolysis: acetylcholine-esterase (AChE) being the responsible enzyme that cleaves ACh to choline and acetate. To inhibit AChE is therefore a straightforward procedure to stimulate cholinergic neurotransmission in the CNS. AChE inhibitors are first-line drugs for the treatment of AD and may be useful for other types of dementia and/or psychiatric disorders. In AD, cholinergic dysfunction is closely related to the clinical symptoms and it seems likely that AD cannot be treated successfully unless the cholinergic dysfunction is somehow corrected [122]. Microdialysis studies have been instrumental to characterize the in vivo-actions of AChE inhibitors. In this regard it is of importance to point out that drug development programs may fail because compounds that inhibit AChE in vitro may not be sufficiently brain-permeable in vivo, or they may compartmentalize in a way that prevents AChE inhibition [123]. The starting point in the drug development process in this area was the development of plant products with AChE inhibitory potency such as the well-known physostigmine, an alkaloid from the Calabar bean (Physostigma venenosum) which causes a dose-dependent increase in the extracellular level of ACh in brain after systemic administration [124]. While physostigmine has a very short half-life in humans, it was successfully developed conceptually to the long-acting agent rivastigmine which has a pseudo-irreversible binding mode to AChE. Other plant constituents with AChE inhibitory potency are huperzine A and galanthamine, a marketed drug that is also an allosteric activator of nicotinic transmission [125]. The most successful AChE inhibitor on the market, donepezil, is a synthetic drug developed by Eisai Corp. and inhibits AChE by an allosteric mechanism [125]. Among the marketed drugs, donepezil is the only one that is specific for AChE and does not affect ACh levels in AChE-deficient mice [126]. All of the AChE inhibitors reliably increase ACh levels in rodent brain [127], although relative high doses have been used in some studies to obtain adequate results. Unfortunately, no methods are available to measure concentrations of ACh in human brain, so animal experiments must suffice at this time as a proof-of-principle.
Butyrylcholinesterase (BChE), also called plasma cholinesterase, is also present in the CNS (mainly in astrocytes) and can rapidly hydrolyze ACh. Microdialysis studies in AChE-deficient mice demonstrated that BChE can to some extent substitute for AChE when AChE is absent [128]. As AChE levels are low in advanced AD, BChE inhibitors have been suggested as a treatment option for advanced AD. Several promising compounds have been synthesized starting with cymserine as a model drug [129]. Phenserine, for instance, has been shown to bolster Ach levels in vivo and to be active in various behavioral paradigms [122]. BChE inhibitors can increase ACh in the brain – as shown by microdialysis – not only when AChE activity is absent [128], but even when injected systemically into healthy rats [130].
In this context, it may be added that researchers at Daiichi Sankyo have developed dual-mechanism drugs that inhibit both AChE and the 5-HT transporter. These compounds enhance the extracellular concentrations of both ACh and 5-HT in animals [131]. The potential of dual mechanism-drugs for neuropsychiatric disease has recently been emphasized [132]. In the present example, the parallel stimulation of cholinergic and serotonergic transmission was expected to improve cognition synergistically [133].
6. 4 Drugs acting at cholinergic receptors
Central cholinergic stimulation cannot only be obtained through inhibition of ACh hydrolysis, but also through direct stimulation of cholinergic (nicotinic or muscarinic) receptors. While microdialysis studies measuring ACh are not well suited to follow agonist actions at postsynaptic receptors (such as M1 receptors), numerous studies have investigated presynaptic receptors which control ACh release. In these studies, the levels of ACh – as sampled by microdialysis – represent the direct readout of the various presynaptic influences acting upon cholinergic nerve endings. Muscarinic receptors, both M2 and M4, are located presynaptically at cholinergic synapses and inhibit ACh release in a feedback fashion. An increase in ACh levels at the synaptic cleft results in a receptor-mediated blockade of Ach release. Therefore, M2 receptor antagonists tend to increase the release of ACh because they block the negative feedback inhibition of ACh via M2 receptors [124]. A dedicated effort for drug development was undertaken by researchers at Schering-Plough Co. who combined microdialysis and a behavioral (passive avoidance) procedure in rats. Using p.o. application of drugs, this group simultaneously tested (a) the oral bioavailability and brain permeability of their compounds and (b) the pharmacological effect reflected as an increase of ACh in rat brain by their “microdialysis assay”. The group was able to identify compounds that were active in vivo and showed a 100-fold higher affinity towards M2 receptors when compared with M1 and M3 receptors [134,135]. Unfortunately, none of these compounds reached the market, and the concept of M2 antagonists does not seem to be prominently pursued at this time.
It has to be noted that the concept of M2 antagonists has a weakness which relates to the mechanism of action. In vitro and in vivo, M2 receptor blockade only enhances ACh release when ACh levels are high causing massive feedback inhibition of ACh release. Thus, it may be promising to add M2 antagonists to a therapy based on AChE inhibitors to optimize their effectiveness. This idea has been followed in an attempt by Parke-Davis to develop a dual mechanism drug CI-1002/PD 142676 which inhibits both AChE and (at somewhat higher concentration) also M2 receptors. This compound effectively increased ACh in rat cortex and was also active in behavioral assays although further studies were apparently not pursued [136].
Nicotinic receptors were also targeted by new drug developments using microdialysis. Nicotine has well-known cognitive-enhancing effects, and hippocampal and cortical ACh concentrations in aged rats were amplified by nicotinic agonists such as SIB-1553A acting on α4s2 receptors in studies from Merck Co. [137,138]. It is, however, unclear whether these effects are due to enhancement of ACh release because the compounds under study also show binding with serotonergic and histaminergic receptors (see below). Another nicotinic agonist, a compound called TC-1734, was developed by Aventis Pharma. It not only increases ACh and bolsters cognitive functions in memory tests, but also possesses neuroprotective and antidepressant effects which may partly be due to cortical ACh release [139]. Nicotinic α7-receptor agonists were also found to enhance ACh in hippocampus and frontal cortex; their cognitive effects, however, also involve actions on the release of Glu [140] and/or DA [141].
6.5 Heteroreceptor-mediated stimulation of ACh release
Among the various approaches to influence ACh release through heteroceptors, select serotonergic and histaminergic agents have received focused attention in recent drug development studies. Serotonergic-cholinergic interactions are well known to contribute to cognitive processes [133], and antagonists at both 5-HT1A and 5-HT1B receptors were reported to increase ACh levels in microdialysis studies. The 5-HT1A antagonist WAY-101405 increased ACh in rat hippocampus and concomitantly antagonized memory deficits in fear conditioning induced by scopolamine [142]. 5-HT1B antagonists do not increase ACh levels by themselves, but they may be useful as a comedication to antidepressant 5-HT-selective re-uptake inhibitors (SSRIs) because SSRI treatment leads to a decline of ACh levels secondary to the increase of 5-HT levels [143]. The 5-HT6 receptor has been the target of drug development in cognition, schizophrenia and obesity. Procognitive functions of 5-HT6 antagonists concur with increases of ACh release in rat frontal cortex [144,145]. In both studies, 5-HT6 antagonists were also active in cognitive paradigms such as passive avoidance and object recognition tests. The mechanism of action of 5-HT6 antagonists may involve additional neuronal interactions because they were found to interact with glutamatergic and GABAergic systems as well (reviewed by Geldenhuys and Van der Schyf [146]).
