

Abstract
FRA-2/FOSL2 is a basic region-leucine zipper motif transcription factor that is widely expressed in mammalian tissues. The functional repertoire of this factor is unclear, partly due to a lack of knowledge of genomic sequences that are targeted. Here, we identified novel, functional FRA-2 targets across the genome through expression profile analysis in a knockdown transgenic rat. In this model, a nocturnal rhythm of pineal gland FRA-2 is suppressed by a genetically encoded, dominant negative mutant protein. Bioinformatic analysis of validated sets of FRA-2-regulated and -nonregulated genes revealed that the FRA-2 regulon is limited by genomic target selection rules that, in general, transcend core cis-sequence identity. However, one variant AP-1-related (AP-1R) sequence was common to a subset of regulated genes. The functional activity and protein binding partners of a candidate AP-1R sequence were determined for a novel FRA-2-repressed gene, Rgs4. FRA-2 protein preferentially associated with a proximal Rgs4 AP-1R sequence as demonstrated by ex vivo ChIP and in vitro EMSA analysis; moreover, transcriptional repression was blocked by mutation of the AP-1R sequence, whereas mutation of an upstream consensus AP-1 family sequence did not affect Rgs4 expression. Nocturnal changes in protein complexes at the Rgs4 AP-1R sequence are associated with FRA-2-dependent dismissal of the co-activator, CBP; this provides a mechanistic basis for Rgs4 gene repression. These studies have also provided functional insight into selective genomic targeting by FRA-2, highlighting discordance between predicted and actual targets. Future studies should address FRA-2-Rgs4 interactions in other systems, including the brain, where FRA-2 function is poorly understood.
Keywords: AP-1 Transcription Factor, CBP, CREB, Gene Expression, Microarray, Pineal Gland, Rat, FRA-2, Rgs Gene
Introduction
Fos-related antigen 2 (FRA-2/FOSL2) is a member of the FOS/JUN subgroup of bZIP3 transcription factors (TFs) that function in concert with regulatory DNA sequences in target genes (1). The various FOS proteins appear to serve distinct developmental, physiological, and pathological roles (2, 3), and current studies are seeking to define these individual roles and the mechanisms involved. FRA-2 exerts a specific action in bone development (4) and appears to have selective physiological and pathological roles in diverse processes, including photoperiodic regulation (5), some cancers (6), and pulmonary fibrosis (7). This accumulated evidence of functional specialization indicates that mechanisms confer specificity to the actions of the FRA-2 TF relative to other members of the FOS/JUN group.
One aspect of this selectivity that remains to be fully characterized is selective genomic targeting by FRA-2. Selection at the level of cis-sequence specificity is clearly one aspect of this targeting; numerous studies established that different combinations of FOS and JUN proteins (28 dimeric combinations in total) have different binding affinities for activator protein-1 (AP-1) and cAMP-response element (CRE) sequences (8, 9). A study using tethered protein dimers (10) showed that whereas the c-FOS/c-JUN dimer interacts exclusively with AP-1-like sequences (core consensus, tga(g/c)tca), other dimers, including FRA-2/c-JUN, are less selective and also bind CRE elements (core consensus, tgacgtca). However, it is also apparent that there are other levels of selection because about one-third of vertebrate promoters contain consensus AP-1 family sites (AP-1F; MatInspector, Genomatix), but not all these genes are regulated by AP-1 (11).
FRA-2, like FRA-1, differs from FOS and FOSB in lacking a potent C-terminal transactivation domain (12). As may be predicted from this structural difference, FRA-2 can act to suppress transcription, for example at promoters induced by c-JUN homodimers (12). However, in heterodimeric combination with JUND, FRA-2 can enhance transcription relative to JUND homodimers (12). These findings exemplify the varied actions of FRA-2 and underline the critical role that AP-1 protein composition plays in transactivation potential (13).
The role of FRA-2 has been studied using a unique transgenic animal model in which FRA-2 function is perturbed in a tissue-selective manner using a dominant negative (C-terminally truncated) genetic construct (DN-FRA-2 (14)). In this model, DN-FRA-2 is stably expressed in transgenic rats under the control of a conditional promoter (Aanat (15, 16)) that confers cellularly (pinealocyte) and temporally (nocturnal) specific expression to the pineal gland. DN-FRA-2 binds DNA normally, but because it is unable to transactivate target genes, it competes with endogenous FRA-2 for target sites and thereby inhibits endogenous FRA-2 activity. In addition, it has been shown that this construct also knocks down expression of endogenous Fra-2 via competition of autoregulatory feedback (14).
The conditional nature of this transgenic model is of notable importance because it allows examination of the role of FRA-2 throughout life under physiological conditions. In contrast, the study of FRA-2 function in conventional knock-out animal models is limited by neonatal lethality of Fra-2 null alleles (17). The model is also able to reveal true physiological roles of FRA-2 because the pineal gland has an endogenous rhythm of gene expression (linked to rhythms of hormone production) that is regulated by FRA-2 (14, 18); when induced in this animal model, DN-FRA-2 therefore intervenes in an autonomous physiological response. Accordingly, this model is superior to experimental paradigms where gene expression is artificially induced.
