

Abstract
Triplication of chromosome 21 in Down syndrome (DS) results in overexpression of the minibrain kinase/dual-specificity tyrosine phosphorylated and regulated kinase 1A gene (DYRK1A). DYRK1A phosphorylates cytoplasmic tau protein and appears in intraneuronal neurofibrillary tangles (NFTs). We have previously shown significantly more DYRK1A-positive NFTs in DS brains than in sporadic Alzheimer disease (AD) brains. This study demonstrates a gene dosage–proportional increase in the level of DYRK1A in DS in the cytoplasm and the cell nucleus and enhanced cytoplasmic and nuclear immunoreactivity of DYRK1A in DS. The results suggest that overexpressed DYRK1A may alter both phosphorylation of tau and alternative splicing factor (ASF). Two-dimensional electrophoresis revealed modification of ASF phosphorylation in DS/AD and AD in comparison to controls. Altered phosphorylation of ASF by overexpressed nuclear DYRK1A may contribute to the alternative splicing of the tau gene and an increase by 2.68× of the 3R/4R ratio in DS/AD, and a several-fold increase in the number of 3R-tau–positive NFTs in DS/AD subjects compared to in sporadic AD subjects. These data support the hypothesis that phosphorylation of ASF by overexpressed DYRK1A may contribute to alternative splicing of exon 10, increased expression of 3R tau, and early onset of neurofibrillary degeneration in DS.
Keywords: 3-Repeat tau, Alternative splicing factor, Alzheimer disease, Down syndrome, DYRK1A, Neurofibrillary degeneration
INTRODUCTION
Almost all adults with Down syndrome (DS) have Alzheimer disease (AD)-type pathology that is associated with an elevated risk of dementia (1–4). Early onset of neurofibrillary degeneration is considered to be the major causative factor of neuronal dysfunction, loss of neurons, and dementia in DS (5–7). The molecular and cellular mechanisms leading to the early onset of neurofibrillary degeneration in adults with DS are not understood. In contrast to earlier studies suggesting a similar course of pathological changes in sporadic AD and in DS/AD, numerous studies have indicated qualitative and quantitative differences (1, 8–10). The presence of AD-type pathology in almost all individuals with DS who survive to 40 years of age suggests that overexpressed genes located on chromosome 21 contribute to the early onset of brain β-amyloidosis, neurofibrillary degeneration, neuronal loss, and functional deterioration associated with dementia.
In AD, loss of cell function is associated with replacement of the normal neuronal cytoskeleton by paired helical filaments (PHFs) arranged in neurofibrillary tangles (NFTs) in cell bodies and in neuropil threads in the dendritic tree (11-13). Neuronal degeneration is the result of an increased level of cytosolic abnormally phosphorylated tau protein and accumulated PHFs composed mainly of abnormally hyperphosphorylated tau (14, 15). Tau protein is encoded by a single gene consisting of 16 exons. Exons 2, 3, and 10 undergo developmentally regulated alternative splicing, producing 6 tau protein isoforms ranging from 352 to 441 amino acids (16). The C-terminal domain of the tau protein contains the microtubule-binding domains necessary to maintain and stabilize microtubules of the neuronal cytoskeleton (17, 18). Alternative splicing of exon 10 results in tau protein containing either 3 repeats (3R-tau) or 4 repeats (4R-tau) of the microtubule-binding domains. Exclusion of exon 10 results in 3R-tau, whereas inclusion of exon 10 results in 4R-tau. The change in the 1:1 ratio of 3R and 4R in the normal brain may initiate neurofibrillary degeneration. 4R-tau predominates in progressive supranuclear palsy and corticobasal degeneration, whereas more 3R- than 4R-tau has been detected in AD and Pick disease (PD) (19). Studies using combinations of antibodies detecting 3R- (RD3) and 4R- (RD4) tau and antibodies detecting phosphorylated tau revealed that RD4- and pSer422-positive NFTs predominate in sectors CA2–4, whereas RD3- and pSer262-positive NFTs prevail in sector CA1, subiculum, and entorhinal cortex (19). Selective neurofibrillary degeneration in sector CA2 is associated with 4R tauopathies (20).
The minibrain kinase/dual-specificity tyrosine phosphorylated and regulated kinase 1A gene (DYRK1A), located in the critical region of chromosome 21 (21–23) and overexpressed in DS (24, 25), is considered a factor contributing to the early onset of neurofibrillary degeneration in DS.
DYRK1A protein phosphorylates microtubule-associated protein tau at several sites, including Thr181, Ser199, Ser202, Thr205, Thr212, Thr217, Thr231, Ser396, Ser400, Ser404, and Ser422 (26–28). Phosphorylation by DYRK1A primes further phosphorylation of tau by glycogen synthase kinase 3 at Thr181, Ser199, Ser202, Thr205, and Ser208 (26, 27).
Hyperphosphorylation of tau at these sites has been reported in individuals with AD and in adults with DS (26, 29). Tau phosphorylation at Thr212, Ser202, and Ser404 is significantly increased in DYRK1A transgenic mice overexpressing human DYRK1A (28). DYRK1A-induced tau phosphorylation inhibits tau activity to stimulate microtubule assembly and promotes its self-assembly into filaments (26, 28). Both the levels and the activity of DYRK1A are higher in the brains of people with DS as compared to controls or individuals diagnosed with AD (26, 30, 31). A several-fold increase in the number of DYRK1A-positive NFTs in the brains of subjects with DS/AD suggests a gene dosage–dependent contribution of DYRK1A to neurofibrillary degeneration (31).
Recent studies suggest that DYRK1A also contributes to a nuclear molecular mechanism leading to neurofibrillary degeneration in people with DS (32). DYRK1A phosphorylates alternative splicing factor (ASF) at Ser227, Ser234, and Ser238, whereas SR protein kinase1 and 2, Clk/Sty, and DNA topoisomerase-I phosphorylate other sites on ASF (32–36). Phosphorylation of ASF by SR protein kinase 1 (SRPK1) in the cytosol results in ASF relocation to the nucleus, whereas phosphorylation of ASF by Clk/Sty releases ASF from speckles (37, 38) and recruits it into nascent transcripts where ASF regulates alternative splicing. Phosphorylation of ASF by DYRK1A drives ASF back to speckles, decreases its involvement in the regulation of alternative splicing, and inhibits its ability to promote tau exon 10 inclusion. This phenomenon results in an increase in 3R-tau and an imbalance of 3R/4R-tau ratio and facilitates neurofibrillary degeneration. These data indicate that the overexpression of DYRK1A in DS brains may contribute to the early onset of neurofibrillary degeneration directly through the hyperphosphorylation of tau (26, 28, 31) and indirectly through the phosphorylation of ASF (32).
Here, we tested the hypotheses that in DS, in comparison with sporadic AD, 1) the level of nuclear DYRK1A is increased proportionally to gene overexpression, and 2) overexpression of nuclear DYRK1A contributes to a cascade of molecular events resulting in the early onset of neurofibrillary degeneration and an increased number of 3R-tau–positive NFTs.
MATERIALS AND METHODS
Tissues, Clinical and Neuropathological Evaluation
Tissue samples from the brains of 26 subjects with similar stages of neurofibrillary pathology were used for immunocytochemical and morphometric studies. There were 12 DS subjects 41 to 71 years of age (4 females, 8 males) and 14 subjects 68 to 94 years of age (7 females, 7 males) with histories of dementia and postmortem-confirmed AD. Frozen samples from the cerebral frontal cortex of 6 subjects with DS/AD, 6 with sporadic AD, and 6 control subjects were used for fractionation and Western blot analysis of DYRK1A and ASF (Table 1). The brains of subjects with changes associated with mechanisms of death (respiratory brain) or autolytic changes and long postmortem interval were removed during neuropathological prescreening. Prescreening based on evaluation of Western blots resulted in the removal of tissue samples with evidence of protein degradation. All brain tissue samples were identified by case number and examined blind to clinical and demographic information.
Table 1.
Control, Down Syndrome and Alzheimer Disease Brain Tissue Samples Used for Biochemical Studies
| Number | Group | Age (y) | Sex | PMI (h) |
|---|---|---|---|---|
| 1 | Control | 67 | F | 4 |
| 2 | Control | 68 | F | 3 |
| 3 | Control | 78 | M | 4 |
| 4 | Control | 86 | M | 1.5 |
| 5 | Control | 87 | M | 19 |
| 6 | Control | 90 | F | 13 |
| 7 | DS | 51 | M | 2 |
| 8 | DS | 61 | M | 3 |
| 9 | DS | 63 | M | 14 |
| 10 | DS | 65 | M | 4.5 |
| 11 | DS | 65 | M | NA |
| 12 | DS | 67 | M | 3 |
| 13 | AD | 64 | F | NA |
| 14 | AD | 68 | M | 3.5 |
| 15 | AD | 71 | M | 4 |
| 16 | AD | 73 | F | 4.5 |
| 17 | AD | 80 | M | 4.5 |
| 18 | AD | 85 | F | 6 |
AD = Alzheimer disease; DS = Down syndrome; PMI (hours), postmortem interval (hours). F = female; M = male; NA = not applicable; y = years.
One brain hemisphere from each subject was cut into 1-cm-thick frontal slabs. Diagnostic tissue samples were removed and fixed in formalin for routine neuropathological examination; the majority of tissue was frozen at −80°C for biochemistry. The other hemisphere was fixed in 10% buffered formalin for 6 weeks to several months and then dissected into 1-cm-thick frontal slabs. The tissue blocks were dehydrated in a graded series of ethanol for 5 days in 70% ethanol, then 2 days in 80% ethanol, and finally 1 week in 96% ethanol. Dehydrated tissue was infiltrated with polyethylene glycol (PEG) 400 (Merck #807 485) for 6 days (2 changes of 3 days each at room temperature [RT]) and with PEG 1000 for another 6 days (2 changes of 3 days each at 42°C). Slabs were embedded in fresh PEG 1000 (39) and stored in 4°C. Tissue blocks were then cut at 18°C into 50-μm-thick serial sections. They were stored in 70% ethyl alcohol at RT and used for immunocytochemical and morphometric studies.