Histaminergic neurons – in addition to their role in wakefulness and vigilance – have cognitive effects that seem to be mediated largely by modulation of cholinergic neurons via H1, H2 and/or H3 receptors. H3 histamine receptors are presynaptic receptors situated on cholinergic neurons which mitigate ACh release. Accordingly, H3 antagonists such as ABT-239 were found to increase ACh release in rat frontal cortex and hippocampus and were found to improve social memory [147]. Similarly, the H3 antagonist GSK 189254 increased ACh release in rat cingulate cortex and hippocampus and improved the performance of rats in passive avoidance, water maze and object recognition procedures [148]. Another H3 antagonist, JNJ-10181457, was found to increase ACh release in rat cortex, while also antagonizing scopolamine-induced deficits in behavioral assays including delayed non-matching to position and reversal learning [149]. While H3 antagonists also influence cortical levels of catecholamines, precognitive effects of these compounds are usually ascribed to their effects on ACh release in all cited studies.
A range of other compounds have been shown to modulate ACh release in microdialysis studies in vivo, including cannabinoids, purines such as adenosine, and many neuropeptides such as orexin and galanin [150]. These studies are not discussed here for lack of space and limited drug development efforts in these areas. Finally, microdialysis was also used to characterize drugs with unknown mechanisms of action, such as nootropic drugs including nimodipine, denbufylline, nefiracetam and linopirdine (DuP 996) [151].
6.7 Outlook
Microdialysis of ACh is now a routine procedure that can be established in experienced neurochemical laboratories within a few months. Analytical methods are available for reproducible determination of ACh in low femtomolar amounts, and many parameters of cholinergic function (as far as they are reflected in ACh levels) have been defined. Levels of ACh for example in striatal or hippocampal tissue are meaningful parameters and can be interpreted on the basis of much basic research into the role of ACh in motor function, motivation and addiction, attention and arousal, and cognitive processes. Microdialysis of ACh is uniquely suited to investigate ACh release in vivo. Occasionally, microdialysis is the only meaningful approach such as when ACh release from grafted neurons is monitored in vivo [152]. Nevertheless, at this time, there is a lack of studies that address ACh in the human brain. Such studies are urgently required to validate rodent findings for the therapeutic situation.
7 EXPERT OPINION
A reading of this review clarifies and emphasizes the versatility, utility and importance of in vivo microdialysis as an emerging mainstay technique for preclinical neuropharmacological research. In particular, elucidation of the neurochemical effects and mechanisms of action of established drugs and investigational new drug candidates through in vivo brain microdialysis is a field of investigation that is only now coming into its own. Exploitation of the power of this technique for drug discovery should proceed with vigor in the near future. With tremendous progress underway in the elucidation of drug-targetable mechanisms of addiction, pathways of neurodegeneration in AD, PD, amytrophic lateral sclerosis and other neurodegenerayive diseases, and an increasing understanding of the neuromolecular basis for many psychiatric diseases, targeted drug design aimed at these conditions has become a rational endeavor. Although in vivo brain microdialysis – like any other technique – has caveats that should be considered by the investigator, these issues can be mitigated with proper precautionary measures, good technique and diligent execution of safeguards. A particularly important advantage offered by this technique is the ability to measure neurotransmitters and their metabolites in awake, freely moving animals both pre- and post-drug administration. The size range and types of animals that can be used as models in investigations are also notable, varying from animals as small as the mouse (including the prospect of using transgenics) to large primates. Brain microdialysis – by virtue of its use of extremely accurate stereotaxic probe implantation – offers the capacity to measure neurotransmitters in discreet brain regions and permits investigators to locally apply selected pharmacological agents to the same brain regions via reverse dialysis. This approach allows immediate assessment of drug action at the pharmacological locus and establishes a contemporaneous neurochemical profile of investigational new drug candidates. Another key feature of in vivo brain microdialysis is the measurement of dialysate samples in conjunction with behavioral assays such as cognition or hyperlocomotion. Such approaches offer tremendous insights into the pharmacological profile and possible therapeutic applications of novel investigative agents that affect a variety of brain functions. If developed successfully, the increasing availability of on-line in vivo brain microdialysis protocols and informative tools aimed at making this technique useful for high-throughput screening of novel compounds opens the capacity to rapidly move investigational new drug candidates into the clinical trial stages and to market, thereby speeding up drug discovery efforts for neurological diseases that are in dire need of successful therapies.
ARTICLE HIGHLIGHTS.
In vivo microdialysis is a sampling technique with pre-clinical and clinical applications useful to assay the chemical contents of extracellular fluid.
Microdialysis has been extensively used over the past two decades to study the physiological and pharmacological changes of several low molecular weight substances in extraneuronal fluid.
Rodent brain in vivo microdialysis has enabled neurophysiologists, neurochemists and neuropharmacologists to measure neurochemical changes in discrete brain regions in freely moving, awake animals in response to pharmacological and physiological stimuli.
An extremely important application of this technique is its use in the measurement of drug-induced changes in concentrations of monoamine and amino acid neurotransmitters and Ach, and their respective metabolites.
In vivo microdialysis finds extensive use in the neurochemical characterization of neuropharmacological agents such as drugs of abuse, for neurotherapeutic agents such as antipsychotic agents and antidepressants, and for drugs used to improve neurocognitive deficits as well as learning and memory.
The in vivo microdialysis technique has been a boon to researchers in the development of novel classes of drugs with potential use in the treatment of neuropsychiatric disorders such as addiction, anxiety, attention deficit hyperactivity disorder, depression and schizophrenia as well as neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease. It has developed into a powerful tool in the development of therapeutic strategies for such diseases.
Acknowledgments
The assistance of Cindy Moore, Lisa Dirling and Natalie Sashkin is greatly appreciated.
Footnotes
Declaration of Interest
AS Darvesh, CJ Van der Schyf, RT Carroll and WJ Geldenhuys are all supported by Northeastern Ohio Universities Colleges of Medicine and Pharmacy (NEUOCOM). RT Caroll and WJ Geldenhuys are also supported by the Stark Community Foundation. GA Gudelsky is supported by a U.S. Public Health Service NIH Grant DA07427 while CK Meshul was supported by NIH grants R01 NS060662 and R01 AA011877 and by the Department of Veterans Affairs. Finally, J Klein was supported by Novartis.