In previous studies with the DN-FRA-2 model, we have shown that FRA-2 mediates both positive and negative transcriptional regulation of genes (14). These opposite modes of regulation by FRA-2 are intriguing because studies have shown that the nocturnal AP-1 binding complex in the rat pineal gland is largely composed of FRA-2/JUND that is inconsistent, prima facie, with multiple modes of regulation (14). FRA-2 phosphorylation in the nocturnal pineal gland is also progressive and homogeneous (14), arguing against a role for different protein isoforms underlying differential regulation. We therefore hypothesize that differential regulation of target gene expression is mediated at the level of a cis-acting sequence. To pursue this, we have now sought to explain the complexity of gene regulation by FRA-2 through further analysis using transcriptome-scale expression profiling. This strategy was adopted to obtain a dataset of FRA-2-regulated genes for bioinformatic analysis of cis-regulatory sequences. The results of these studies provide insight into selective genomic targeting of FRA-2 and reveal discordance between predicted and real targets.
EXPERIMENTAL PROCEDURES
Animal Care and Sampling
Animal studies were conducted in accordance with both Home Office regulations (United Kingdom) and local ethical review. Transgenic (Sprague-Dawley) rats of the DN-FRA-2 line (14) were maintained under standard laboratory conditions in a 14:10 light/dark cycle (lights on: 05.00 h). For microarray analysis, 12 rats were killed at each sample point, and dissected pineal glands were pooled (four pineal glands in each pool, in triplicate) and stored at −70 °C. The four sample points were as follows: wild-type, 12.00 h (WT12) and transgenic, 12.00 h (TG12); wild-type, 24.00 h (WT24) and transgenic, 24.00 h (TG24). Therefore, a total of 48 animals were used in this initial array analysis. For candidate gene validation, a similar sampling regimen was employed, but only two pineal glands were pooled in each sample (for sample numbers see Table 1). Pineal glands and brain cortex (parietal, layers I–VI) from additional wild-type animals were sampled to provide material for the EMSA and ChIP analyses.
TABLE 1.
Validation of novel FRA-2-regulated and nonregulated pineal genes in transgenic rats
Values are mean ± S.E. of Northern blot mRNA levels calculated as percentage of maximum within each individual co-blotted group of four conditions (12WT and 12TG and 24WT and 24TG; note that the timing/genotype of maximum values varied for some genes). Each individual RNA sample was extracted from a pair of pineal glands (two rats) and multiple replicates of each group of four samples were used: n = 4 (Atf4, Cox6a2, Rgs4, Mt1a, Opn1sw, and Crem); n = 3 (Dbp, E4BP4, and Id1); n = 2 (Per2 and Syt4). Numbers in superscript refer to statistically distinct groups determined by ANOVA combined with Duncan's post hoc test (p < 0.05). Statistical analysis was not conducted where n = 2. ND indicates no detectable mRNA band.
| Gene | 12.00 h |
24.00 h |
||
|---|---|---|---|---|
| Wild-type | Transgenic | Wild-type | Transgenic | |
| FRA-2 repressed genes | ||||
| Atf4 | 31.2 ± 3.21 | 30.0 ± 2.21 | 56.0 ± 8.12 | 100.0 ± 03 |
| Cox6a2 | 20.7 ± 3.81 | 89.2 ± 10.82 | 18.6 ± 1.51 | 83.1 ± 8.72 |
| Rgs4 | 95.9 ± 3.31 | 98.4 ± 1.61 | 36.8 ± 6.22 | 76.8 ± 8.53 |
| FRA-2 enhanced genes | ||||
| Mt1a | ND | ND | 100.0 ± 01 | 50.3 ± 9.02 |
| Opn1sw | 100.0 ± 01 | 26.6 ± 3.42 | 43.0 ± 6.13 | 12.0 ± 1.94 |
| FRA-2 non-regulated genes | ||||
| Crem/ICER | ND | ND | 97.7 ± 2.31 | 93.7 ± 4.41 |
| Dbp | 44.4 ± 3.31 | 47.8 ± 6.01 | 98.0 ± 2.02 | 92.4 ± 5.12 |
| E4bp4 | 48.8 ± 3.21 | 46.8 ± 3.81 | 100 ± 02 | 95.1 ± 7.12 |
| Id1 | 15.0 ± 2.01 | 18.0 ± 4.51 | 98.0 ± 2.32 | 96.0 ± 4.82 |
| Per2 | 20.0 ± 5.0 | 21.5 ± 3.5 | 100 ± 0 | 89.1 ± 5.9 |
| Syt4 | 31.6 ± 5.1 | 32.4 ± 2.1 | 89.7 ± 10.3 | 95.5 ± 4.5 |
Microarray Analysis
Total cellular RNA was extracted from pooled pineal glands as described previously (18). Aliquots of each RNA sample were supplied to the Wales Gene Park Expression Profiling Service (Cardiff University) where RNA quality was verified using Agilent RNA6000 chips. 10 mg of each sample was used to generate cRNA targets using Affymetrix GeneChip® protocols and reagents (Affymetrix, Santa Clara, CA). Each sample was then probed with Affymetrix Rat Genome 230A microarray chips (15,923 probe sets; 10,174 genes). Hybridization and washing were performed using a GeneChip® fluidics station 400 (Affymetrix). Following microarray scanning, the average signal intensity of each array was scaled to 100 (Microarray Suite 5.0, Affymetrix). Microarray data are available at the Entrez Gene Expression Omnibus, National Center for Biotechnology Information (GEO series accession number GSE12344).