Functional assessment staging (FAST) was used to evaluate the progression of the clinical course of sporadic AD (40). Control subjects were classified as FAST stage 1 (normal adult without functional decrement documented in available medical records including antemortem evaluations) or stage 2 (normal aged adult with subjective deficit only in cognition-related functioning such as recalling names). At the time of their demise, 3 AD subjects were at FAST stage 3 (deficits noted in executive functioning such as demanding employment settings corresponding to mild cognitive impairment); 1 subject was at FAST stage 4 (mild AD with deficits in performance of complex tasks of daily life); 2 subjects were at FAST stage 5 (moderate AD manifested with deficient performance in choosing proper attire); 3 subjects were at FAST stage 6 (moderately severe AD manifested with 5 sub-stages including decreased ability to dress properly and to handle the mechanics of bathing and toileting, and finally with urinary and fecal incontinence, respectively); 5 subjects were at FAST stage 7 (severe AD with 6 sub-stages including incipient averbalism, limitation of speech to a single word, loss of ambulation, and, subsequently, loss of the ability to sit up independently, to smile, and to hold up or move the head, respectively).
In sporadic AD, the duration of the disease from FAST stage 3 to the onset of FAST stage 7f (loss of ability to hold head up or to move the head) is estimated to be 19 years on average (40, 41). The presence of early pathological findings of AD during the approximately 15-year-long FAST stage 2 indicates that the total duration of the disease is about 34 years. The disease duration for DS subjects has not been established, but the presence of AD-type pathology in all examined DS subjects from 41 to 71 years of age suggests a comparable duration of AD in both cohorts. Patients with sporadic AD were examined and diagnosed by clinicians at the Aging and Dementia Research Center (ADRC) at the New York University Medical Center. Patients with DS were examined in local settings and were evaluated with standardized protocols at the ADRC or at the NYS Institute for Basic Research in Developmental Disabilities.
Neuropathological evaluation included gross brain description and examination of sections from 16 brain regions stained for neurofibrillary degeneration with antibody Tau-1 and for β-amyloidosis with monoclonal antibody (mAb) 4G8. AD-type pathology was evaluated using criteria developed at a consensus conference organized by the National Institute on Aging and the Reagan Institute of the National Alzheimer Disease Association (42). All control subjects younger than 42 years were free of AD pathology. All older control subjects were affected with incipient neurofibrillary degeneration corresponding to Braak et al stage I or II (43, 44) and were free of amyloid plaques. AD subjects represented a broad spectrum of changes from stage III to neocortical stage VI, and all subjects with DS were affected by both neurofibrillary degeneration corresponding to stage III to VI and β-amyloidosis.
Immunohistochemistry
DYRK1A was detected with mouse mAb 7F3 against the amino-terminus of DYRK1A, produced at the NYS Institute for Basic Research in Developmental Disabilities) (immunogen: 1–160 aa of rat Dyrk1A; epitope: 74–78 of 754 aa isoform; 1 mg/ml, diluted 1:50) (45). ASF was detected with mouse mAb SF2/ASF, sc-33652 (IgG2b; 200 μg/ml diluted 1:400; epitope: near the N-terminus of the ASF protein; Santa Cruz Biotechnology Inc., Santa Cruz, CA). Mouse mAb RD3 (clone 8E6/C11, culture supernatant; Upstate, Temecula, CA) (46), diluted 1:800 was used to detect the 3R-tau protein. Bovine thyroglobulin-conjugated synthetic peptide corresponding to amino acids 209–224 of human tau used for immunization is the flanking junction coded by exons 9 and 11 in the absence of exon 10 (Upstate). To expose epitopes reacting with mAb RD3, the sections immersed in citric acid were microwaved (2 × 2 minutes), rinsed, and then treated with 88% formic acid for 15 minutes. Endogenous peroxidase in the sections was blocked with 0.2% H2O2 in methanol. To block nonspecific binding, the sections were treated with 10% fetal bovine serum in phosphate buffer solution (PBS) for 30 minutes. The primary antibodies were diluted in 10% fetal bovine serum in PBS and were incubated with sections overnight at 4°C. The sections were washed and treated for 30 minutes with biotinylated sheep anti-mouse IgG antibody diluted 1:200 (RPN 1001V1). The sections were treated with an extravidin-peroxidase conjugate (1:200) for 1 hour, and the product of the reaction was visualized with diaminobenzidine (0.5 mg/ml with 1.5% H2O2 in PBS). Application of the mouse-specific HRP/DAB detection kit (Ab 64259; Abcam Inc., Cambridge, MA) significantly improved the consistency of immunodetection of DYRK1A in postmortem material. This kit was used to characterize differences in DYRK1A distribution in the frontal cortex of control, DS/AD, and AD subjects. After immunostaining, the sections were lightly counterstained with hematoxylin.
Confocal Microscopy
To characterize the intracellular distribution of DYRK1A and ASF, 50-μm-thick sections from the frontal cortex were treated with citric buffer, pH 6.0, at 87°C for 30 minutes and immunostained with mAb 7F3 to detect DYRK1A and rabbit polyclonal antibody (pAb) Ab 38017 diluted 1:150 (Abcam) to detect ASF. To characterize distribution of ASF and neurofibrillary tangles, sections were treated with alkaline phosphatase and immunostained with mAb Tau-1 and rabbit pAb Ab 38017. Secondary antibodies were affinity-purified donkey anti-mouse IgG labeled with Alexa Fluor 555, and anti-rabbit IgG labeled with Alexa Fluor 488 (both from Invitrogen Corp., Grand Island, NY). Images were generated using Nikon C1 confocal microscope and EZC1 image analysis software.
Brain Nuclei Isolation and Evaluation of Quality of Nuclear Fraction
To evaluate the DYRK1A levels in the nuclei of DS, AD, and control subjects, the nuclei were isolated from brain tissue samples according to the method of Blobel and Potter (47), with minor modifications. In brief, samples of the frontal cortex were homogenized in 2 volumes of ice-cold 0.25M sucrose in TKM buffer (50mM Tris-HCl, pH 7.5, 25 mM KCl, 5 mg MgCl2, and Complete cocktail of protease inhibitors; Roche Diagnostics, Indianapolis, IN). The filtered homogenate was mixed with 2 volumes of 2.3 M sucrose in TKM and applied into a polyallomer tube on a 1-volume cushion of 1.8 M sucrose in TKM buffer. After centrifugation for 30 minutes at 124,000 g at 4°C, the supernatant and material floating on the top was transferred to another tube. The 2.1 M sucrose interphase containing nuclei was collected for analysis. The nuclear pellet was taken up in 0.25 M TKM buffer, supplemented with 25% glycerol, and stored at −20°C.
To evaluate the purity of the nuclear fraction, isolated nuclei were fixed with a mixture of 2% paraformaldehyde and 2% glutaraldehyde in sodium cacodylate buffer, spun down, rinsed in the same buffer and fixed with 2% osmium in cacodylate buffer for 1 hour. To enhance contrast, samples were treated with 1% tannic acid for 1 hour, rinsed and dehydrated with rising concentrations of ethyl alcohol and propylene oxide, and embedded in Epon 812. Sections 0.35-μm-thick were stained with aldehyde fuchsin and toluidine blue. Ultrathin sections were stained with uranyl acetate and examined in a Hitachi 7500 electron microscope.
To evaluate the preservation of nuclear ASF and DYRK1A in the isolated nuclei, 3 μl of suspended nuclei were fixed with 4.5% buffered formalin for 20 minutes. After fixative washing with 3 changes of water, nonspecific binding was blocked with 5% fetal bovine serum in PBS for 30 minutes. mAb SF2/ASF and Alexa Fluor 488 were used to evaluate the preservation of nuclear ASF, and mAb7F3 linked with Alexa Fluor 568 was used to evaluate the preservation of nuclear DYRK1A.
Gel Electrophoresis, Immunoblotting and Immunoprecipitation
Proteins were resolved on 7.5% (DYRK1A detection) or 10% SDS–PAGE (ASF detection) with Tris-Tricine running buffer, and electroblotted onto 0.2-μm nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA). Membranes were blocked with 5% fat-free milk in PBST buffer (0.05% Tween-20, PBS, pH 7.4) and incubated overnight at 4°C with mAb 8D9 diluted to 0.2 μg/ml to detect DYRK1A and mAb SF2/ASF diluted 1:1,000. Total tau was detected with mAb Tau-5 diluted 1:1,000 (Millipore, Temecula, CA). 3R and 4R tau isoforms were detected with mAbs RD3 and RD4 (Upstate, Temecula, CA) diluted 1:2,000 and 1:200, respectively. MAb AC-15 diluted 1:50,000 (Sigma, Saint Louis, MO) and mAb Nup62 diluted 1:5,000 (BD Biosciences, San Diego, CA) were used to detect β-actin and nucleoporin p62, respectively. Incubation with horseradish peroxidase conjugated with anti-mouse antibody (GE Healthcare, Piscataway, NJ) was performed for 1 hour at RT. Membranes were developed with the HyGlo chemiluminescent detection reagents (Denville Scientific, Metuchen, NJ). 1DScan EX software (BD Biosciences, Rockville, MD) was used for quantitative analysis of chemiluminescent data. EZ-Run Rec Protein Ladder standards (Fisher BioReagents, Pittsburgh, PA) were applied to estimate the size of proteins in the gels. The difference between the groups was determined with T-test with KS normalization using Prism 4 software (GraphPad Software, Inc., La Jolla, CA)
Immunoprecipitation was applied to test the ability of DYRK1A and ASF to form complexes in the human brain. Nuclear fraction from the frontal cortex of control and DS subjects was extracted with RIPA buffer (50 mM Tris-HCl, pH7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, and Complete protease inhibitors) and clarified by centrifugation at 16,000 × g for 15 minutes. Extract was precipitated overnight at 4°C with mAb 8D9 to detect DYRK1A and with mAb SF2/ASF to detect ASF. Immuno-complexes were captured on protein-G–conjugated Dynabeads (Invitrogen Corp.). Products of precipitation were subjected to Western blot analysis and immunostaining with 8D9 and SF2/ASF antibodies.