References
- 1.Bourne JA. Intracerebral microdialysis: 30 years as a tool for the neuroscientist. Clin Exp Pharmacol Physiol. 2003;30:16–24. doi: 10.1046/j.1440-1681.2003.03789.x. [DOI] [PubMed] [Google Scholar]
- 2.Marsden CA. Measurement of neurotransmitter release by intracranial dialysis. In: Ungerstedt U, editor. Measurement of neurotransmitter release in vivo. John Wiley & Sons; New York: 1984. pp. 81–105. [Google Scholar]
- 3.Ungerstedt U, Pycock C. Functional correlates of dopamine neurotransmission. Bull Schweitz Akad Med Wiss. 1974;1278:1–13. [PubMed] [Google Scholar]
- 4.Ungerstedt U. Microdialysis-principles and applications for studies in animals and man. J Int Med. 1991;230:365–73. doi: 10.1111/j.1365-2796.1991.tb00459.x. [DOI] [PubMed] [Google Scholar]
- 5.Benveniste H. Brain Microdialysis. J Neurochem. 1989;52:1667–79. doi: 10.1111/j.1471-4159.1989.tb07243.x. [DOI] [PubMed] [Google Scholar]
- 6.Chaurasia CS. In vivo microdialysis sampling: theory and applications. Biomed Chromat. 1999;13:317–32. doi: 10.1002/(SICI)1099-0801(199908)13:5<317::AID-BMC891>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 7.Hansen DK, Davies MI, Lunte SM, et al. Pharmacokinetic and metabolism studies using microdialysis sampling. J Pharm Sci. 1999;88:14–27. doi: 10.1021/js9801485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen KC, Höistad M, Kehr J, et al. Theory relating in vitro and in vivo microdialysis with one or two probes. J Neurochem. 2002;81:108–21. doi: 10.1046/j.1471-4159.2002.00793.x. [DOI] [PubMed] [Google Scholar]
- 9.Höcht C, Opezzo JAW, Taira CA. Applicability of reverse microdialysis in pharmacological and toxicological studies. J Pharmacol Toxicol Met. 2007;55:3–15. doi: 10.1016/j.vascn.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 10.Pan YF, Feng J, Cheng QY, et al. Intracerebral microdialysis technique and its application on brain pharmacokinetic-pharmacodynamic study. Arch Pharm Res. 2007;30:1635–45. doi: 10.1007/BF02977335. [DOI] [PubMed] [Google Scholar]
- 11.Schultz KN, Kennedy RT. Time-resolved microdialysis for in vivo neurochemical measurements and other applications. Ann Rev Anal Chem. 2008;1:627–61. doi: 10.1146/annurev.anchem.1.031207.113047. [DOI] [PubMed] [Google Scholar]
- 12.Chefer VI, Thompson AC, Zapata A. Overview of brain microdialysis. Curr Prot Neurosci. 2009:7.1.1–28. doi: 10.1002/0471142301.ns0701s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van der Zeyden M, Oldenziel WH, Rea K, et al. Microdialysis of GABA and glutamate: analyziz, interpretation and comparison with microsensors. Pharmacol Biochem Behav. 2008;90:135–47. doi: 10.1016/j.pbb.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 14.Lanckmans K, Sarre S, Smolders I, et al. Quatitative liquid chromatography/mass spectrometry for the analysis of microdialysates. Talanta. 2008;74:458–69. doi: 10.1016/j.talanta.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 15.Cheng GW, Hsu KC, Lee CF, et al. On-line microdialysis coupled with liquid chromatography for biomedical analysis. J Chromat Sci. 2009:624–30. doi: 10.1093/chromsci/47.8.624. [DOI] [PubMed] [Google Scholar]
- 16.Perry M, Li Q, Kennedy RT. Review of recent advances in analytical techniques for the determination of neurotransmitters. Analytica Chimica Acta. 2009;653:1–22. doi: 10.1016/j.aca.2009.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bourin M, David DJ, Jolliet P, et al. Mechanism of action of antidepressants and therapeutic perspectives. Therapie. 2002;57:385–96. [PubMed] [Google Scholar]
- 18.Guiard BP, Lanfumey L, Gardier AM. Microdialysis approach to study serotonin outflow in mice following selective serotonin reuptake inhibitors and substance P (neurokinin 1) receptor antagonist administration: a review. Curr Drug Targets. 2006;7:187–201. doi: 10.2174/138945006775515428. [DOI] [PubMed] [Google Scholar]
- 19.Beyer CE, Cremers TI. Do selective serotonin reuptake inhibitors acutely increase frontal cortex levels of serotonin? Eur J Pharmacol. 2008;580:350–54. doi: 10.1016/j.ejphar.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 20.Kennedy SH, Rizvi SJ. Emerging drugs for major depressive disorder. Expert Opin Emerg Drugs. 2009;14:439–53. doi: 10.1517/14728210903107751. [DOI] [PubMed] [Google Scholar]
- 21.Racagni G, Popoli M. The pharmacological properties of antidepressants. Int Clin Psychopahrmacol. 2010;25:117–31. doi: 10.1097/YIC.0b013e3283311acd. [DOI] [PubMed] [Google Scholar]
- 22.National Institute on Drug Abuse. NIDA InfoFacts. 2010 http://nida.nih.gov/DrugPages.
- 23.Yamamoto BK, Moszczynska A, Gudelsky GA. Amphetamine toxicities Classical and emerging mechanisms. Ann NY Acad Sci. 2010;1187:101–21. doi: 10.1111/j.1749-6632.2009.05141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci. 1995;15:4102–08. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: A review. Prog Neurobiol. 2005;75:406–33. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 26.Rothman RB, Baumann MH. Monoamine transporters and psychostimulant drugs. Eur J Pharmacol. 2003;479:23–40. doi: 10.1016/j.ejphar.2003.08.054. [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto BK, Nash JF, Gudelsky GA. Modulation of methylenedioxymethamphetamine-induced striatal dopamine release by the interaction between serotonin and GABA in the substantia nigra. J Pharmacol Exp Ther. 1995;273:1063–70. [PubMed] [Google Scholar]
- 28.Shankaran M, Yamamoto BK, Gudelsky GA. Mazindol attenuates the 3,4-methylenedioxymethamphetamine-induced formation of hydroxyl radicals and long-term depletion of serotonin in the striatum. J Neurochem. 1999;72:2516–22. doi: 10.1046/j.1471-4159.1999.0722516.x. [DOI] [PubMed] [Google Scholar]
- 29.Bowyer JF, Newport GD, Slikker W, Jr, et al. An evaluation of l-ephedrine neurotoxicity with respect to hyperthermia and caudate/putaman microdialysate levels of ephedrine, dopamine, serotonin, and glutamate. Toxicol Sci. 2000;55:133–42. doi: 10.1093/toxsci/55.1.133. [DOI] [PubMed] [Google Scholar]
- 30.Gough B, Imam SZ, Blough B, et al. Comparative effects of substituted amphetamines (PMA, MDMA, and METH) on monoamines in rat caudate. A microdialysis study. Ann NY Acad Sci. 2002;965:410–20. doi: 10.1111/j.1749-6632.2002.tb04182.x. [DOI] [PubMed] [Google Scholar]