Further analysis of the microarray data and selection of candidate gene lists were conducted using GeneSpring software (7.0; Silicon Genetics, Redwood City, CA). The aim of this in silico analysis was to select genes (transcripts) that appeared to be differentially regulated in the DN-FRA-2 transgenic model and therefore provide candidates for validation and further bioinformatic and biological analysis. Stringent selection criteria were applied to generate short candidate gene lists; a consequence of this approach is that the candidate gene lists (supplemental Table S2) should not be considered as replete with respect to the 230A GeneChip features (see comment under “Results”). Using GeneSpring 7.0, the scaled microarray data were first normalized (to mean of WT12) using default settings. The data were then filtered (raw (average difference) value >50 in at least two samples) using c-Jun (19) as a “guide gene” to direct an appropriate level of filtering that would remove genes detected at negligible levels but retain documented, rhythmic pineal gland transcripts. This filtered gene list of 8607 genes was then used to generate four lists of genes that exhibited differential expression with respect to either strain (TG versus WT) or time (12.00 h versus 24.00 h). These gene lists were generated using either a fold-difference approach for strain (1.5-fold up- or down-regulated in at least two samples) or a statistical test for time (t test with multiple testing correction, assuming equal variances, applying Benjamini and Hochberg false discovery rate, significance accepted for p < 0.05). Finally, the four gene lists were merged using the GeneSpring Venn diagram tool, generating six candidate gene lists that represent each of the six possible differential gene expression conditions: TG > WT, 12 > 24; TG > WT, 12 < 24; TG > WT, 12 = 24; TG < WT, 12 > 24; TG < WT, 12 < 24; TG < WT, 12 = 24 (supplemental Table S2). The selection of individual genes for validation by Northern blot analysis was conducted subjectively, selecting both novel examples from the candidate gene lists (see above) and genes that are known to exhibit rhythmic pineal expression.
Candidate Gene Validation/Northern Analysis
Northern blots were used for validation because some pineal transcripts are unique to this tissue due to alternative promoter usage, e.g. Slc15a1 (20). Total cellular RNA was prepared from a pool of two pineal glands at each sample point (for sample numbers see Table 1), and specific mRNAs were detected and quantified by Northern blot analysis as described previously (18). cDNA probes were labeled by random priming using [32P]dCTP (GE Healthcare). Northern blots were stripped (boiling 0.1% SDS, three times for 2 min) and re-probed with an 18 S cDNA (DecaTemplateTM, Ambion, Austin, TX). Densitometric analysis of mRNA levels was performed using ImageQuantTM software (version 3.0, Amersham Biosciences), correcting values against the level of 18 S RNA. Statistical comparison of the experimental groups was conducted using ANOVA with Duncan's post hoc test where appropriate (p < 0.05 significance level; SPSS 13, SPSS Inc., Chicago).
Bioinformatic Analysis
Genomic DNA sequences were obtained from current Ensembl genome builds. Some specific gene promoter sequences were obtained from the Genomatix data base (Matbase, Genomatix Software Gmbh, Munich, Germany) (21). Sequence alignment and comparative genomic analysis were conducted using features of the UCSC Genome Browser: Multiz alignment (22) and PhastCons conservation (23). The empirically determined transcriptional start site (TSS) for human RGS4 was obtained from the Eukaryotic Promoter Database (Release 91 (24)), and the location of the mouse and human Rgs4 promoters was confirmed using MPromDb (bioinformatics. med.ohio-state.edu/MPromDb (25)). Our Northern blot analysis (see “Results”) is consistent with a single transcript that uses the Rgs4 “TSS1” structure (26). The presence of common cis-regulatory modules in sets of co-regulated genes was analyzed using ModuleMiner (27), which scans the whole genome, optimizing co-regulated/expressed gene sets relative to all others. Genomatix software (MatInspector) was used to identify individual transcription factor consensus binding elements. Images of consensus sequence logos were downloaded from the Genomatix site and modified in Photoshop (CS2, Adobe Systems Inc., CA).
Chromatin Immunoprecipitation (ChIP) Analysis
ChIP analysis was conducted by modifying a published protocol (28) to incorporate the ChIP-IT express kit (Active Motif). For cross-linked chromatin isolation, paired pineal glands were rapidly isolated from the skull and bisected prior to incubation in 1% formaldehyde in PBS for 15 min at room temperature. Following formaldehyde removal, the tissue was washed twice in ice-cold PBS containing both protease and phosphatase inhibitor mixtures (P8340 and P2850; Sigma). The tissue was then disrupted with a Dounce homogenizer (two times for 10 s) in a buffer containing 10 mm HEPES-KOH, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, and & P2850 mixtures) and centrifuged at 14,000 rpm for 10 s. The homogenate was then resuspended in shearing buffer (Active Motif) and incubated on ice for 10 min. Chromatin was then sheared to 200–500 bp (verified by agarose gel electrophoresis) using a closed system ultrasonic cell disruptor (Bioruptor 200, Diagenode s.a., Liege, Belgium). Samples were centrifuged at 14,000 rpm for 10 min at 4 °C, and the supernatant was stored at −80 °C. An aliquot of this material was retained as “input” DNA. The remaining chromatin sample was divided; one-half was immunoprecipitated with the “test” antiserum, and the second half was used for a mock immunoprecipitation with a control serum (see below). For quantitation, ChIP assay values were compared with assay values obtained with the input DNA. ChIP assays were conducted according to the manufacturer's protocol (Active Motif) using antisera to FRA-2 (14), RNA polymerase II (Active Motif), and CBP (A-22, Santa Cruz Biotechnology). For this application, equal amounts of two FRA-2 antisera directed against different epitopes of the FRA-2 protein were used (2605 and 2607 see (14)). Mock immunoprecipitation reactions were performed using either control IgG (ChIP-IT Control Kit, Active Motif) or FRA-2 preimmune serum (14). A 140-bp region of the rat Rgs4 promoter that contains the conserved tgcgtca site was amplified (GoTaq, Promega) using primers Rgs4-C1F and Rgs4-C1R (supplemental Table S1). For comparison, a 223-bp sequence from the rat β-actin gene (primers, ChIP-IT control kit, Active Motif) was also amplified from the IP and mock IP samples. Semi-quantitative analysis of FRA-2 and RNA polymerase II chip assays was conducted via standard PCR amplification (Go-Taq) and agarose gel electrophoresis. Amplified DNA bands were resolved on 2% agarose gels, and images were obtained using Genesnap software (Syngene, Frederick, MD). PCR band sizes were verified using a low molecular mass DNA ladder (HyperLadder V, Bioline Ltd., London, UK), and band intensity was estimated using GeneTools software (Syngene). The resultant levels of amplified Rgs4 promoter sequence were compared with levels of amplified ChIP input DNA.