Phosphorylated and Dephosphorylated ASF
Cerebral cortices from 6 control subjects (67 to 87 years of age), 6 DS subjects (61 to 65 years of age), and 6 AD subjects (68 to 85 years of age) were homogenized. Protein aliquots (500 μg) from the brain homogenates were precipitated with 80% acetone at −20°C for 20 minutes. Proteins (250 μg) were then subjected to high-resolution 2-dimensional polyacrylamide gel electrophoresis. The Bio-Rad system using 70 mm, pH 3-10, immobilized pH gradient strips and 10% polyacrylamide gels (Bio-Rad) was applied. Samples were dissolved in a buffer containing 7 M urea, 2 M thiourea, 65 nM dithiothreitol, 0.125% (v/v) Biolytes 3-10, 2% CHAPS, and 0.1% Bromophenol Blue. For the first dimension, 250 μg of protein was applied to a dehydrated immobilized pH gradient strip; isoelectric focusing was carried out at RT. Before the separation of proteins by SDS gel electrophoresis (and to achieve a disulfide reduction), the isoelectric-focusing gel strips were equilibrated for 15 minutes in a buffer consisting of 37.5 mM Tris-HCl at pH 8.8, 6 M urea, 2% (w/v) SDS, 30% (w/v) glycerol, 0.5% dithiothreitol, and 0.1% Bromophenol Blue. To achieve carbamoylmethylation, the gel strips were re-equilibrated for 15 minutes in the same buffer containing 2% iodoacetamide, used as a replacement of dithiothreitol. The second dimension separation was carried out by placing the strips on 10% polyacrylamide gels, after which gels were electroblotted onto nitrocellulose membranes (Bio-Rad) and reacted with the antibodies.
For dephosphorylation, protein extract aliquots (250 μg) were subjected to dephosphorylation by lambda protein phosphatase (λPP) (New England BioLabs, Ipswich, MA). The reaction was conducted for 2 hours at 30°C with 1,000 U of enzyme in 250 μL of buffer containing 50 mM HEPES, pH 7.5, 0.1 mM EDTA, 5 mM DTT, and 0.01% Brij-35.
For Western blot detection of ASF, mAb SF2/ASF was used at concentration of 0.1 μg/mL. Polyclonal anti-mouse antibody linked to horseradish peroxidase (GE Healthcare) was used at 0.2 μg/mL. Membranes were developed with HyGlo chemiluminescent detection reagents (Denville Scientific).
Morphometry
For immunocytochemistry-based morphometric analysis, 7 brain regions indicative of the progression of the topographic expansion of neurofibrillary degeneration were selected, including entorhinal cortex (layers II and III–VI, entorhinal stage), amygdala, pyramidal cell layer in sectors CA1 and CA4 (limbic stage), and layer III in 3 neocortical regions, i.e. middle temporal gyrus, inferior frontal gyrus, and occipital cortex (layer III in Brodmann area 17). The 3 neuropathologists who morphometrically examined these brain regions were blind to the clinical and demographic records.
Numbers of neurons with NFTs and ghost tangles were determined using an image analyzer with StereoInvestigator (MicroBrightfield, Williston, VA) software providing a uniform counting-frame distribution within the region of interest (ROI). A computer-controlled automated stage provided an unbiased sampling of test areas. The borders of the examined structures, layers, and sectors were delineated and the counting grid and counting frames superimposed over the section images. Structures were examined at a final magnification of 1,480× for the ×40 lens, and a magnification 2,300× for the 60× lens. The shape and size of the grid were selected to adapt to the size and shape of the ROI. The size of the grid was from 200 × 300 μm for the second layer of the entorhinal cortex to 800 × 800 μm for layers III–VI in this cortex. The counting frame size was 60 × 60 μm for the entorhinal cortex, and 80 × 80 μm for other ROIs. The average number of counting frames was determined by the size of the structure and the severity of atrophy, and it ranged from 29 to 33 for the entorhinal cortex and sector CA4, and from 103 to 115 for the amygdala and layer III in the neocortical subdivisions (Table 2).
Table 2.
Morphometric Procedures Used for Estimation of Numerical Density of RD3-Positive Neurofibrillary tangles
| Region examined | Magnification | Grid Size (μm) |
Counting Frame Size (μm) |
Counting Frames per Region/Case |
|
|---|---|---|---|---|---|
| Objective lens (x) |
Final (x) |
||||
| EC II L | 40 | 1,480 | 200 × 300 | 60 × 60 | 33 |
| EC III-VI L | 40 | 1,480 | 800 × 800 | 60 × 60 | 29 |
| Amygdala | 40 | 1,480 | 600 × 600 | 80 × 80 | 104 |
| CA1 | 40 | 1,480 | 400 × 400 | 80 × 80 | 49 |
| CA4 | 40 | 1,480 | 300 × 300 | 80 × 80 | 31 |
| Middle temporal gyrus, III layer | 63 | 2,300 | 300 × 300 | 80 × 80 | 104 |
| Inferior frontal gyrus, III layer | 63 | 2,300 | 300 × 300 | 80 × 80 | 115 |
| Occipital cortex (BA17), III layer | 63 | 2,300 | 300 × 300 | 80 × 80 | 103 |
BA17 = Brodmann area 17; EC, entorhinal cortex; CA, cornu Ammonis.
Because MAb RD3 detects both intracellular NFTs and extracellular ghost tangles, these were counted separately. Intracellular NFTs correspond collectively to stages 0, 1, and 2, and ghost tangles correspond to stage 3 in the temporal history of NFTs (48, 49). The numerical densities (n/mm2) of RD3-positive neurons and ghost tangles were estimated in 4 sections from each brain for subjects with DS/AD and sporadic AD.
Analysis of variance was used to examine differences in the numerical density of RD3-positive NFTs and ghost tangles in 7 brain structures reflecting the major stages of topographic expansion of neurofibrillary degeneration in 2 groups: 12 adults with DS (41 to 71 years of age), and 14 subjects with sporadic AD and functional deterioration corresponding to FAST stages 3–7f.
RESULTS
Evaluation of Nuclear Fraction Purity
The nuclear and cytoplasmic fractions were isolated from the frontal cortex. Light microscopy revealed that the nuclear fraction was free of non-nuclear components (Fig. 1A, B). Fragments of cytoplasm attached to nuclei identified in ultrastructural evaluation were so small that the nuclear fraction could be considered as almost free of cytoplasmic components (Fig. 1C). Large nuclei of neurons with relatively homogenous nucleoplasm and prominent round nucleolus reflect the morphology of nuclei of pyramidal neurons, whereas smaller nuclei with homogenous nucleoplasm and undetectable nucleolus are most likely nuclei of glial cells. Immunofluorescence with mAb SF2/ASF and Alexa Fluor 488–conjugated secondary antibody and Alexa Fluor 568–linked mAb7F3 revealed preservation of ASF and DYRK1A in isolated nuclei (Fig. 1D, E).
Figure 1.

(A, B) The nuclear fraction isolated from frontal cortex was rich in nuclei and almost free of other tissue components in an Epon section stained with toluidine blue (A). Only a narrow rim of fragments of cytoplasm was detected around some nuclei by light microscopy (B). The proportion between neuronal (red arrowheads) and glial (blue arrowheads) nuclei in the nuclear fraction of brain homogenates reflects the proportion between neurons and glial cells in the cerebral cortex. (C) Electron microscopy revealed only tiny fragments of cytoplasmic membranes and matrix attached to nuclear envelope. (D, E) Immunofluorescence with mAb SF2/ASF and Alexa Fluor 488–conjugated secondary antibody (green) (D) and with Alexa Fluor 568 (red) linked mAb7F3 (E) demonstrates preserved distribution patterns of alternative splicing factor (ASF) and DYRK1 in isolated nuclei.
Estimation of DYRK1A and ASF Levels on Western Blots
In tissue homogenates and nuclear and cytoplasmic fractions from the frontal cortex of control, DS, and AD subjects, DYRK1A was detected as 2 main bands with a molecular weight in the range of 94 kD and a third band with molecular weight below 84 kD (Fig. 2A, C, E). Quantitative analysis of 6 samples per group revealed that in the homogenates and the nuclear and cytoplasmic fractions, the levels of DYRK1A were 1.68, 1.67 (p < 0.001), and 1.5 (p < 0.01) times higher, respectively, in the DS cases vs. controls. The difference between DYRK1A levels in homogenates and the nuclear and cytoplasmic fractions of control and AD subjects was not significant (Fig. 2B, D, F). Application of mAb SF2/ASF to Western blots of the homogenates and nuclear and cytoplasmic fractions from the frontal cortex revealed ASF in the range of 34 kD (Fig. 2A, C, E). The differences between levels of ASF in the control, DS, and AD subjects were not significant (Fig. 2B, D, F).
Figure 2.

Western blots of frontal cortex homogenates, and nuclear and cytoplasmic fractions from 6 control (Con), Down syndrome (DS), and Alzheimer disease (AD) subjects. (A-F) Blots were immunostained with mAb 8D9 to detect DYRK1A (A, C, E). In DS, the level of DYRK1A in frontal cortex homogenates, and nuclear and cytoplasmic fractions was 1.68 × (p < 0.0001), 1.67 × (p < 0.0001), and 1.51× (p < 0.01) higher, respectively, than in the control subjects (B, D, F). The differences between AD and controls were not significant. Western blots with mAb SF2/ASF revealed alternative splicing factor (ASF) in the range of 34 kDa (A, C, E), with no significant difference between levels of ASF in homogenates, nuclear and cytoplasmic fractions of control, DS and AD subjects (B, D, F). The loads per lane were 20 μg of homogenates and cytoplasmic fractions protein, and 10 μg of nuclear fraction protein. The protein loading in homogenates and cytoplasmic fractions was verified by β-actin blots (mAb AC-15) and in nuclear fractions by p62 blot (mAb Nup62). The nearly complete absence of cytoplasmic β-actin (C) confirmed the purity of the nuclear fraction, whereas uniform levels of nuclear marker (Nup62) confirmed nuclei preservation.
On Western blots immunostained with mAb AC-15, β-actin was detected in the range of 42 kD, and levels in tissue homogenates and cytoplasmic fraction from the frontal cortex of control, DS, and AD subjects were comparable (Fig. 2A, E). The absence of β-actin in the nuclear fractions of control, DS, and AD subjects reflect the purity of the nuclear fraction (Fig. 2C). Immunostaining of the nuclear fraction with Nup62 revealed a band in the range of 62 kD. There was no difference between levels of this marker of the nuclear fraction in the control, DS, and AD groups (Fig. 2C).