- 31.Shoblock JR, Sulliven EB, Maisonneuve IM, Glick SD. Neurochemical and behavioral differences between d-methamphetamine and d-amphetamine in rats. Psychopharmacol. 2003;165:359–69. doi: 10.1007/s00213-002-1288-7. [DOI] [PubMed] [Google Scholar]
- 32.Shankaran M, Gudelsky GA. Effect of 3,4-methylenedioxymethamphetamine (MDMA) on hippocampal dopamine and serotonin. Pharmcol Biochem Behav. 1998;61:361–66. doi: 10.1016/s0091-3057(98)00103-8. [DOI] [PubMed] [Google Scholar]
- 33.Bankson MG, Yamamoto BK. Serotonin-GABA interactions modulate MDMA-induced mesolimbic dopamine release. J Neurochem. 2004;91:852–59. doi: 10.1111/j.1471-4159.2004.02763.x. [DOI] [PubMed] [Google Scholar]
- 34.Nair SG, Gudelsky GA. Protein kinase c inhibition differentially affects 3,4-methylenedioxymethamphetamine-induced dopamine release in the striatum and prefrontal cortex of the rat. Brain Res. 2004;1013:168–73. doi: 10.1016/j.brainres.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 35.Gudelsky GA, Yamamoto BK. Actions of 3,4-methylenedioxiymethamphetamine (MDMA) on cerebral dopaminergic, serotonergic and cholinergic neurons. Pharmacol Biochem Behav. 2008;90:198–207. doi: 10.1016/j.pbb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shioda K, Nisijima K, Yoshino T, Kato S. Effect of risperidone on acute methamphetaime-induced hyperthermia in rats. Drug Alcohol Depend. 2010;111:241–49. doi: 10.1016/j.drugalcdep.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 37.Kuczenski R, Segal DS, Cho AK, et al. Hippocampal norepinephrine, caudate dopamine and serotonin, and behavioral responses to the stereoisomers of amphetamine and methamphetamine. J Neurosci. 1995;15:1308–17. doi: 10.1523/JNEUROSCI.15-02-01308.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gough B, Ali SF, Slikker W, Jr, et al. Acute effects of 3,4-methylenedioxymethamphetamine (MDMA) on monoamines in rat caudate. Pharmacol Biochem Behav. 1991;39:619–23. doi: 10.1016/0091-3057(91)90137-q. [DOI] [PubMed] [Google Scholar]
- 39.Gudelsky GA, Nash JF. Carrier-mediated release of serotonin by 3,4-methylenedioxymethamphetamine: implications for serotonin-dopamine interactions. J Neurochem. 1996;66:243–49. doi: 10.1046/j.1471-4159.1996.66010243.x. [DOI] [PubMed] [Google Scholar]
- 40.Baumann MH, Clark RD, Rothman RB. Locomotor stimulation produced by 3,4-methylenedioxymethamphetamine (MDMA) is correlated with dialysate levels of serotonin and dopamine in rat brain. Pharmacol Biochem Behav. 2008;90:208–17. doi: 10.1016/j.pbb.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rothman RB, Baumann MH. Balance between dopamine and serotonin release modulates behavioral effects of amphetamine-type drugs. Ann NY Acad Sci. 2006;1074:245–60. doi: 10.1196/annals.1369.064. [DOI] [PubMed] [Google Scholar]
- 42.Rothman RB, Blough BE, Baumann MH. Dual dopamine/serotonin releasers: potential treatment agents for stimulant addiction. Exp Clin Psychopharmacol. 2008;16:458–74. doi: 10.1037/a0014103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rothman RB, Blough BE, Baumann MH. Dual dopamine-5-HT releasers: potential treatment agents for cocaine addiction. Trends Pharmacol Sci. 2006;27:612–18. doi: 10.1016/j.tips.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 44.Rothman RB, Partilla JS, Baumann MH, et al. Neurochemical neutralization of methamphetamine with high-affinity nonselective inhibitors of biogenic amine transporters: a pharmacological strategy for treating stimulant abuse. Synapse. 2000;35:222–27. doi: 10.1002/(SICI)1098-2396(20000301)35:3<222::AID-SYN7>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 45.Dackis CA, Gold MS. New concepts in cocaine addiction: the dopamine depletion hypothesis. Neurosci Biobehav Rev. 1985;9:469–77. doi: 10.1016/0149-7634(85)90022-3. [DOI] [PubMed] [Google Scholar]
- 46.Czoty PW, Ginsberg BC, Howell LL. Serotonergic attenuation of the reinforcing and neurochemical effects of cocaine in squirrel monkeys. J Pharmacol Exp Ther. 2002;300:831–37. doi: 10.1124/jpet.300.3.831. [DOI] [PubMed] [Google Scholar]
- 47.Negus SS, Mello NK, Blough BE, et al. Monoamine releasers with varying selectivity for dopamine/norepinephrine versus serotonin release as candidate “agonist” medications for cocaine dependence: studies in assays of cocaine discrimination and cocaine self-administration in rhesus monkeys. J Pharmacol Exp Ther. 2007;320:627–36. doi: 10.1124/jpet.106.107383. [DOI] [PubMed] [Google Scholar]
- 48.Durston S. A review of the biological bases of ADHD: what have we learned from imaging studies? Ment Retard Dev Disabil Res Rev. 2003;9:184–95. doi: 10.1002/mrdd.10079. [DOI] [PubMed] [Google Scholar]
- 49.Durston S, Tottenham NT, Thomas KM, et al. Differential patterns of striatal activation in young children with and without ADHD. Biol Psychiatry. 2003;53:871–78. doi: 10.1016/s0006-3223(02)01904-2. [DOI] [PubMed] [Google Scholar]
- 50.Solanto MV. Dopamine dysfunction in AD/HD: integrating clinical and basic neuroscience research. Behav Brain Res. 2002;130:65–71. doi: 10.1016/s0166-4328(01)00431-4. [DOI] [PubMed] [Google Scholar]
- 51.Berridge CW, Devilbiss DM, Andrzejewski ME, et al. methylphenidate preferentially increases catecholamine neurotransmission with the prefrontal cortex at low doses that enhance cognitive function. Biol Psychiatry. 2006;60:1111–20. doi: 10.1016/j.biopsych.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 52.Bymaster FP, Katner JS, Nelson DL, et al. Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology. 2002;27:699–711. doi: 10.1016/S0893-133X(02)00346-9. [DOI] [PubMed] [Google Scholar]
- 53.Swanson CJ, Perry KW, Koch-Krueger S, et al. Effect of the attention deficit/hyperactivity disorder. Drug atomoxetine on extracellular concentrations of norepinephrine and dopamine in several brain regions of the rat. Neuropharmacology. 2006;50:755–60. doi: 10.1016/j.neuropharm.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 54.Horner WE, Johnson DE, Schmidt AW, et al. Methylphenidate and atomoxetine increase histamine release in rat prefrontal cortex. Eur J Pharmacol. 2007;558:96–7. doi: 10.1016/j.ejphar.2006.11.048. [DOI] [PubMed] [Google Scholar]
- 55.Liu LL, Yang J, Lei GF, et al. Atomoxetine increases histamine release and improves learning deficits in an animal model of attention deficit hyperactivity disorder: the spontaneously hypertensive rat. Basic Clin Pharmacol Toxicol. 2008;102:527–32. doi: 10.1111/j.1742-7843.2008.00230.x. [DOI] [PubMed] [Google Scholar]