To obtain a quantitative measure of CBP occupancy at the Rgs4 promoter in DN-Fra-2 and wild-type rats, QPCR analysis of CBP ChIP was conducted using a real time PCR system (Mx3005P, Stratagene, La Jolla, CA). A similar region of the rat Rgs4 promoter was amplified using primers designed for QPCR (Rgs4-C2F and Rgs4-C2R; supplemental Table S1). Amplification reactions were performed using Brilliant® Core reagents according to the manufacturer's instructions (Stratagene), and products were detected using SYBR Green®. Each PCR was conducted in duplicate. Threshold (Ct) values for ChIP and input chromatin were obtained directly from the MxPro software (Stratagene) and %-of-Input values were calculated using the Superarray ChIP-QPCR data analysis template (Superarray Bioscience Corp., Frederick, MD).
Rgs4 Promoter Analysis
rRgs4 promoter function was investigated in PC12 cells, a rat cell line that has been shown to express Rgs4 mRNA and protein (29, 30). Novel Rgs4 promoter plasmids derived from pGL4.11 (Promega) were constructed by ligating rat Rgs4 promoter fragments (−167 to +6 and −426 to +6 of TSS) into the pGL4.11 multiple cloning site. These fragments were amplified (Extract N′Amp, Sigma) from rat genomic DNA using PCR primers (Rgs4-167F, Rgs4-R, Rgs4-m167F, Rgs4-426F, and Rgs4-m426F; supplemental Table S1). Rgs4-m167F and Rgs4-m426F contain modifications to the predicted AP-1 sites (tgcgtca and tgactca, respectively) creating the mutant sequences ttcgaaa and ttcgaga, respectively. Amplified products were cut with SacI and HindIII and ligated into SacI/HindIII-cut pGl4.11 using T4DNA ligase (Promega). Transformed bacteria were expanded, and plasmids were extracted using the PureYield kit (Promega). Plasmid sequences were determined by standard DNA sequencing (Prism ready reaction dye-deoxy terminator cycle sequencing kit (PerkinElmer Life Sciences)) and an ABI prism automated DNA sequencer (377, PerkinElmer Life Sciences).
PC12 cells (CRL-1721.1; LGC Standards, Teddington, Middlesex, UK) were grown in DMEM (Invitrogen) with 10% horse serum, 5% FBS (Invitrogen) and 1× antibiotic/antimycotic (Invitrogen) at 37 °C and 5% CO2. Cells were transfected (TransFast protocol, Promega) with the Rgs4 promoter constructs together with the reference plasmid pGL4.75(hRluv/CMV (50:1 molar ratio; Promega)) and maintained for 30 h. After this time, cells were lysed, and both Firefly and Renilla luciferase assays were conducted according to the manufacturer's protocol (Dual-Luciferase reporter assay system, Promega). Relative luminescence values were measured on a Luminometer (model TD-20/20, Turner Biosystems, Sunnyvale, CA). Each transfection was replicated 8-fold (four replicates in two separate experiments). Statistical analysis was conducted with SPSS16 for Mac using one-way ANOVA together with a post hoc Duncan's test. Significance was accepted for p < 0.05.