We have previously shown in whole cellular extracts that DYRK1A and ASF co-immunoprecipitated each other when no detergent was included in the immunoprecipitation buffer (32). However, when we extracted proteins from nuclear fraction using RIPA buffer and then performed co-immunoprecipitation of DYRK1A and ASF using RIPA buffer, we did not detect ASF in DYRK1A precipitates or DYRK1A in ASF precipitates in either control or DS subjects (Fig. 3).
Figure 3.
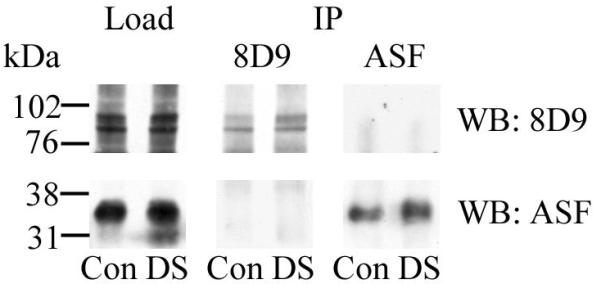
Co-immunoprecipitation (IP) of DYRK1A and alternative splicing factor (ASF) from nuclear extracts of control (Con) and Down syndrome (DS) subjects. Aliquots of nuclear extracts were immunoprecipitated with mAb 8D9 for DYRK1A and with mAb SF2/ASF for ASF. The presence of both target proteins was confirmed in Western blots (WB). DYRK1A was not detected in ASF precipitates (upper panel) and ASF was not detected in DYRK1A precipitates (bottom panel). Ten μg of the nuclear fraction of control and DS subjects were loaded in each lane.
Phosphorylated and Dephosphorylated ASF in DS/AD, Sporadic AD, and Control Subjects
Two-dimensional electrophoresis of brain homogenates dephosphorylated with λPPase revealed in all 6 samples of control, DS/AD, and AD subjects that ASF was detected in an almost identical narrow pI range from 7.6 to 7.8 (Fig. 4A). Quantitative analysis of 6 samples in each group that were not dephosphorylated revealed a pI shift by 0.25 units in the control group, 0.48 units in the DS/AD group, and 0.84 units in the sporadic AD group. The differences between the shifts detected in the DS/AD and AD groups were statistically significant vs. the control group (Fig. 4B).
Figure 4.

Phosphorylation patterns of alternative splicing factor (ASF) in brain homogenates from 6 control (Con), Down syndrome/Alzheimer disease (DS/AD), and sporadic AD subjects. Samples were treated with lambda protein phosphatase (λPP); both dephosphorylated (+, A) and untreated (−, A) samples were resolved by 2-D gel electrophoresis. In all samples, dephosphorylated ASF was detected at pI range 7.6 to 7.8. (A) Panel shows the results of electrophoresis of homogenates from 1 control, DS/AD, and AD subject. (B) In untreated samples from control individuals, the majority of ASF is found at pI range 7.4 to 7.8, with a pI shift by 0.25 units. In untreated samples from subjects with DS/AD, almost 50% of ASF is detected in a more acidic range, (from 7.0 to 7.4), with a pI shift of 0.48 units. In untreated samples from subjects with sporadic AD, the majority of ASF was found in a pI range from 6.4 to 7.4, revealing a shift by 0.84 units. The pI shift observed in the DS/AD and AD groups was statistically significant when compared to the range observed in the control group (***p < 0.0001).
Immunocytochemistry of DYRK1A and ASF in the Frontal Cortex
Immunostaining with mAb 7F3 of serial sections from the frontal lobe of control, DS/AD, and AD subjects revealed DYRK1A in the neuron nucleus and cytoplasm, including the cell body and the dendritic tree. Immunoreactivity in both the neuron nucleus and cytoplasm was stronger and more consistent in the frontal cortex of the 8 DS/AD subjects than in the other 4 DS/AD subjects and all control or AD subjects (Fig. 5). Some inconsistencies in DYRK1A immunodetection in the DS/AD and the 2 other cohorts appear to reflect death-related changes, mainly ischemia/hypoxia, and postmortem tissue degradation. In contrast to the nuclear and cytoplasmic immunoreactivity of DYRK1A in the frontal cortex, immunoreactivity with mAb SF2/ASF was almost exclusively limited to neuron nuclei. Immunodetection of ASF was much less affected by mechanisms of death or postmortem changes than that of DYRK1A. There were cell type– and region-specific differences in the amount and distribution of ASF in neurons and glia (Supplemental Text, Supplemental Fig. 1). There was a gradual reduction of ASF in neurons in late stages of neurofibrillary degeneration (Supplemental Text, Supplemental Fig. 2), and there were considerable differences between the distribution of DYRK1A and ASF in the examined brain regions (Supplemental Text and Supplemental Fig. 3).
Figure 5.

Immunostaining of the frontal lobe cortex of control (C), Down syndrome/Alzheimer disease (DS), and Alzheimer disease (AD) subjects with mAb 7F3 illustrates comparable immunoreactivity for DYRK1A in the neurons of control and AD subjects, and greater immunoreactivity in DS/AD subject. There is no difference in the pattern of alternative splicing factor (ASF) immunoreactivity (mAb FS2/ASF) in the frontal cortex of control, DS/AD, and AD subjects. The third row shows a pyramidal neuron in the frontal cortex of a 67 y old DS subject with 3-color immunostaining. DYRK1A detected with mAb 7F3 is red, and ASF is green. Merged image with nuclei counterstained blue with TO-PRO-3I is shown in the right panel. Fine granular immunoreactivity for DYRK1A is partially colocalized with a fine granular ASF reaction product in the neuron nucleus.
Confocal microscopy revealed DYRK1A in neurons in the cytoplasm and in the nucleus. The nuclear DYRK1A detected with mAb 7F3 had an appearance of multiple fine granules, most of which did not colocalize with ASF detected with pAb Ab 38017 (Fig. 5).
Determination of the Total Tau and the Ratio of 3R and 4R Tau in Brain Homogenates
In homogenates from the frontal cortex of 6 control subjects, the total tau, 3R tau, and 4R tau detected with mAbs Tau-5, RD3, and RD4, respectively, were found mainly in prominent bands in the range between 50 and 70 kDa (Fig. 6). In DS/AD cases there was a very strong signal for total tau and 3R and 4R were detected above the range observed in the control subjects, consistent with tau aggregation in NFTs in this cohort. Quantitative analysis of Western blots revealed a significant increase in the 3R-tau/4R-tau ratio in the DS/AD group vs. control and AD subjects (Fig. 6B).
Figure 6.

Western blots of frontal cortex homogenates from 6 control (Con), Down syndrome/Alzheimer disease (DS), and Alzheimer disease (AD) subjects immunostained with mAb Tau-5 to detect total tau, and antibodies specific for 3R and 4R tau isoforms. (A) No significant difference was observed between the relative ratio of 3R and 4R isoforms in the control and AD groups. (B) In the DS group, the ratio was significantly higher (2.68 ×). The loads of homogenate protein per lane were 2 μg for total tau and 3R isoform, and 20 μg for 4R isoform.
Number of 3-R-Tau–Positive NFTs and Ghost Tangles in DS and AD
RD3 detects both intracellular NFTs, corresponding to stages 0, 1, and 2 of neurofibrillary degeneration (48, 49) and ghost tangles (Fig. 7, upper panels). Confocal examination of the frontal cortex sections immunostained with rabbit pAb Ab 38017 for ASF and mouse mAb Tau-1 for abnormally phosphorylated tau, revealed no colocalization of ASF in NFTs (Fig. 7, lower panels). DS/AD brains characteristically showed greater numbers of RD3-positive NFTs vs. brains of subjects with sporadic AD (Fig. 8).
Figure 7.

MAb RD3 detects not only intracellular neurofibrillary tangles (NFTs) at 3 stages (0, 1, 2) of maturation, but also ghost tangles (GT) in different stages of degradation. In stage 0 (pretangles), RD3-positive granular material appears in a portion of cytoplasm in neurons with normal morphology. In stage 1 (early tangles), immunoreactive material aggregates in small, rod-shaped bundles of fibrils. Large, flame-shaped, RD3-positive inclusions filling almost the entire cell cytoplasm correspond to stage 2 (mature tangles). GTs (extracellular NFTs) represent the end stage (stage 3) of neurofibrillary degeneration. They are strongly RD3-positive, loosely arranged clusters of extracellular bundles of abnormal fibers exceeding by 2 to 3 times the size of degraded neuron. In the lower row, 3-color immunostaining of tau protein (red; mAb Tau1), alternative splicing factor (ASF) (green, pAb Ab 38017), and nuclei (blue) in the frontal cortex of a 67-year-old DS subject does not show colocalization of ASF and NFTs.
Figure 8.

Late stage neurofibrillary degeneration in the brain of a 57-year-old subject with Down syndrome (DS) and an 81-year-old subject with moderately severe Alzheimer disease (AD), functional assessment staging 6d. Immunostaining with mAb RD3 demonstrates many more NFTs and neuropil threads with 3R tau in DS than in sporadic AD in all 6× examined brain structures including the entorhinal cortex (EC, III layer), sector CA1, and amygdala (Amy), and III layer in the temporal (TC), frontal (FC) and occipital cortex (OC).
The estimated numerical density of NFTs (stages 0, 1, and 2 of neurofibrillary degeneration in neurons with a detectable nucleus) revealed significantly more NFTs in all examined brain structures in DS subjects than in sporadic AD subjects (p < 0.0001). The numerical density of RD3-positive NFTs was from 1.2× (in sector CA1) to 3.7× (in the frontal cortex) higher in the brains of cases with DS than sporadic AD (Table 3, Fig. 9).
Table 3.