- 56.Heal DJ, Smith SL, Kulkarni RS, et al. New perspectives from microdialysis studies in freely-moving, spontaneously hypertensive rats in the pharmacology of drugs in the treatment of ADHD. Pharmacol Biochem Behav. 2008;90:184–87. doi: 10.1016/j.pbb.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 57.Heal DJ, Cheetham SC, Smith SL. The neuropharmacology of ADHD drugs in vivo: Insights on efficacy and safety. Neuropharmacol. 2009;57:608–18. doi: 10.1016/j.neuropharm.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 58.Moreland RB, Patel M, Hsieh GC, et al. A-412997 is a selective dopamine D4 agonist in rats. Pharmacol Biochem Behav. 2005;82:140–47. doi: 10.1016/j.pbb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 59.Woolley ML, Waters KA, Reavill C, et al. Selective dopamine D4 receptor agonist (A-412997) improves cognitive performance and stimulates motor activity without influencing reward-related behavior in rat. Behav Pharmacol. 2008;19:765–76. doi: 10.1097/FBP.0b013e32831c3b06. [DOI] [PubMed] [Google Scholar]
- 60.Carroll RT, Jr, Sharmeen L. United States, WARNER-LAMBERT LLC; 9201 Tabor Road, Morris Plains, NJ, US: COMBINATION OF ATOMOXETINE AND A 5HT1A RECEPTOR AGONIST FOR TREATING ADHD AND OTHER DISORDERS. http://www.freepatentsonline.com/y2007/0219201.html. [Google Scholar]
- 61.Dounay AB, Barta NS, Campbell BM, et al. Design, synthesis, and pharmacological evaluation of phenoxy pyridyl derivatives as dual norepinephrine reuptake inhibitors and 5-HT1A partial agonists. Bioorg Med Chem Lett. 2010;20:1114–7. doi: 10.1016/j.bmcl.2009.12.023. [DOI] [PubMed] [Google Scholar]
- 62.Ichikawa J, Meltzer HY. Relationship between dopaminergic and serotonergic neuronal activity in the frontal cortex and the action of typical and atypical antipsychotic drugs. Eur Arch Psychiatry Clin Neurosci. 1999;249S:90–98. doi: 10.1007/pl00014190. [DOI] [PubMed] [Google Scholar]
- 63.Kuroki T, Nagao N, Nakahara T. Neuropharmacology of second-generation antipsychotic drugs: a validity of the serotonin-dopamine hypothesis. In: Di Giovanni G, Di Matteo V, Esposito E, editors. Serotonin-dopamine interaction: experimental evidence and therapeutic relevance, Progress in Brain Research. Vol. 172. Elsevier Science BV; Amsterdam: 2008. pp. 199–212. [DOI] [PubMed] [Google Scholar]
- 64.López-Gil X, Artigas F, Adell A. Unraveling monoamine receptors involved in the action of typical and atypical antipsychotics on glutamatergic and serotonergic transmission in prefrontal cortex. Curr Pharm Des. 2010;16:502–15. doi: 10.2174/138161210790361416. [DOI] [PubMed] [Google Scholar]
- 65.Davis KL, Kahn RS, Ko G, et al. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. 1991;148:1474–86. doi: 10.1176/ajp.148.11.1474. [DOI] [PubMed] [Google Scholar]
- 66.Meltzer HT, Huang M. In vivo actions of atypical antipsychotic drug on serotonergic and dopaminergic systems. In: Di Giovanni G, Di Matteo V, Esposito E, editors. Serotonin-dopamine interaction: experimental evidence and therapeutic relevance, Progress in Brain Research. Vol. 172. Elsevier Science BV; Amsterdam: 2008. pp. 177–97. [DOI] [PubMed] [Google Scholar]
- 67.Moghaddam B, Bunney BS. Acute effects of typical and atypical antipsychotic drugs on the release of dopamine from prefrontal cortex, nucleus accumbens, and striatum of the rat: an in vivo microdialysis study. J Neurochem. 1990;54:1755–60. doi: 10.1111/j.1471-4159.1990.tb01230.x. [DOI] [PubMed] [Google Scholar]
- 68.Pehek EA, Yamamoto BK. Differential effects of locally administered clozapine and haloperidol on dopamine efflux in the rat prefrontal cortex and caudate-putamen. J Neurochem. 1994;63:2118–24. doi: 10.1046/j.1471-4159.1994.63062118.x. [DOI] [PubMed] [Google Scholar]
- 69.Kuroki T, Meltzer HY, Ichikawa J. Effects of antipsychotic drugs on extracellular dopamine levels in rat medial prefrontal cortex and nucleus accumbens. J Pharmacol Exp Ther. 1999;288:774–81. [PubMed] [Google Scholar]
- 70.Deutch AY, Cameron DS. Pharmacological characterization of dopamine systems in the nucleus accumbens core and shell. Neurosci. 1992;46:49–56. doi: 10.1016/0306-4522(92)90007-o. [DOI] [PubMed] [Google Scholar]
- 71.Liégeois JF, Ichikawa J, Meltzer HY. 5-HT2A receptor antagonism potentiates haloperidol-induced dopamine release in rat medial prefrontal cortex and inhibits that in the nucleus accumebns in a dose-dependent manner. Brain Res. 2002;947:157–65. doi: 10.1016/s0006-8993(02)02620-3. [DOI] [PubMed] [Google Scholar]
- 72.Li Z, ichikawa J, Huang M, et al. ACP-103, a 5-HT2A/2C inverse agonist, potentiates haloperidol-induced dopamine release in rat medial prefrontal cortex and nucleus accumbens. Pyschopharmacol. 2005;183:144–53. doi: 10.1007/s00213-005-0170-9. [DOI] [PubMed] [Google Scholar]
- 73.Bonaccorso S, Meltzer HY, Li Z, Dai J, Alboszta AR, Ichikawa J. SR46349-B, a 5-HT2A/2C receptor antagonist, potentiates haloperidol-induced dopamine release in rat medial prefrontal cortex and nucleus accumbens. Neuropsychopharmacology. 2002;27:430–41. doi: 10.1016/S0893-133X(02)00311-1. [DOI] [PubMed] [Google Scholar]
- 74.Ichikawa J, Ishii H, Bonaccorso S, et al. 5-HT(2A) and D92) receptor blockade increases cortical DA release via 5-HT91A) receptor activation: a possible mechanism of atypical antipsychotic-induced cortical dopamine release. J Neurochem. 2001;76:1521–31. doi: 10.1046/j.1471-4159.2001.00154.x. [DOI] [PubMed] [Google Scholar]
- 75.Youngren KD, Inglis FM, Pivirotto PJ, et al. Clozapine preferentially increases dopamine release in the rhesus monkey prefrontal cortex compared with the caudate nucleus. Neuropsychopharmacol. 1999;20:403–12. doi: 10.1016/S0893-133X(98)00082-7. [DOI] [PubMed] [Google Scholar]
- 76.Hagger C, Buckley P, Kenny JT, et al. Improvement in cognitive functions and psychiatric symptoms in treatment-refractory schizophrenic patients receiving clozapine. Biol Psychiatry. 1993;34:702–12. doi: 10.1016/0006-3223(93)90043-d. [DOI] [PubMed] [Google Scholar]
- 77.Shirazi-Southall S, Rodroguez DE, Nomikos GG. Effects of typical and atypical antipsychotics and receptor selective compounds on acetylcholine efflux in the hippocampus of the rat. Neuropsychopharmacol. 2002;26:583–94. doi: 10.1016/S0893-133X(01)00400-6. [DOI] [PubMed] [Google Scholar]