EMSA Analysis
Electrophoretic mobility shift assay analysis (EMSA) was conducted as described previously (18), but annealed oligonucleotides (170 fmol; Sigma) containing the −155 Rgs4 AP-1R element (Rgs4-EF and Rgs4-ER, supplemental Table S1) were 3′-biotinylated rather than radiolabeled. Nuclear protein extracts (27) were quantified using the QuickStartTM Bradford reagent (Bio-Rad), and equalized aliquots added to EMSA binding reactions conducted in 10 mm Tris-HCl, 50 mm KCl, 1 mm dithiothreitol, 0.1 mm EDTA, 2.5% glycerol, 5 mm MgCl2. Following binding, the reactions were resolved on 6% native polyacrylamide gels in 0.5× TBE at 150 V for ∼45 min. DNA was then transferred to a nylon membrane (Hybond-XL; GE Healthcare) using either a standard denaturation and blotting procedure (18) or electrotransfer (30 mins, 300 mA; Mini Trans-Blot, Bio-Rad). The membranes were then baked at 80 °C for 45 min. Biotin-labeled EMSA bands were detected with chemiluminescence (Pierce). Gel loading/transfer was verified by inspection of unbound probe bands. The specificity of DNA-protein interactions was investigated by conducting the binding reactions in the presence of a molar excess of annealed nonbiotinylated oligonucleotides as follows: (i) Rgs4 AP-1R oligonucleotides (as above); (ii) mutant Rgs4 AP-1R oligonucleotides (Rgs4-mEF and Rgs4-mER; supplemental Table S1). The presence of specific proteins to the binding complexes was investigated using multiple protein antisera as follows: anti-FRA-2 (Ab2605 and Ab2607 (21)); anti-CREB-1 (sc186, Santa Cruz Biotechnology, Santa Cruz, CA; 9197, Cell Signaling Technology, Beverly, MA); anti-phospho(p)-CREB-1 (Ser-133) (sc-7978, Santa Cruz Biotechnology; 9191, Cell Signaling Technology); anti-CBP (A-22, Santa Cruz Biotechnology) or as a control rabbit IgG (Active Motif, Carlsbad, CA).
Western Blot Analysis
Nuclear or whole cell protein extracts were analyzed as described previously (18). Primary antibodies used were anti-CBP (A-22), anti-CREB-1 (9197), anti-p-CREB-1(9191), anti-FRA-2 (2607 (14)), and anti-GAPDH (ab9845, Abcam). Protein bands were detected with enhanced chemiluminescence (ECL Plus, GE Healthcare) and quantitated using ImageQuantTM 3.0 (GE Healthcare).
RESULTS
Expression profiling with the Affymetrix 230A rat genome microarray (15866 probe sets) demonstrated that a large number of gene transcripts are dysregulated in the DN-FRA-2 rat model. The full set of (predicted) FRA-2 targets are listed in supplemental Table S2, and a subset of Northern blot-validated targets are listed in Table 1. Representative Northern blots are shown in Fig. 1 and supplemental Fig. S1. Our (statistically verified) validation procedure also involved Northern blot analysis of five nonregulated transcripts (Table 1) and confirmed the microarray/GeneSpring-based prediction of FRA-2 targets. These targets (supplemental Table S2) include proteins of diverse molecular function and biological process, with cellular locations from plasma membrane to nucleus. ModuleMiner analysis confirmed that no Gene Ontology terms are over-represented in the FRA-2-regulated gene set; therefore FRA-2 does not appear to be associated with one particular aspect of cellular function. To gain an understanding of the general molecular mechanisms used by FRA-2, we next conducted bioinformatic analysis of validated target genes.
FIGURE 1.

Validation of Rgs4 regulation in DN-FRA-2 transgenic rats. A, representative Northern analysis of Rgs4 transcript and 18 S RNA control expression in pineal glands sampled from DN-FRA-2 transgenic rats (TG) and wild-type controls (WT) during the day (12.00 h) and night (24.00 h). 18 S and 28 S mark the location of ribosomal RNA bands. B, summated Northern blot densitometric data from four independent blots as in A. Values (mean ± S.E.) are percentage of maximum level in each blot, corrected against the 18 S level. Numbers denote independent groups as derived from ANOVA and Duncan's test (p < 0.05). C, consensus sequence logos for the AP-1R (V$BACH2.01) PWM, and AP-1F (V$AP-1.01) PWM. D, schematic illustration of rat Rgs4 gene structure based on Ensembl transcript ENSRNOT00000003774. Note that the length of both introns and untranslated region of exon 5 (open box) are truncated. Exon lengths are shown above each exon. The 5′-flanking region is partially expanded to show the position and sequence of the −155 AP-1R element relative to the transcriptional start site. Below, sequence is further expanded to show relative positions of the AP-1R element, an associated CRE half-site, and an ETSF site (see text).
In Silico Analysis of Candidate Gene Promoters
FRA-2 actions may be either direct or indirect, i.e. involve intermediating TFs that themselves directly target gene promoters. One potential intermediary factor (CREM/ICER (31)) is not regulated in the DN-FRA-2 model (Table 1); however, by using in silico analysis, we sought to obtain positive evidence of conserved sequences that could mediate direct genomic actions of FRA-2.
Large scale (−10,000 bp from TSS) regulatory module homology analysis (ModuleMiner (27)) showed that AP-1 elements were over-represented in the regulated group of genes versus validated nonregulated genes (Table 2). Further division of the regulated genes into positively and negatively regulated subgroups generated an additional interesting finding, namely that only repressed genes exhibited an enrichment of AP-1 sites. This finding might indicate that some (positively regulated) genes may be indirectly regulated.
TABLE 2.