Average Numerical Density (n/mm2) of RD3-positive Intracellular Neurofibrillary Tangles and Ghost Tangles in Down Syndrome and Alzheimer Disease Subjects
| NFT Type |
Group | EC II L |
EC III-VI L |
CA1 | CA4 | Amygdala | Temporal Cortex |
Frontal Cortex |
Occipital Cortex |
|---|---|---|---|---|---|---|---|---|---|
| Intra- cellular NFTs |
DS | 92 (13.2) p < 0.0001 |
117.1 (10.3) p < 0.0001 |
136.4 (7.3) p < 0.003 |
61.0 (5.2) p < 0.0001 |
167.9 (4.3) p < 0.0001 |
120.6 (4.4) p < 0.0001 |
94.1 (3.6) p < 0.0001 |
58.4 (3.2) p < 0.0001 |
| AD | 37 (6.7) |
73 (7.4) |
110.2 (5.2) |
19.7 (2.4) |
70.9 (3.0) |
78.8 (2.9) |
25.3 (1.8) |
27.7 (2.3) |
|
| Ratio of DS:AD |
2.5 | 1.6 | 1.2 | 3.0 | 2.4 | 1.5 | 3.7 | 2.1 | |
| Ghost tangles |
DS | 439 (27.2) p < 0.05 |
179 (13.8) p < 0.02 |
68.0 (5.9) p < 0.0001 |
52.3 (5.0) p < 0.0001 |
40.5 (2.0) p < 0.0001 |
37.9 (2.3) p < 0.0001 |
24.7 (1.6) p < 0.0001) |
1.7 (0.5) p < 0.002 |
| AD | 509.1 (22.8) |
225 (14.3) |
106.0 (8.0) |
27.3 (3.2) |
70.0 (3.5) |
17.2 (1.4) |
1.4 (0.4) |
0.1 (0.0) |
|
| Ratio of DS:AD |
0.9 | 0.8 | 0.6 | 1.9 | 0.6 | 2.2 | 17.6 | 17 |
12 Down Syndrome (DS) and 14 sporadic Alzheimer disease (AD) cases were evaluated; EC, entorhinal cortex; CA, cornu Ammonis; L = layer; NFT = neurofibrillary tangle; p values in the DS rows are vs. AD. Numbers in parentheses are SD.
Figure 9.

(A) The mean numerical density (n/mm2) of intracellular RD3-positive neurofibrillary tangles (NFTs) is higher in Down syndrome (DS) than in Alzheimer disease (AD) subjects in all examined structures (p < 0.003), but with significant regional differences. The numerical density of RD3-positive NFTs was higher in DS (black bars; n = 12; SE) than in sporadic AD (open bars; n = 14; SE) subjects by 2.5× in layer II and 1.6× in layers III-VI in the entorhinal cortex; by 1.2× in sector CA1 and by 3.0× in sector CA4; by 2.4 in the amygdala; and by 1.5× in the temporal, 3.7× in the frontal, and 2.1× in the occipital cortex. (B) In the frontal and occipital cortex affected in late stages of neurofibrillary degeneration, the mean numerical density of RD3-positive ghost tangles was strikingly higher (about 17×) in DS than in AD subjects (p < 0.002). The difference was relatively small in CA4 and temporal cortex (1.9× and 2.2×, respectively), which were affected in the mid-stages of neurofibrillary degeneration. The opposite trend was noted in structures characterized by early onset of neurofibrillary changes and severe neuronal loss. The ratio between the numerical density of ghost tangles in DS and AD was 0.8–0.9× in the entorhinal cortex, and 0.6× in sector CA1 and the amygdala.
The numerical density of RD3-positive ghost tangles provides a measure of the number of neurons that died with neurofibrillary degeneration. In the neocortex, which is affected relatively late in the course of neurofibrillary degeneration, the numerical density of ghost tangles was 2.2× higher in the temporal cortex and 17× higher in the frontal and occipital cortices of subjects with DS vs. the sporadic AD cases. However, in the entorhinal cortex, sector CA1, and amygdala, structures that are affected in early stages of neurofibrillary degeneration and that show the most severe neuronal loss, the numerical density of RD3-positive ghost tangles was less in the DS group than in the AD group (Table 3).
DISCUSSION
Overexpression of Nuclear DYRK1A in DS
We have previously shown that the level and the activity of DYRK1A in brain homogenates of DS subjects are approximately 1.5× higher than in control samples (26, 30, 31). In the present study, Western blot analysis revealed that an extra copy of the DYRK1A gene in DS is associated with gene dosage–proportional increases in the levels of DYRK1A in nuclear and cytoplasmic fractions. Immunocytochemistry of well-preserved samples of frontal cortex demonstrates enhanced immunoreactivity with mAb 7F3 in DS subjects compared to that in sections from control and AD subjects. The detected increase in the nuclear level of DYRK1A implies that there is an abnormal DYRK1A interaction with nuclear targets and that this interaction may have diverse structural and functional consequences. DYRK1A phosphorylates several transcription factors, including NFAT (50, 51), CREB (52), FKHR (27), and Gli-1 (53), and splicing factors including ASF (32), cyclin L2 (54), and SF3b. Phosphorylation of these factors by overexpressed DYRK1A may affect their trafficking and expression of their downstream targets and may contribute to the complex DS phenotype.
DYRK1A is considered to be a nuclear kinase because it contains the nuclear targeting sequence, is present in nucleoplasm, and is concentrated in nuclear speckles (55-57). Kaczmarski et al have confirmed the nuclear localization of DYRK1A (unpublished data), but in contrast to the expected prevalence of nuclear DYRK1A, fractionation revealed that only 12% of human brain DYRK1A is detected in the nuclear fraction, 78% is associated with an insoluble cytoskeletal fraction, and 10% is associated with a soluble cytoplasmic fraction. The presence of 12% of the total cellular DYRK1A in the nuclear fraction reflects a 10% to 12% contribution of nuclei to the cell volume and suggests that the DYRK1A concentration is comparable in the cell nucleus and cytoplasm. These proportions support reports that demonstrate that DYRK1A phosphorylates numerous substrates in the cytosol, cytoskeleton, synapses, and nucleus. Therefore, the overexpression of DYRK1A observed in DS may result in modification of the phosphorylation of many substrates, thereby contributing to abnormal cell development, maturation, aging, and degeneration (58).
Studies of cultured cells with overexpression of DYRK1A and ASF have revealed co-immunoprecipitation of DYRK1A and ASF in model conditions (32), and suggest that DYRK1A and ASF may form complexes. However, the absence of ASF in DYRK1A precipitates and DYRK1A in ASF precipitates in nuclear fractions of control and DS subjects indicates that these nuclear proteins do not form complexes detectable by co-immunoprecipitation in human tissue samples. The negative results of co-immunoprecipitation we observed suggest that under native conditions, complexes of DYRK1A and ASF may be transient or unstable, or that their concentrations are below the detection level of the applied methods.
It is possible that overexpressed DYRK1A may contribute to the shift of the ASF isoelectric point observed in DS (which is different than in controls and AD) and to the change in the ratio of 3R and 4R tau and the prevalence of 3R tau in the DS cohort.
Distribution of DYRK1A in Normal and Pathological Conditions
Our previous biochemical study revealed almost identical DYRK1A levels in the frontal, temporal, and occipital cortices (17–18 ng/mg of total proteins) in control brains. For adults with DS, the level of DYRK1A was higher in all examined cortical regions than in control cases but varied topographically from 25 ng/mg in the frontal, 21 ng/mg in the temporal, and 20 ng/mg in the occipital cortex to only 15 ng/mg in the cerebellar cortex (30). Immunocytochemistry expanded these data by showing striking brain structure-specific and neuron type-specific differences in the distribution of DYRK1A detected with mAb 7F3 (31, 45). In the present study, enhanced immunoreactivity with mAb 7F3 was detected in sections from the frontal cortex in 8 of 12 DS subjects. Inter-individual differences in the examined material might be in part be related to the mechanisms of death and postmortem tissue degradation. Immunostaining of 50-um-thick sections may contribute to the higher proportion of DYRK1A-immunoreactive neurons detected in our material than in other studies (59). Increased DYRK1A immunoreactivity has been reported in the cytoplasm and nuclei of scattered neurons of the entorhinal cortex, hippocampus, and neocortex in neurodegenerative diseases associated with tau phosphorylation, including AD, DS, and PD. The percentage of neurons with increased DYRK1A immunoreactivity shows significant differences across individuals and brain structures. The percentage of DYRK1A-positive nuclei in the frontal cortex was only 0.5% in controls, 10% in AD, and 5% in PD. The percentage of DYRK1A-positive nuclei in the dentate gyrus granule layer was found to be 0.5% in control and AD cases but 60% in PD (59), and significant changes in DYRK1A expression during development (45) indicate that structure-specific, age-associated, and disease-associated factors modify the amount and distribution of DYRK1A. DYRK1A immunoreactivity was also observed in reactive astrocytes, including those in the amyloid plaque corona. However, DYRK1A is undetectable in oligodendrocytes (59). Several studies suggest that the cytoplasmic and nuclear level of DYRK1A is locally regulated (31, 45, 59) and that local levels of overexpressed DYRK1A might be a factor co-determining the cell susceptibility to age/AD-associated neurofibrillary degeneration in DS (26, 31, 32).
Increase of 3R-tau in NFTs in DS
The application of several highly specific anti-3R and -4R tau antibodies, including the RD3 antibody, revealed the presence of 3R tau in many extracellular tangles but in fewer pretangles or mature intracellular NFTs. Tangles in brain structures affected early and severely by neurofibrillary degeneration such as in the entorhinal cortex, sector CA1, and subiculum were predominantly RD3-positive (19, 60). Sectors CA2–4 showed predominantly 4R-tau containing the pSer422 epitope (19). Selective neurofibrillary degeneration of sector CA2 was reported in 4R-tauopathies (20). RD3-positive NFTs had a distribution similar to that of NFTs positive for pSer262, AT8, and Gallyas-impregnated NFTs (19).
The most striking feature of neurofibrillary degeneration in DS is a several-fold higher numerical density of RD-3-positive NFTs in neurons in all examined brain structures as compared to sporadic AD. This suggests that the contribution of R3 tau to neurofibrillary degeneration is much more significant in the brains of people overexpressing DYRK1A, and that this contribution varies substantially among brain structure (from 1.2× in sector CA1 to 3.7× in the frontal cortex). A further reflection of this process in living cells is a several-fold increase in the number of RD3-positive ghost tangles in all neocortical regions and sector CA4. The number of RD3-positive NFTs in living neurons is higher in DS than in AD subjects in all structures affected in the early and late stages of AD, but the number of extracellular NFTs is reduced in DS subjects, suggesting that the rate of degradation of RD3-positive ghost tangles is faster in DS than in sporadic AD.