- 78.Ichikawa J, Dai J, O’Laughlin IA, et al. Atypical, but not typical, antipsychotic drugs increase cortical acetylcholine release without an effect in the nucleus accumbens or striatum. Neuropsychopharmacol. 2002;26:325–39. doi: 10.1016/S0893-133X(01)00312-8. [DOI] [PubMed] [Google Scholar]
- 79.Huang M, Li Z, Ichikawa J, et al. Effects of divalproex and atypical antipsychotic drugs on dopamine and acetylcholine efflux in rat hippocampus and prefrontal cortex. Brain Res. 2006;1099:44–55. doi: 10.1016/j.brainres.2006.04.081. [DOI] [PubMed] [Google Scholar]
- 80.Amargós-Bosch M, López-Gil X, Artigas F, et al. Clozapine and olanzapine, but not haloperidol, suppress serotonin efflux in the medial prefrontal cortex elicited by phencyclidine and ketamine. Int J Neuropsychopharmacol. 2006;9:565–73. doi: 10.1017/S1461145705005900. [DOI] [PubMed] [Google Scholar]
- 81.López-Gil X, Babot Z, Amargós-Bosch M, et al. Clozapine and haloperidol differently suppress the MK-801-increased glutamatergic and serotonergic transmission in the medial prefrontal cortex of the rat. Neuropsychopharmacol. 2007;32:2087–97. doi: 10.1038/sj.npp.1301356. [DOI] [PubMed] [Google Scholar]
- 82.Linn GS, Negi S, Gerum SV, et al. Reversal of phencyclidine-induced prepulse inhibition deficits by clozapine in monkeys. Psychopharmacol. 2003;169:234–39. doi: 10.1007/s00213-003-1533-8. [DOI] [PubMed] [Google Scholar]
- 83.Bourdelais AJ, Deutch AY. The effects of haloperidol and clozapine on extracellular GABA levels in the prefrontal cortex of the rat: an in vivo microdialysis study. Cereb Cortex. 1994;4:69–77. doi: 10.1093/cercor/4.1.69. [DOI] [PubMed] [Google Scholar]
- 84.See RE, Berglind WJ, Krentz L, et al. Convergent evidence from microdialysis and presynaptic immunolabeling for the regulation of gamma aminobutyric acid release in the globus pallidus following acute clozapine or haloperidol administration in rats. J Neurochem. 2002;82:172–80. doi: 10.1046/j.1471-4159.2002.00974.x. [DOI] [PubMed] [Google Scholar]
- 85.Freund TF, Powell JF, Smith AD. Tyrosine hydroxylase-immunoreactive boutons in synaptic contact with identified striatonigral neurons, with particular reference to dendritic spines. Neurosci. 1984;13:1189–1215. doi: 10.1016/0306-4522(84)90294-x. [DOI] [PubMed] [Google Scholar]
- 86.Lacey CJ, Boyes J, Gerlach O, et al. GABA-B receptors at glutamatergic synapses in the rat striatum. Neurosci. 2005;136:1083–95. doi: 10.1016/j.neuroscience.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 87.Meshul CK, Stallbaumer RK, Taylor B, et al. Haloperidol-induced synaptic changes in striatum are associated with glutamate synapses. Brain Res. 1994;648:181–95. doi: 10.1016/0006-8993(94)91117-7. [DOI] [PubMed] [Google Scholar]
- 88.Lindefors N, Ungerstedt U. Bilateral regulation of glutamate tissue and extracellular levels in caudate-putamen by midbrain dopamine neurons. Neurosci Lett. 1990;115:248–52. doi: 10.1016/0304-3940(90)90463-j. [DOI] [PubMed] [Google Scholar]
- 89.Reid MS, Herrera-Marschitz M, Kehr J, et al. Striatal dopamine and glutamate release: effects of intranigral injections of substance P. Acta Physiol Scand. 1990;140:527–37. doi: 10.1111/j.1748-1716.1990.tb09030.x. [DOI] [PubMed] [Google Scholar]
- 90.Touchon JC, Holmer HK, Moore C, et al. Apomorphine-induced alterations in striatal and substantia nigra glutamate following unilateral loss of striatal dopamine. Exp Neurol. 2005;193:131–40. doi: 10.1016/j.expneurol.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 91.Calabresi P, Mercuri NB, Sancesario G, et al. Electrophysiology of dopamine-denervated striatal neurons. Brain. 1993;116:433–52. [PubMed] [Google Scholar]
- 92.Schultz W, Ungerstedt U. Short-term increase and long-term reversion of striatal cell activity after degeneration of the nigrostriatal dopamine system. Exp Brain Res. 1978;33:159–71. doi: 10.1007/BF00238057. [DOI] [PubMed] [Google Scholar]
- 93.Meshul CK, Emre N, Nakamura CM, et al. Time-dependent changes in striatal glutamate synapses following a 6-hydroxydopamine lesion. Neurosci. 1999;88:1–16. doi: 10.1016/s0306-4522(98)00189-4. [DOI] [PubMed] [Google Scholar]
- 94.Garcia-Arencibia M, Ferraro L, Tanganelli S, et al. Enhanced striatal glutamate release after the administration of rimonabant to 6-hydroxydopamine-lesioned rats. Neurosci Letts. 2008;438:10–13. doi: 10.1016/j.neulet.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 95.Jonkers N, Sarre S, Ebinger G, et al. MK801 suppresses the L-DOPA-induced increase of glutamate in striatum of hemi-Parkinson rats. Brain Res. 2002;926:149–55. doi: 10.1016/s0006-8993(01)03147-x. [DOI] [PubMed] [Google Scholar]
- 96.Walker RH, Koch RJ, Sweeney JE, et al. Effects of subthalamic nucleus lesions and stimulation upon glutamate levels in the dopamine-depleted rat striatum. Neuroreport. 2009;20:770–75. doi: 10.1097/WNR.0b013e32832ad556. [DOI] [PubMed] [Google Scholar]
- 97.Bianchi L, Galeffi F, Bolam JP, et al. The effect of 6-hydroxydopamine lesions on the release of amino acids in the direct and indirect pathways of the basal ganglia: a dual microdialysis probe analysis. Eur J Neurosci. 2003;18:856–868. doi: 10.1046/j.1460-9568.2003.02795.x. [DOI] [PubMed] [Google Scholar]
- 98.Marti M, Mela F, Bianchi C, et al. Striatal dopamine-NMDA receptor interactions in the modulation of glutamate release in the substantia nigra pars reticulata in vivo: opposite role for D1 and D2 receptors. J Neurochem. 2002;83:635–644. doi: 10.1046/j.1471-4159.2002.01169.x. [DOI] [PubMed] [Google Scholar]
- 99.Robelet S, Melon C, Guillet B, et al. Chronic L-DOPA treatment increases extracellular glutamate levels and GLT1 expression in the basal ganglia in a rat model of Parkinson’s disease. Eur J Neurosci. 2004;20:1255–1266. doi: 10.1111/j.1460-9568.2004.03591.x. [DOI] [PubMed] [Google Scholar]
- 100.Holmer HK, Keyghobadi M, Moore C, et al. L-Dopa-induced reversal in striatal glutamate following partial depletion of nigrostriatal dopamine with MPTP. Neurosci. 2005;136:333–341. doi: 10.1016/j.neuroscience.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 101.Robinson S, Freeman P, Moore C, et al. Acute and subchronic MPTP administration differentially affects striatal glutamate synaptic function. Expe Neurol. 2003;180:73–86. doi: 10.1016/s0014-4886(02)00050-x. [DOI] [PubMed] [Google Scholar]