Analysis of cis-regulatory module conservation in FRA-2-regulated gene sets
Analysis was conducted on microarray-derived, validated gene sets using ModuleMiner (see under “Experimental Procedures”). Input sequence indicates human-mouse conserved noncoding sequences, 10 kb 5′ of TSS (default setting), based on Ensembl 36. Abbreviations used are as follows: PWM, position weight matrix; V$AP1_01, etc., are codes for the Transfac-defined PWM groups of sequences; V indicates vertebrate.
| Gene set | Individual PWM (top 5) | PWM weight | Top PWM groupa | Top PWM group weight |
|---|---|---|---|---|
| All FRA-2 regulated genes | V$AP1_Q2_01 | 1.08 | V$AP1_01 | |
| V$CDX_Q5 | 0.869 | V$AP1_Q2_01 | ||
| V$NFY_Q6_01 | 0.659 | V$AP1_Q4_01 | 1.76 | |
| V$ALPHACP1_01 | 0.591 | V$BACH2_01 | ||
| V$AP1_01 | 0.382 | |||
| FRA-2 repressed genes | V$AP1_01 | 0.955 | ||
| V$ALPHACP1_01 | 0.855 | V$AP1_01 | ||
| V$LEF1_Q2 | 0.765 | V$AP1_Q6_01 | 1.87 | |
| V$AP1_Q6_01 | 0.735 | V$AP1_Q6 | ||
| V$PAX4_03 | 0.55 | |||
| FRA-2 enhanced genes | V$AMEF2_Q6 | 0.97 | ||
| RUSH1-a | 0.776 | |||
| V$AP4_01 | 0.699 | V$AMEF2_Q6 | 0.97 | |
| V$NFY_Q6_01 | 0.563 | |||
| V$ALPHACP1_01 | 0.514 | |||
| FRA-2-nonregulated genes | V$ER_Q6_02 | 0.955 | V$NFY_Q6_01 | |
| Arnt-Ahr | 0.605 | NF-Y | ||
| V$KROX_Q6 | 0.594 | V$NFY_01 | 0.988 | |
| V$AP2_Q6 | 0.409 | V$NFY_Q6 | ||
| V$NFY_01 | 0.295 |
a TOP PWM GROUP is a ModuleMiner output that provides a summed score for the most represented PWM group.
However, analysis of the validated genes employing a more proximal (−2000 to +100) sequence and MatInspector, which distinguishes different classes of AP-1 sites, delivered a less clear outcome (Table 3). We found the following: (i) the presence of AP-1 family (AP-1F, Fig. 1C) and CRE sequences in the majority (but not all) of both FRA-2-regulated and nonregulated genes; (ii) no obvious distinction in the distribution of these sites between FRA-2-regulated and nonregulated genes; (iii) no clear distinction in the distribution of these sites between FRA-2-positively and -negatively regulated genes. At the same time, MatInspector analysis revealed the presence of some interesting alternative sites in the regulated genes set; the most common (8 of 9 genes) AP-1 sites were of the AP-1-related (AP-1R, Fig. 1C) matrix family. This was of interest because this family of elements includes the sequence tgcgtca (the highly conserved AP-1F “A” at position 3 is substituted by a “C”, Fig. 1C) that is present in the FRA-2-regulated Nr4a1 gene and binds FRA-2 (18). The tgcgtca sequence (present in Nr4a1, FRA-2, and Rgs4) includes, on the reverse DNA strand, a CRE half-site which, in some sequence contexts, can bind CREB with high affinity (32). In Rgs4, this CRE site has been predicted to be a functional CRE (CRE-TATA module) as defined by whole genome bioinformatics and ChIP-Chip analysis (33, 34). The potential role of this AP-1R site in mediating the effects of FRA-2 was therefore addressed in further extensive studies of Rgs4, which is robustly expressed in the pineal gland as a single transcript of ∼3 kb (Fig. 1A).
TABLE 3.
Search for AP1/CRE elements in FRA-2-regulated and nonregulated genes
Consensus elements were identified over the region −2000 to +100 bp relative to TSS using MatInspector (see text). AP1F and Bach2.01 are Transfac notations for different classes of AP-1 and AP-1-like elements. Repressed and enhanced refer to the mode of FRA-2 regulation identified using the DN-FRA-2 rat model. Up, down, and over refer to the position (upstream, downstream, and overlapping) of identified elements relative to CRE within this region of the gene sequences.
| Genes | AP1F elements relative to CRE | Bach2.01 elements relative to CRE | tgcgtca sequence |
|---|---|---|---|
| Validated FRA-2 regulated genes (repressed) | |||
| Atf-4 | Four up and down of eight CRE | two up and down of eight CRE | 0 |
| Cox6a2 | Two up of one CRE | None | 0 |
| Dio2 | One up and down of five CRE | Two up of one CRE | 0 |
| Nr4a1 | No AP1F, six CRE | Four down and over six CRE | 4 |
| Rgs4 | Three up and down of four CRE | Two down and over two CRE | 1 |
| Validated FRA-2 regulated genes (enhanced) | |||
| Cd24 | Two up of three CRE | One up and down of three CRE | 0 |
| Fra2(Fosl2) | None | Two down and over six CRE | 2 |
| Mt1a | One up and down of five CRE | Two up of one CRE | 0 |
| Opn1sw | None | One upstream of three CRE | 0 |
| Validated FRA-2 nonregulated genes | |||
| Aanat | One up and down of five CRE | None | 0 |
| Dbp | None | One down of two CRE | 0 |
| E4bp4 (Nfil3) | One up and down of two CRE | None | 0 |
| Crem (ICER) | Two up and down of four CRE | One down of four CRE | 0 |
| Id1 | Three up and down of six CRE | Four up and down of six CRE | 0 |
| Per2 | Three up & down of 5 CRE | One up & down of 5 CRE | 0 |
| Syt4 | None | Three up and down of eight CRE | 0 |
Rgs4 as a Model FRA-2-regulated Gene
The Rgs4 AP-1R site, which starts at position −155 (relative to TSS in rat, Fig. 1D), is 100% conserved across rat, mouse, and human genomic sequences and is generally highly conserved in mammalian species and many higher vertebrate species (supplemental Fig. S2). Furthermore, this is the only AP-1R element in the rat Rgs4 genomic locus between −5 kb of the 5′-flanking sequence to +5kb of the 3′-flanking sequence (chromosome 13, 85,533,882–85,540,173; Ensembl rat genome build Feb. 2006). No other AP-1 or CRE elements were found to be positionally conserved (in rat, mouse, and human) across this region. A consensus AP-1F (tgactca) sequence in the rat 5′-flanking sequence at position −415 is conserved neither in mouse, human (no AP-1F elements in −1500 to +100 region, MatInspector), nor other mammals. The −155AP-1R site of the rat Rgs4 promoter is therefore a promising candidate sequence for mediating the regulatory action of FRA-2.