Topographic progression of neurofibrillary degeneration and neuronal loss are considered direct evidence of a link between tau fibrillization, loss of neuron function, and death (61–63). Recent studies suggest that tau acquires complex oligomeric conformations that may be toxic. Granular-shaped protofilaments of tau are detected in vitro (64) and in human brain in Braak stages 0 and 1, suggesting that the granular tau oligomers may be present before NFTs are detectable (61, 64). Similar oligomers have been identified in rodent models of tauopathies (65) and cell cultures (66). Oligomerization appears to be not only an essential step of tau aggregation, leading to the formation of abnormal filaments but also a toxic factor affecting neuron function. Evidence that oligomeric tau protein may contain a 3- or 4-repeat of the microtubule-binding domain’s isoform (67) suggests that alternative splicing favoring the 3R tau production observed in DS may involve both 3R aggregated oligomers and 3R-positive NFTs. Although a several-fold increase in the number of 3R-positive NFTs has been shown in the examined DS cohort, a hypothetical increase of 3R-positive oligomers and their presumed toxicity requires confirmation. On the other hand, Andorfer et al showed that in human tau transgenic mice, PHF-containing neurons appear “healthy” in terms of nuclear morphology (68), but speculations that the polymerization of hyperphosphorylated tau into fibrils is neuroprotective conflict with the neuronal nucleus misplacement and deformation and loss of nuclear ASF shown in this study.
Contribution of Overexpressed DYRK1A to Early Onset of Neurofibrillary Degeneration in DS
The leading hypothesis of this study is that phosphorylation of ASF by overexpressed DYRK1A destabilizes the regulatory mechanisms controlling the balance between 3R and 4R tau, thereby contributing to the early onset of neurofibrillary degeneration with a prevalence of 3R-positive NFTs in DS. Shi et al showed that enhanced phosphorylation of ASF by overexpressed DYRK1A contributes to a cascade of molecular mechanisms, leading to an increased 3R tau level (32). In that study, a cell culture overexpressing system revealed that DYRK1A not only phosphorylates ASF but also forms complexes with ASF. However, co-immunoprecipitation of DYRK1A and ASF did not reveal detectable amounts of DYRK1A and ASF complexes in control and DS frontal cortex nuclear extracts. The difference between the results of co-immunoprecipitation of human brain nuclear extracts and the results of the study of the cell culture overexpression systems may reflect differences in protein levels and in the methods of their extraction and detection. Detection of partial colocalization by confocal microscopy indicates a portion of nuclear DYRK1A may also be linked to ASF in human brain.
Here, the average ages in the DS and sporadic AD groups were 55 and 83 years, respectively, but these cohorts had a similar spectrum of neurofibrillary pathology. The several-times-higher number of 3R tau–positive intracellular NFTs in the substantially younger DS subjects suggests that nuclear overexpression of DYRK1A could be the factor contributing to the early onset of neurofibrillary degeneration.
The presence of a surprisingly consistent amount of nuclear ASF during several steps of neurofibrillary degeneration (including deposition of rod-like inclusions in the cell body, growth of a fibrillar ring around the cell nucleus, and the filling of the cell body with NFT) indicates that ASF might be involved in regulation of tau transcription nearly throughout the process of intracellular NFT formation until very late stages of cell degeneration. Detection of DYRK1A with pAb G19 in a larger percentage of intracellular NFTs in subjects with DS than in those with sporadic AD supports the hypothesis that in DS, an increased percentage of neurons expresses DYRK1A to the late stages of cell degeneration (31); in some neurons both ASF and DYRK1A are present in affected cells up to the late stage of intracellular NFT formation. However, the loss of ASF only in neurons with very neurofibrillary changes appears not to be sufficient to modify the ASF level detected in Western blot analysis of tissue from DS/AD or AD cases. The death of a neuron with NFTs results in extracellular deposition of the ghost tangles, preserving the original properties with respect to the presence of 3R tau (shown herein) but modified due to the degradation of DYRK1A that is exposed to the extracellular environment, as shown in a previous report (31).
Several reports have indicated that DYRK1A can contribute to different forms of degeneration. DYRK1A phosphorylates and binds α-synuclein (69), and Septin4 (SEPT4) (70). Complexes of these 3 proteins may contribute to the cytoplasmic aggregation/fibrillization observed in PD, Lewy body dementia, and multiple system atrophy (71, 72). DYRK1A is also present in the NFTs of subjects with AD. The number of DYRK1A-positive NFTs is several times higher in the NFTs of DS subjects overexpressing DYRK1A than in subjects with sporadic AD (31). SEPT4 has been detected in NFTs, neuropil threads, and dystrophic neurites in amyloid plaques in AD (73). Binding of DYRK1A to SEPT4 and the presence of SEPT4 and DYRK1A in NFTs and Lewy bodies suggests that the DYRK1A/SEPT4 tandem may play a significant role in both tauopathies and α-synucleinopathies. The increased number of 3R-tau-positive NFTs and neuropil threads appears to be strong evidence that DYRK1A-regulated transcription contributes to neurofibrillary degeneration, together with cytoplasmic tau phosphorylation (26, 27). Thus, inhibition of excessive activity of DYRK1A might result in cytoplasmic and nuclear pathways that could prevention or delay several forms of neurodegeneration.
Overexpressed DYRK1A as a Risk Factor for Neurofibrillary Degeneration
The distribution patterns of DYRK1A and ASF suggest that 1) the level of nuclear DYRK1A could be an indicator of neuronal susceptibility to neurofibrillary degeneration, 2) factors enhancing the expression of DYRK1A may contribute to neurofibrillary degeneration, and 3) factors reducing excessive DYRK1A levels may reduce the risk of onset of neurofibrillary degeneration in susceptible neuronal populations. This may indicate that high levels of DYRK1A increase the risk of neurofibrillary degeneration and that this risk increases in structures with a higher ratio of DYRK1A to ASF. DYRK1A phosphorylates APP at Thr668 in vitro and in mammalian cells and might contribute to the enhancement of β-amyloidosis and neurofibrillary degeneration (74). On the other hand, other studies demonstrate that Aβ loading in neuroblastoma cells and in transgenic mice increases the DYRK1A expression, resulting in hyperphosphorylation of tau at Thr212 (26, 28, 74, 75).
The present results confirm our hypothesis that overexpression of DYRK1A results in a gene dosage–proportional increase in the level of DYRK1A in neuronal nuclei in DS vs. control and those sporadic AD subjects. Results of 2-dimensional gel electrophoresis of ASF before and after reaction with λPPase indicate that in the controls only a small fraction of ASF is phosphorylated and detected in the range of pI 7.4 to 7.6. The difference between ASF phosphorylation in DS/AD and in control brains is reflected in a striking increase of the signal in the range between 7.0 and 7.4. The third pattern of phosphorylation of ASF (with pI ranging from 6.4 to 7.6), as revealed in sporadic AD, indicate that ASF is phosphorylated differently in DS/AD than in sporadic AD and in control subjects.
ASF activity and intracellular trafficking are tightly regulated by sites and degree of phosphorylation. Phosphorylation by SRPK1 drives ASF from the cytosol to the nucleus. Phosphorylation by Clk/Sty causes the release of ASF from speckles. DYRK1A phosphorylates the ASF mainly at Ser-227, Ser-234, and Ser-238 (32). Although 4 other kinases (SRPK1, SRPK2, Clk/Sty, or DNA topoisomerase) phosphorylate ASF (32–36), none of the sites phosphorylated by DYRK1A is known to be phosphorylated by these kinases. DYRK1A phosphorylation drives nuclear ASF into speckles. ASF phosphorylation by overexpressed DYRK1A in trisomic subjects (32) determines the difference between ASF phosphorylation in sporadic AD and DS/AD. These data support the hypothesis that in DS the overexpressed DYRK1A may phosphorylate ASF and modulate the ASF role in alternative splicing of tau, resulting in prevalence of 3R-tau protein. These changes might increase the number of 3R-tau–positive NFTs and contribute to early and severe neuronal loss in DS.
The relative levels of tau 3R and tau 4R mRNA in AD brain provided inconsistent results. Higher levels of 3R tau than 4R tau were reported in AD brain (76). Application of semiquantitative methods of RNA analysis revealed an increase in 3R tau mRNA in AD (77), no changes in the relative isoform composition, or changes only in some cases (78-80). Application of real-time PCR to directly evaluate the 4R/3R mRNA ratio in individual postmortem brain samples confirmed that the differences in the 4R/3R ratio in the cortex of AD and control subjects are not statistically significant (81).
Several studies of frontotemporal dementia and Parkinsonism linked to chromosome 17 have shown a link between abnormal proportions of tau protein isoforms, an increase in exon 10 retention, and a several-fold excess of 4R tau mRNA over 3R tau mRNA (81-83). These data support the possibility that dysregulation of tau splicing may be a contributing factor to tauopathies, including DS/AD. By contrast, Connell et al suggest that although modified splicing may be a contributing factor to neurofibrillary degeneration in some forms of tauopathies, post-translational events are likely to be the significant factors contributing to the tau isoform composition in PD and sporadic tauopathies (81). It appears that a combination of factors changing the level of tau isoforms and post-translational modification of tau, such as phosphorylation, may determine the pattern of tau pathology in DS. This and other studies indicate that overexpressed DYRK1A may play a role in both mechanisms contributing to the early onset of neurofibrillary degeneration in DS.
In summary, the present findings and recent studies suggest that DYRK1A overexpression may result in a gene dosage–dependent modification of mechanisms leading to or contributing to early onset of neurofibrillary degeneration in DS, including 1) DYRK1A phosphorylation of tau protein at 11 sites, 2) DYRK1A stimulation of a several-fold increase in the rate of tau protein phosphorylation by glycogen synthase kinase 3, 3) a several-fold greater increase in the number of DYRK1A-positive NFTs in the brains of people with DS/AD than in individuals with sporadic AD, and 4) altered phosphorylation of ASF by overexpressed nuclear DYRK1A leads to alternative splicing of exon 10, imbalance between 3R/4R-tau (32), and a several-fold greater increase in the number of 3R-tau–positive NFTs in the brains of people with DS/AD than in those with sporadic AD.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Maureen Marlow for editorial corrections, Elaine Marchi for help in lab work coordination, and Jadwiga Wegiel, Cathy Wang, and En Wu Zhang for histology and immunocytochemistry. The tissue was provided by the Aging and Dementia Research Center at New York University Medical Center, the Brain and Tissue Bank at the New York State Institute for Basic Research in Developmental Disabilities, and the Brain and Tissue Bank for Developmental Disorders at the University of Maryland, supported by NICHD contract # NO1-HD-4-3368 and NO1-HD-4-3383. The methods applied in this study were approved by the Institutional Review Board at the New York State Institute for Basic Research in Developmental Disabilities.