- 102.Petroske E, Meredith GE, Callen S, et al. Mouse model of Parkinsonism: a comparison between subacute MPTP and chronic MPTP/probenecid treatment. Neurosci. 2001;106:589–601. doi: 10.1016/s0306-4522(01)00295-0. [DOI] [PubMed] [Google Scholar]
- 103.Dervan AG, Meshul CK, Beales M, et al. Astroglial plasticity and glutamate function in a chronic mouse model of Parkinson’s disease. Exp Neurol. 2004;190:145–156. doi: 10.1016/j.expneurol.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 104.Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–75. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- 105.Raju DV, Shah DJ, Wright TM, et al. Differential synaptology of vGluT2-containing thalamostriatal afferents between the patch and matrix compartments. J Comp Neurol. 2006;499:321–243. doi: 10.1002/cne.21099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fremeau RT, Jr, Troyer MD, Pahner I, et al. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- 107.Bacci JJ, Kachidian P, Kerkerian-Le Goff L, et al. Intralaminar thalamic nuclei lesions: widespread impact on dopamine denervation-mediated cellular defects in the rat basal ganglia. J Neuropath Exp Neurol. 2004;63:20–31. doi: 10.1093/jnen/63.1.20. [DOI] [PubMed] [Google Scholar]
- 108.Raju DV, Ahern TH, Shah DJ, et al. Differential synaptic plasticity of the corticostriatal and thalamostriatal systems in an MPTP-treated monkey model of parkinsonism. Eur J Neurosci. 2008;27:1647–1658. doi: 10.1111/j.1460-9568.2008.06136.x. [DOI] [PubMed] [Google Scholar]
- 109.Touchon JC, Moore C, Frederickson J, Meshul CK. Lesion of subthalamic or motor thalamic nucleus in 6-hydroxydopamine treated rats: effects on striatal glutamate and apomorphine-induced contralateral rotations. Synapse. 2004;51:287–98. doi: 10.1002/syn.10306. [DOI] [PubMed] [Google Scholar]
- 110.Holmer HK, Keyghobadi M, Moore C, et al. Dietary restriction affects striatal glutamate in the MPTP-induced mouse model of nigrostriatal degeneration. Synapse. 2005;57:100–112. doi: 10.1002/syn.20163. [DOI] [PubMed] [Google Scholar]
- 111.Perisike M, Zimmermann M, Klein J, et al. Quantitative determination of acetylcholine and choline in microdialysis samples by MALDI-TOF-MS. Anal Chem. 2010;82:922–29. doi: 10.1021/ac902130h. [DOI] [PubMed] [Google Scholar]
- 112.Löffelholz K, Klein J. Precursors: choline and glucose. In: Giacobini E, Pepeu G, editors. The Brain Cholinergic System. Informa/Taylor and Francis; London: 2006. pp. 99–105. [Google Scholar]
- 113.Sarter M, Parikh V. Choline transporters, cholinergic transmission and cognition. Nature Rev Neurosci. 2005;6:48–56. doi: 10.1038/nrn1588. [DOI] [PubMed] [Google Scholar]
- 114.Day JC, Kornecook TJ, Quirion R. Application of in vivo microdialysis to the study of cholinergic systems. Methods. 2001;23:21–39. doi: 10.1006/meth.2000.1103. [DOI] [PubMed] [Google Scholar]
- 115.Phillis JW. Acetylcholine release from the central nervous system:a 50-year retrospective. Crit Rev Neurobiol. 2005;17:161–217. doi: 10.1615/critrevneurobiol.v17.i3-4.30. [DOI] [PubMed] [Google Scholar]
- 116.Gold P. Acetylcholine modulation of neural systems involved in learning and memory. Neurobiol learn Mem. 2003;80:194–210. doi: 10.1016/j.nlm.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 117.Henn C, Löffelholz K, Klein J. Stimulatory and inhibitory effects of ethanol on hippocampal acetylcholine reelase. Naunyn-Schmeideberg’s Arch Pharmacol. 1998;357:640–47. doi: 10.1007/pl00005219. [DOI] [PubMed] [Google Scholar]
- 118.Buchholzer M, Dvorak C, Chatterjee SS, et al. Dual modulation of striatal acetylcholine release by hyperforin, a constituent of St. John’s wort. J Pharmacol Exp Ther. 2002;301:714–19. doi: 10.1124/jpet.301.2.714. [DOI] [PubMed] [Google Scholar]
- 119.Westernick BHC. Brain microdialysis and its application for the study of animal behavior. Behav Brain Res. 1995;70:103–24. doi: 10.1016/0166-4328(95)80001-8. [DOI] [PubMed] [Google Scholar]
- 120.Pepeu G, Giovannini MG. Changes in acetylcholine extracellular levels during cognitive processes. Learn Mem. 2004;11:21–27. doi: 10.1101/lm.68104. [DOI] [PubMed] [Google Scholar]
- 121.Kopf SR, Buchholzer ML, Hilgert M, et al. Glucose plus choline improve passive avoidance behavior and increase hippocampal acetylcholine release in mice. Neurosci. 2001;103:365–71. doi: 10.1016/s0306-4522(01)00007-0. [DOI] [PubMed] [Google Scholar]
- 122.Klein J. Phenserine. Expert Opin Investig Drugs. 2007;16:1087–87. doi: 10.1517/13543784.16.7.1087. [DOI] [PubMed] [Google Scholar]
- 123.Hilgert M, Nöldner M, Chatterjee SS, et al. KA-672 inhibits rat brain acetylcholinesterase in vitro, but not in vivo. Neurosci Lett. 1999;263:193–96. doi: 10.1016/s0304-3940(99)00149-4. [DOI] [PubMed] [Google Scholar]
- 124.Liu JK, Kato T. Effect of physostigmine on relative acetylcholine output induced by systemic treatment with scopolamine in in vivo microdialysis of rat frontal cortex. Neurochem Int. 1994;24:589–586. doi: 10.1016/0197-0186(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 125.Giacobini E. Cholinesterase inhibitors: new roles and therapeutic alternatives. Pharmacol Res. 2004;50:433–40. doi: 10.1016/j.phrs.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 126.Naik RS, Hartmann J, Kiewert C, et al. Effects of rivastigmine and donepezil on brian acetylcholine levels in acetylcholinesterase-deficient mice. J Pharm Pharmaceut Sci. 2009;12:79–85. doi: 10.18433/j3mk59. [DOI] [PubMed] [Google Scholar]
- 127.Pepeu G, Giovannini MG. Cholinesterase inhibitors and memory. Chem-Biol Interact. 2009;187:403–08. doi: 10.1016/j.cbi.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 128.Hartmann J, Kiewert EG, Duysen EG, et al. Excessive hippocampal acetylcholine levels in acetylcholinesterase-deficient mice are moderated by butyrylcholinesterase activity. J Neurochem. 2007;100:1421–29. doi: 10.1111/j.1471-4159.2006.04347.x. [DOI] [PubMed] [Google Scholar]
- 129.Yu QS, Holloway HW, Utsuki T, et al. Synthesis of novel phenserine-based selective inhibitors of butyrylcholinesterase for Alzhemier’s disease. J Med Chem. 1999;42:1855–61. doi: 10.1021/jm980459s. [DOI] [PubMed] [Google Scholar]
- 130.Greig NH, Utsuki T, Ingram DK, et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc Natl Acad Sci USA. 2005;102:17213–18. doi: 10.1073/pnas.0508575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Toda N, Kaneko T, Kogen H. Development of an efficient therapeutic agent for Alzheimer’s disease: design and sysmthesis of dual inhibitors of acetylcholinesterase and serotonin transporter. Chem Pharm Bull. 2010;58:273–87. doi: 10.1248/cpb.58.273. [DOI] [PubMed] [Google Scholar]