Ex Vivo ChIP Analysis of Rgs4 Promoter Occupancy
To confirm that FRA-2 regulation of Rgs4 is associated with a direct interaction of FRA-2 with the rat Rgs4 genomic locus, we next conducted ChIP analysis on rat pineal gland chromatin, targeting a region of the Rgs4 promoter that contains the AP-1R element discussed above. Using this approach, we found a specific enrichment of proximal Rgs4 promoter sequence following chromatin immunoprecipitation with a FRA-2 antiserum as compared with a control (FRA-2) preimmune serum (Fig. 2). Control ChIP assays also showed relative enrichment of both this Rgs4 sequence and a β-actin sequence when using a RNA polymerase II antiserum (Fig. 2).
FIGURE 2.
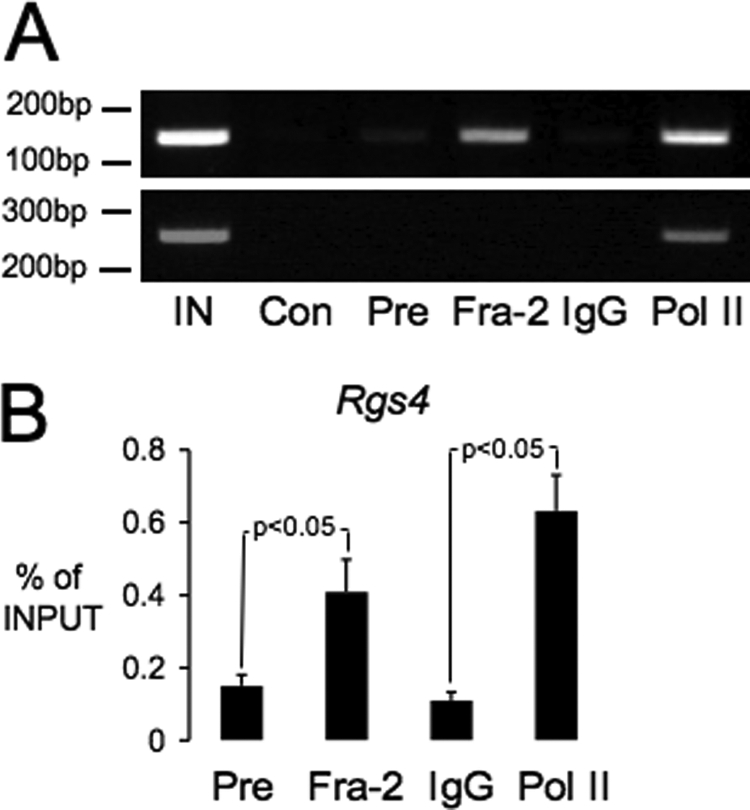
Association of FRA-2 with Rgs4 promoter sequence in vivo. ChIP assays were conducted using chromatin extracted from rat pineal glands; gene promoter sequences were amplified by PCR and visualized by ethidium bromide staining of agarose gels. A, ChIP analysis reveals enrichment of pineal chromatin when precipitated with FRA-2 and RNA polymerase II (Pol II) antisera compared with a FRA-2 preimmune serum and IgG. IN, input chromatin; Con, water PCR control. Note that input chromatin is diluted relative to ChIP chromatin. A parallel assay conducted with primers specific for the control β-actin gene revealed enrichment only with polymerase II antisera. B, summated data from multiple Rgs4 ChIP assay. Values are expressed as % of input DNA (mean ± S.E. n = 3 assays from three individual groups of rats). Group means that are significantly different (p < 0.05) are indicated (paired Student's t test).
Functional Analysis of the Rgs4 Promoter
Following our demonstration that the Rgs4 promoter is directly associated with FRA-2, we next conducted a functional investigation of Rgs4 promoter sequence via transfection assays in cultured cells. These experiments were designed to independently test the function of both the −155 AP-1R and the −415 AP-1F sequences. PC12 cells were selected for this analysis because it is a rat cell line that expresses Rgs4 (30), FRA-2 (35), and p-CREB (36). The nuclear environment in these cells therefore has characteristics of the nocturnal rat pineal gland in which both FRA-2 and p-CREB activity are strongly induced (14, 37, 38).