This study is supported in part by funds from the New York State Office For People With Developmental Disabilities (formerly the Office of Mental Retardation and Developmental Disabilities) and by grants from the National Institutes of Health (NIH) National Institute of Child Health and Human Development, R01 HDO43960 (JW), PO1-HDO35897 (WS), and HDO38295 (YWH; the NIH National Institute of Aging, AG03051 (BR), AG08051 (TW), and R01 AG027429 (C-XG); the Alzheimer’s Association, IIRG-05-13095 (C-XG) and NIRG-08-91126 (FL); and the Jerome Lejeune Foundation (YWH).
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Wisniewski HM, Wegiel J, Popovitch ER. Age-associated development of diffuse and thioflavin-S-positive plaques in Down syndrome. Dev Brain Dysfunction. 1994;7:330–9. [Google Scholar]
- 2.Leverenz J, Raskind MA. Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: A regional quantitative analysis. Exp Neurol. 1998;150:296–304. doi: 10.1006/exnr.1997.6777. [DOI] [PubMed] [Google Scholar]
- 3.Sadowski M, Wisniewski HM, Tarnawski M, et al. Neuronal loss in the entorhinal cortex of aged subjects with Down syndrome. Acta Neuropathol. 1999;97:156–64. doi: 10.1007/s004010050968. [DOI] [PubMed] [Google Scholar]
- 4.Wegiel J, Wisniewski HM, Dziewiatkowski J, et al. Differential susceptibility to neurofibrillary pathology among patients with Down syndrome. Dementia. 1996;7:135–41. doi: 10.1159/000106868. [DOI] [PubMed] [Google Scholar]
- 5.Lai F, Williams RS. A prospective study of Alzheimer disease in Down syndrome. Arch Neurol. 1989;46:849–58. doi: 10.1001/archneur.1989.00520440031017. [DOI] [PubMed] [Google Scholar]
- 6.Zigman WB, Schupf N, Sersen E, et al. Prevalence of dementia in adults with and without Down syndrome. Am J Ment Retard. 1995;100:403–12. [PubMed] [Google Scholar]
- 7.Holland AJ, Hon J, Huppert FA, et al. Population-based study of the prevalence and presentation of dementia in adults with Down’s syndrome. Br J Psychol. 1998;172:493–8. doi: 10.1192/bjp.172.6.493. [DOI] [PubMed] [Google Scholar]
- 8.Allsop D, Haga SI, Haga C. Early senile plaques in Down’s syndrome brains show a close relationship with cell bodies of neurons. Neuropathol Appl Neurobiol. 1989;15:531–42. doi: 10.1111/j.1365-2990.1989.tb01252.x. [DOI] [PubMed] [Google Scholar]
- 9.Mukaetova-Ladinska EB, Harrington CR, Roth M, et al. Distribution of Tau protein in Down’s syndrome: Quantitative differences from Alzheimer’s disease. Dev Brain Dysfunction. 1995;7:311–29. [Google Scholar]
- 10.Teller JK, Russo C, DeBusk LM, et al. Presence of soluble amyloid ß-peptide precedes amyloid plaque formation in Down’s syndrome. Nature Med. 1996;2:93–5. doi: 10.1038/nm0196-93. [DOI] [PubMed] [Google Scholar]
- 11.Kidd M. Alzheimer’s disease: An electron microscopical study. Brain. 1964;87:307–20. doi: 10.1093/brain/87.2.307. [DOI] [PubMed] [Google Scholar]
- 12.Terry RD, Gonatas NK, Weiss M. Ultrastructural studies in Alzheimer’s presenile dementia. Am J Pathol. 1964;44:269–97. [PMC free article] [PubMed] [Google Scholar]
- 13.Braak H, Braak E, Grundke-Iqbal I, et al. Occurrence of neuropil threads in the senile human brain and in Alzheimer’s disease: A third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett. 1986;65:351–5. doi: 10.1016/0304-3940(86)90288-0. [DOI] [PubMed] [Google Scholar]
- 14.Grundke-Iqbal I, Iqbal K, Quinlan M, et al. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–9. [PubMed] [Google Scholar]
- 15.Iqbal K, Alonso AC, Chen S, et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 16.Goedert M, Jakes R, Spillantini MG, et al. Assembly of microtubule associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–3. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- 17.Goedert M, Spillantini MG, Jakes R, et al. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–26. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 18.Lee G, Neve RL, Kosik KS. The microtubule binding domain of tau protein. Neuron. 1989;2:1615–24. doi: 10.1016/0896-6273(89)90050-0. [DOI] [PubMed] [Google Scholar]
- 19.Kitamura T, Sugimori K, Sudo S, et al. Relationship between microtubule-binding repeats and morphology of neurofibrillary tangle in Alzheimer’s disease. Acta Neurol Scand. 2005;112:327–34. doi: 10.1111/j.1600-0404.2005.00488.x. [DOI] [PubMed] [Google Scholar]
- 20.Ishizawa T, Ko LW, Cookson N, et al. Selective neurofibrillary degeneration of the hippocampal CA2 sector is associated with four-repeat tauopathies. J Neuropathol Exp Neurol. 2002;61:1040–7. doi: 10.1093/jnen/61.12.1040. [DOI] [PubMed] [Google Scholar]
- 21.Guimera J, Casas C, Pucharcos C, et al. A human homologue of Drosophila minibrain (MNB) is expressed in the neuronal regions affected in Down syndrome and maps to the critical region. Hum Mol Genet. 1996;9:1305–10. doi: 10.1093/hmg/5.9.1305. [DOI] [PubMed] [Google Scholar]
- 22.Rahmani Z, Blouin J, Creaau-Goldberg N, et al. Critical role of the D21S55 region on chromosome 21 in the pathogenesis of Down syndrome. Proc Natl Acad Sci U S A. 1989;86:5958–62. doi: 10.1073/pnas.86.15.5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song W-J, Sternberg LR, Kasten-Sportes C, et al. Isolation of human and murine homologues of the Drosophila minibrain gene: Human homolog maps to 21q22.2 in the Down syndrome critical region. Genomics. 1996;38:331–9. doi: 10.1006/geno.1996.0636. [DOI] [PubMed] [Google Scholar]
- 24.Guimera J, Casas C, Estivill X, et al. Human Minibrain homologue (MNBH/DYRK1): Characterization, alternative splicing, differential tissue expression, and overexpression in Down syndrome. Genomics. 1999;57:407–18. doi: 10.1006/geno.1999.5775. [DOI] [PubMed] [Google Scholar]
- 25.Kentrup H, Becker W, Heukelbach J, et al. Dyrk, a dual specificity protein kinase with unique structural features whose activity is dependent on tyrosine residues between subdomains VII and VIII. J Biol Chem. 1996;271:3488–95. doi: 10.1074/jbc.271.7.3488. [DOI] [PubMed] [Google Scholar]
- 26.Liu F, Liang Z, Wegiel J, et al. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 2008;22:3224–33. doi: 10.1096/fj.07-104539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woods YL, Cohen P, Becker W, et al. The kinase DYRK phosphorylates protein-synthesis initiation factor elF2Bε at Ser539 and the microtubule-associated protein tau at Thr212: Potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem J. 2001;355:609–15. doi: 10.1042/bj3550609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ryoo SR, Jeong HK, Radnaabazar C, et al. DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. J Biol Chem. 2007;2828:34850–7. doi: 10.1074/jbc.M707358200. [DOI] [PubMed] [Google Scholar]
- 29.Gong CX, Liu F, Grundke-Iqbal I, et al. Post-translational modifications of tau protein in Alzheimer’s disease. J Neural Transm. 2005;112:813–38. doi: 10.1007/s00702-004-0221-0. [DOI] [PubMed] [Google Scholar]
- 30.Dowjat WK, Adayev T, Kuchna I, et al. Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with Down syndrome. Neurosci Lett. 2007;413:77–81. doi: 10.1016/j.neulet.2006.11.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wegiel J, Dowjat K, Kaczmarski W, et al. The role of overexpressed DYRK1A protein in the early onset of neurofibrillary degeneration in Down syndrome. Acta Neuropathol. 2008;116:391–407. doi: 10.1007/s00401-008-0419-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi J, Zhang T, Zhou C, et al. Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J Biol Chem. 2008;283:28660–9. doi: 10.1074/jbc.M802645200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gui JF, Tronchère H, Chandler SD, et al. Purification and characterization of a kinase specific for the serine- and arginine-rich pre-mRNA splicing factors. Proc Natl Acad Sci U S A. 1994;91:10824–8. doi: 10.1073/pnas.91.23.10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, Kudoh J, Shinatni A, et al. Identification of two novel 5′ noncoding exons in human MNB/DYRK gene and alternatively spliced transcripts. Biochem Biophys Res Commun. 1998;250:704–10. doi: 10.1006/bbrc.1998.9392. [DOI] [PubMed] [Google Scholar]
- 35.Colvill K, Pawson T, Andrews B, et al. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J. 1996;15:265–75. [PMC free article] [PubMed] [Google Scholar]
- 36.Rossi F, Labourier E, Forne T, et al. Specific phosphorylation of SR proteins by mammalian DNA topoisomerase I. Nature. 1996;381:80–2. doi: 10.1038/381080a0. [DOI] [PubMed] [Google Scholar]
- 37.Koizumi J, Okamoto Y, Onogi H, et al. The subcellular localization of ASF is regulated by direct interaction with SR protein kinases (SRPKs) J Biol Chem. 1999;274:11125–31. doi: 10.1074/jbc.274.16.11125. [DOI] [PubMed] [Google Scholar]
- 38.Ngo JCK, Chakrabarti S, Ding J-H, et al. Interplay between SRPK and Clk/Sty kinases in phosphorylation of the splicing factor ASF/SF2 is regulated by a docking motif in ASF/SF2. Mol Cell. 2005;20:77–89. doi: 10.1016/j.molcel.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 39.Iqbal K, Braak H, Braak E, et al. Silver labeling of Alzheimer neurofibrillary changes and brain ß-amyloid. J Histotechnol. 1993;16:335–42. [Google Scholar]
- 40.Reisberg B. Functional assessment staging (FAST) Psychopharmacol Bull. 1988;24:653–9. [PubMed] [Google Scholar]