- 132.Van der Schyf CJ, Geldenhuys WJ, Youdim MB. Multifunctional drugs with different CNS targets for neuropsychiatric disorders. J Neurochem. 2006;99:1033–48. doi: 10.1111/j.1471-4159.2006.04141.x. [DOI] [PubMed] [Google Scholar]
- 133.Cassel JC, Jeltsch H. Serotonergic modulation of cholinergic function in the central nervous system: cognitive implications. Neurosci. 1995;69:1–41. doi: 10.1016/0306-4522(95)00241-a. [DOI] [PubMed] [Google Scholar]
- 134.Carey GJ, Billard W, Binch H, III, et al. SCH 57790, a selective muscarinic (M2) receptor antagonist, releases acetylcholine and produces cognitive enhancement in laboratory animals. Eur J Pharmacol. 2001;431:189–200. doi: 10.1016/s0014-2999(01)01440-6. [DOI] [PubMed] [Google Scholar]
- 135.Wang Y, Chackalamannil S, Hu Z, et al. Improving the oral efficacy of CNS drug candidates: discovery of highly orally efficacious piperidinyl piperidine M2 muscarinic receptor antagonists. J Med Chem. 2002;45:5415–18. doi: 10.1021/jm0255163. [DOI] [PubMed] [Google Scholar]
- 136.Emmerling MR, Gregor VE, Schwarz RD, et al. PD 142676 (CI 1002), a novel anticholinesterase and muscarinic antagonist. Mol Neurobiol. 1994;9:93–106. doi: 10.1007/BF02816108. [DOI] [PubMed] [Google Scholar]
- 137.Bontempi B, Whelan KT, Risbrough VB, et al. SIB-1553A, (+/−)-4-[[2-(1-methyl-2-pyrrolidinyl)ethyl]thio]phenol hydrochloride, a subtype-selective ligand for nicotinic acetylcholine receptors with putative cognitive-enhancing properties: effects on working and reference memory performances in aged rodents and nonhuman primates. J Pharmacol Exp Ther. 2001;299:297–306. [PubMed] [Google Scholar]
- 138.Rao TS, Reid RT, Correa LD, et al. In vivo pharmacological characterization of (+/−)-4-[2-(1-methyl-2-pyrrolidinyl)ethyl]thiophenol hydrochloride (SIB-1553A), a novel cholinergic ligand: microdialysis studies. Brain Res. 2003;986:71–81. doi: 10.1016/s0006-8993(03)03174-3. [DOI] [PubMed] [Google Scholar]
- 139.Gatto Gj, Bohme GA, Calfwell WS, et al. TC-1734: an orally active neuronal nicotinic acetylcholine receptor modulator with antidepressant, neuroprotective and long lasting cognitive effects. CNS Drug Rev. 2004;10:147–66. doi: 10.1111/j.1527-3458.2004.tb00010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Biton B, Bergis OE, Galli F, et al. SSR180711, a novel selective alpha7 nicotinic receptor partial agonist: (1) bindingand functional profile. Neuropsychopharmacology. 2007;32:1–16. doi: 10.1038/sj.npp.1301189. [DOI] [PubMed] [Google Scholar]
- 141.Sydserff S, Sutton EJ, Song, et al. Selective alpha7 nicotinic receptor activation by AZD0328 enhances cortical dopamine release and improves learning and attentional processes. Biochem Pharmacol. 2009;78:880–88. doi: 10.1016/j.bcp.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 142.Hirst WD, Andree TH, Aschmies S, et al. Correlating efficacy in rodent cognition models with in vivo 5-hydroxytryptamine1a receptor occupancy by a novel antagonist, (R)-N-(2-methyl-(4-indolyl-1-piperazinyl)ethyl)-N-(2-pyridinyl)-cyclohexane carboxamide (WAY-101405) J Pharmacol Exp Ther. 2008;325:134–45. doi: 10.1124/jpet.107.133082. [DOI] [PubMed] [Google Scholar]
- 143.Liao Y, Böttcher H, Harting J, et al. New selective and potent 5-HT (1B/1D) antagonists: chemistry and pharmacological evaluation on N-piperazinylphenyl biphenylcarboxamides and biphenylsulfonamides. J Med Chem. 2000;43:517–525. doi: 10.1021/jm990397l. [DOI] [PubMed] [Google Scholar]
- 144.Riemer C, Borroni E, Lever-Trafit B, et al. Influence of the 5-HT6 receptor on acetylcholine release in the cortex: pharmacological characterization of 4-(2-bromo-6-pyrrolidin-1-ylpyridine-4-sulfonyl)phenylamine, a potent and selective 5-HT6 receptor antagonist. J Med Chem. 2003;46:1273–76. doi: 10.1021/jm021085c. [DOI] [PubMed] [Google Scholar]
- 145.Hirst WD, Stean TO, Rogers DC, et al. SB-399885 is a potent selective 5-HT6 receptor antagonist with cognitive enhancing properties in aged rat water maze and novel object recognition models. Eur J Pharmacol. 2006;553:109–19. doi: 10.1016/j.ejphar.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 146.Geldenhuys WJ, Van der Schyf CJ. The serotonin 5-HT6 receptor: a viable drug target for treating cognitive deficits in Alzheimer’s disease. Expert Rev Neurother. 2009;9:1073–85. doi: 10.1586/ern.09.51. [DOI] [PubMed] [Google Scholar]
- 147.Fox GB, Esbenshade TA, Pan JB, et al. Pharmacological properties of ABT-239 [4-(2-{2-[(2R)-2-Methylpyrrolidinyl]ethyl}-benzofuran-5-yl)benzonitrile]:II. Neurophysiological characterization and broad preclinical efficacy in cognition and schizophrenia of a potent and selective histamine H3 receptor antagonist. J Pharmacol Exp Ther. 2005;313:176–90. doi: 10.1124/jpet.104.078402. [DOI] [PubMed] [Google Scholar]
- 148.Medhurst AD, Atkins AR, Beresford IJ. GSK189254, a novel H3 receptor antagonist that binds to histamine H3 receptors in Alzheimer’s disease brain and improves cognitive performance in preclinical models. J Pharmacol Exp Ther. 2007;321:1032–1045. doi: 10.1124/jpet.107.120311. [DOI] [PubMed] [Google Scholar]
- 149.Galici R, Boggs JD, Alusio L, et al. JNJ-10181457, a selective non-imidazole histamine H(3) receptor antagonist, normalizes acetylcholine neurotransmission and has efficacy in translational rat models of cognition. Neuropharmacology. 2009;56:1131–1137. doi: 10.1016/j.neuropharm.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 150.Fadel JR. Regulation of cortical acetylcholine release: insights from in vivo microdialysis studies. Behav Brain Res. 2010 doi: 10.1016/j.bbr.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Earl RA, Zaczek R, Teleha CA, et al. 2-fluoro-4-pyridinylmethyl analogues of linopirdine as orally active acetylcholine release-enhancing agents with good efficacy and duration of action. J Med Chem. 1998;41:4615–22. doi: 10.1021/jm9803424. [DOI] [PubMed] [Google Scholar]
- 152.Hilbert M, Hartmann J, Jeltsch H, et al. Modulation of hippocampal acetylcholine release after 192IgG-saporin lesions and neuronal grafting. Neurochem Res. 2003;28:467–72. doi: 10.1023/a:1022852819018. [DOI] [PubMed] [Google Scholar]