- 41.Reisberg B, Franssen E, Shah MA, et al. Clinical diagnosis of dementia: A review. In: Maj M, Sartorius N, editors. Dementia. John Wiley and Sons, Ltd.; Chichester, United Kingdom: 2000. pp. 69–115. [Google Scholar]
- 42.The National Institute on Aging and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. Neurobiol Aging. 1997;18:1–2. 1997. [PubMed] [Google Scholar]
- 43.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 44.Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wegiel J, Kuchna I, Nowicki K, et al. Cell type- and brain structure-specific patterns of distribution of minibrain kinase in human brain. Brain Res. 2004;1010:69–80. doi: 10.1016/j.brainres.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 46.Yoshida M. Cellular tau pathology and immunohistochemical study of tau isoforms in sporadic tauopathies. Neuropathology. 2006;26:457–70. doi: 10.1111/j.1440-1789.2006.00743.x. [DOI] [PubMed] [Google Scholar]
- 47.Blobel G, Potter VR. Nuclei from rat liver: Isolation method that combines purity with high yield. Science. 1966;154:1662–5. doi: 10.1126/science.154.3757.1662. [DOI] [PubMed] [Google Scholar]
- 48.Bancher C, Brunner C, Lassmann H, et al. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer’s disease. Brain Res. 1989;477:90–9. doi: 10.1016/0006-8993(89)91396-6. [DOI] [PubMed] [Google Scholar]
- 49.Bancher C, Brunner C, Lassman H, et al. Tau and ubiquitin immunoreactivity at different stages of formation of Alzheimer neurofibrillary tangles. Prog Clin Biol Res. 1989;317:837–48. [PubMed] [Google Scholar]
- 50.Beals CR, Clipstone NA, Ho SN, et al. Nuclear localization of NF-Atc by a calcineurin-dependent, cyclosporine sensitive intramolecular interaction. Genes and Dev. 1997;11:824–34. doi: 10.1101/gad.11.7.824. [DOI] [PubMed] [Google Scholar]
- 51.Graef IA, Mermelstein PG, Stankunas K, et al. L-Type calcium channels and GSK-3 regulate the activity of NF-Atc4 in hippocampal neurons. Nature. 1999;401:703–8. doi: 10.1038/44378. [DOI] [PubMed] [Google Scholar]
- 52.Yang EJ, Ahn YS, Chung KC, et al. Protein kinase Dyrk1A activates cAMP response element-binding protein during neuronal differentiation in hippocampal progenitor cells. J Biol Chem. 2001;276:39819–24. doi: 10.1074/jbc.M104091200. [DOI] [PubMed] [Google Scholar]
- 53.Mao J, Maye P, Kogerman P, et al. Regulation of Gli1 transcriptional activity in the nucleus by Dyrk1. J Biol Chem. 2002;277:35156–61. doi: 10.1074/jbc.M206743200. [DOI] [PubMed] [Google Scholar]
- 54.de Graaf K, Hekerman P, Spelten O, et al. Characterization of cyclin L2, a novel cyclin with an arginine/serine-rich domain: Phosphorylation by DYRK1A and colocalization with splicing factors. J Biol Chem. 2004;279:4612–24. doi: 10.1074/jbc.M310794200. [DOI] [PubMed] [Google Scholar]
- 55.Alvarez M, Estivill X, de la Luna S. Dyrk1A accumulates in splicing speckles through a novel targeting signal induces speckles disassembly. J Cell Sci. 2003;116:3099–107. doi: 10.1242/jcs.00618. [DOI] [PubMed] [Google Scholar]
- 56.de Graaf K, Czajkowska H, Rottmann S, et al. The protein kinase DYRK1A phosphorylates the splicing factor SF3b1/SAP155 at Thr434, a novel in vivo phosphorylation site. BMC Biochem. 2006;7:7. doi: 10.1186/1471-2091-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaczmarski W, Barua M, Bolton D, et al. Subcellular distribution of posttranslationally modified dual-specificity tyrosine phosphorylation-regulated kinase 1A in human and mouse brain. J Neurochem. In press. [Google Scholar]
- 58.Arron JR, Winslow MM, Polleri A, et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441:582–3. doi: 10.1038/nature04678. [DOI] [PubMed] [Google Scholar]
- 59.Ferrer I, Barrachina M, Puig B, et al. Constitutive Dyrk1A is abnormally expressed in Alzheimer disease, Down syndrome, Pick disease, and related transgenic models. Neurobiol Dis. 2005;20:392–400. doi: 10.1016/j.nbd.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 60.Espinoza M, de Silva R, Dickson DW, et al. Differential incorporation of tau isoforms in Alzheimer’s disease. J Alzheimers Dis. 2008;14:1–6. doi: 10.3233/jad-2008-14101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 62.Cras P, Smith MA, Richey PL, et al. Extracellular neurofibrillary tangles reflect neuronal loss and provide further evidence of extensive protein cross-linking in Alzheimer disease. Acta Neuropathol. 1995;89:291–95. doi: 10.1007/BF00309621. [DOI] [PubMed] [Google Scholar]
- 63.Delacourte A, David JP, Sergeant N, et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology. 1999;52:1158–65. doi: 10.1212/wnl.52.6.1158. [DOI] [PubMed] [Google Scholar]
- 64.Maeda S, Sahara N, Saito Y, et al. Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer’s disease. Neurosci Res. 2006;54:197–201. doi: 10.1016/j.neures.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 65.Sahara N, Maeda S, Murayama M, et al. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur J Neurosci. 2007;25:3020–9. doi: 10.1111/j.1460-9568.2007.05555.x. [DOI] [PubMed] [Google Scholar]
- 66.Zhang YU, Xu YF, Chen XQ, et al. Nitration and oligomerization of Tau induced by peroxynitrite inhibit its microtubule-binding activity. FEBS Lett. 2005;579:2421–7. doi: 10.1016/j.febslet.2005.03.041. [DOI] [PubMed] [Google Scholar]
- 67.Sugino E, Nishiura C, Minoura K, et al. Three-/four-repeat-dependent aggregation profile of tau microtubule-binding domain clarified by dynamic light scattering analysis. Biochem Biophys Res Commun. 2009;385:236–40. doi: 10.1016/j.bbrc.2009.05.047. [DOI] [PubMed] [Google Scholar]
- 68.Andorfer C, Acker CM, Kress Y, et al. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–54. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim EJ, Sung Y, Lee HJ, et al. Dyrk1A phosphorylates alpha-synuclein and enhances intracellular inclusion formation. J Biol Chem. 2006;281:33250–7. doi: 10.1074/jbc.M606147200. [DOI] [PubMed] [Google Scholar]
- 70.Sitz JH, Baumgärtel K, Hämmerle B, et al. The Down syndrome candidate dual-specificity tyrosine phosphorylation-regulated kinase 1A phosphorylates the neurodegeneration-related septin 4. Neuroscience. 2008;157:596–605. doi: 10.1016/j.neuroscience.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 71.Ihara M, Tomimoto H, Kitayama H, et al. Association of the cytoskeletal GTP-binding protein Sept4/H5 with cytoplasmic inclusions found in Parkinson’s disease and other synucleinopathies. J Biol Chem. 2003;278:24095–102. doi: 10.1074/jbc.M301352200. [DOI] [PubMed] [Google Scholar]
- 72.Ihara M, Yamasaki N, Hagiwara A, et al. Sept4, a component of presynaptic scaffold and Lewy bodies, is required for the suppression of alpha-synuclein neurotoxicity. Neuron. 2007;53:519–33. doi: 10.1016/j.neuron.2007.01.019. [DOI] [PubMed] [Google Scholar]
- 73.Kinoshita A, Kinoshita M, Akiyama H, et al. Identification of septins in neurofibrillary tangles in Alzheimer’s disease. Am J Pathol. 1998;153:1551–60. doi: 10.1016/S0002-9440(10)65743-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ryoo SR, Cho HJ, Le HW, et al. Dual specificity tyrosine (Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: Evidence for a functional link between Down syndrome and Alzheimer’s disease. J Neurochem. 2008;104:1333–44. doi: 10.1111/j.1471-4159.2007.05075.x. [DOI] [PubMed] [Google Scholar]
- 75.Kimura R, Kamino K, Yamamoto M, et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum Mol Genet. 2007;16:15–23. doi: 10.1093/hmg/ddl437. [DOI] [PubMed] [Google Scholar]
- 76.Ksiezak-Reding H, Shafit-Zagardo B, Yen SH. Differential expression of exons 10 and 11 in normal tau and tau associated with paired helical filaments. J Neurosci Res. 1995;41:583–93. doi: 10.1002/jnr.490410504. [DOI] [PubMed] [Google Scholar]
- 77.Yasojima K, McGeer EG, McGeer PL. Tangled areas of Alzheimer brain have upregulated levels of exon 10 containing tau mRNA. Brain Res. 1999;831:301–5. doi: 10.1016/s0006-8993(99)01486-9. [DOI] [PubMed] [Google Scholar]
- 78.Chambers CB, Lee JM, Troncoso JC, et al. Overexpression of four-repeat tau mRNA isoforms in progressive supranuclear palsy but not in Alzheimer’s disease. Ann Neurol. 1999;46:325–32. doi: 10.1002/1531-8249(199909)46:3<325::aid-ana8>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 79.Boutajangout A, Boom A, Leroy K, et al. Expression of tau mRNA and soluble tau isoforms in affected and non-affected brain areas in Alzheimer’s disease. FEBS Lett. 2004;576:183–9. doi: 10.1016/j.febslet.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 80.Umeda Y, Taniguchi S, Arima K, et al. Alterations in human tau transcripts correlate with those of neurofilament in sporadic tauopathies. Neurosci Lett. 2004;359:151–4. doi: 10.1016/j.neulet.2004.01.060. [DOI] [PubMed] [Google Scholar]
- 81.Connell JW, Rodriguez-Martin T, Gibb GM, et al. Quantitative analysis of tau isoform transcripts in sporadic tauopathies. Mol Brain Res. 2005;137:104–9. doi: 10.1016/j.molbrainres.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 82.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDO-17. Nature. 1998;393:702–4. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 83.Spillantini MG, Murrell JR, Goedert M, et al. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737–41. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.