Analysis of Rgs4 promoter construct expression in PC12 cells revealed that the Rgs4 sequences generated between 20- and 40-fold higher expression levels than the pGL4.11 plasmid alone (Fig. 3A). Mutation of the −155 tgcgtca sequence significantly enhanced expression compared with the wild type sequence (Fig. 3A; p < 0.05, ANOVA and Duncan's post hoc test; F = 31.294, df = 31). The longer (−426) construct was not associated with an altered expression level compared with the −167 wild-type construct, and mutation of the −415 tgactca sequence did not affect expression (Fig. 3A). Our finding that the AP-1R sequence appeared to mediate transcriptional suppression in the basal state was initially surprising, but Western blot analysis (Fig. 3B) revealed high levels of FRA-2 in the PC12 cell cultures used for transfection studies.
FIGURE 3.

Functional analysis of Rgs4 promoter activity in transfected cells. Rgs4 constructs cloned into pGL4.11 were transfected into PC12 cells. Levels of expression were determined by luciferase (Luc) assays and corrected against a co-transfected Renilla luciferase construct. A, expression levels (luciferase activity) of the different constructs relative to empty pGL4.11 (mean ± S.E., n = 8; *, p < 0.05 versus all other groups, ANOVA and post hoc test). The position of the −415 AP-1F, −155 AP-1R, and −42 TATA elements (see text) are indicated by filled symbols and are shown as open symbols where mutated (Rgs4-167m and Rgs4-426m constructs, see text). B, Western blot of whole cell extracts (pineal 24.00 h, 50 μg; PC12 cells, 10 μg) probed sequentially with antisera to FRA-2 and GAPDH. Note the relatively high level of FRA-2 in unstimulated PC12 cells. Arrows indicate two (differentially phosphorylated (14)) FRA-2 bands. Horizontal bars indicate molecular weight markers.
In Vitro Analysis of Rgs4 Promoter Protein Binding
We next examined the composition of protein complexes that bound the Rgs4 promoter. EMSA of interactions between an oligonucleotide probe containing the rat Rgs4 tgcgtca sequence and pineal gland nuclear protein revealed a sequence-specific interaction (Fig. 4). We discovered that two major shifted bands were observed in the presence of pineal protein extracts, and only one of these bands was found with extracts of brain cortex (Fig. 4A). Comparison of multiple pineal extracts obtained at either 12.00 or 24.00 h revealed a 3–4-fold increase in protein binding to the probe at night (Fig. 4B).
FIGURE 4.

Protein composition and specificity of DNA binding activity at the Rgs4 AP-1R sequence in vitro. Representative chemiluminescent images of EMSAs showing bands of biotin-labeled oligonucleotide probe shifted in the presence of nuclear protein extracts (1.2 μg) from either pineal gland (P) or brain cortex (Cx). A, note that two shifted bands (upper and lower arrows) are observed in the pineal gland, whereas only the equivalent upper band is observed in the cortex together with two additional slower migrating bands (arrowheads). Note that both EMSA bands are more abundant in pineal samples extracted at 12.00 versus 24.00 h. Unbound (free) probe is indicated at the gel base. B, summated results of multiple EMSAs comparing the abundance of the upper and lower shifted bands in pineal glands sampled at either 12.00 and 24.00 h. Values are fold-difference compared with the level at 12.00 h (mean ± S.E., n = 6, *, p < 0.05 versus equivalent 12.00-h group, paired Student's t test). C, expanded image of shifted bands (only) showing the effect of different antisera on the abundance of pineal EMSA bands. Two different FRA-2 and two different CREB-1 antisera were compared, and a rabbit IgG was used in the Control lane. Note that the lower EMSA band is completely abrogated by both FRA-2 antibodies, whereas both EMSA bands are diminished by the two CREB-1 antibodies; the CREB-9197 (9197, Cell Signaling Technology) antibody is somewhat more effective in abrogating the bands compared with CREB-1–186 (sc186, Santa Cruz Biotechnology Inc.) antibody. Unbound probe is not shown. D and E, comparison of the effects of CREB-1 and phospho-CREB antisera on the abundance of EMSA bands observed in pineal glands sampled at either 12.00 or 24.00 h. Note that 24.00-h protein extracts were diluted 2-fold to equalize the intensity of binding activity relative to the 12.00-h samples. Expanded images of shifted bands (only) are shown in D, and summated results of multiple EMSAs are shown in E. Note that the CREB-1 antibody (sc186) diminishes the pineal EMSA bands to a similar extent in both groups, whereas the p-CREB antibody (9197, Cell Signaling Technology) significantly diminishes only the 24.00-h group. Histogram values are fold-difference compared with the control (IgG) level (mean ± S.E., n = 4; *, p < 0.05 versus equivalent control group (IgG), paired Student's t test). F and G, comparison of the effects of either unlabeled wild-type Rgs4 AP-1R probe (Rgs4; 4-, 8-, or 16-fold molar excess) with unlabeled mutant Rgs4 AP-1R probe (mutRgs4, similar molar excess) on the abundance of pineal EMSA bands. Expanded images of shifted bands (only) are shown in F, and summated results of multiple EMSAs are shown in G. Note that the unlabeled wild-type probe significantly competes with the labeled probe at all molar concentrations whereas the mutant probe competes significantly only at 16-fold molar excess. Histogram values are fold-difference compared with the control (No competitor, 1st bar in each group; mean ± S.E., n = 3; *, p < 0.05 versus no competitor, ANOVA, and post hoc test.
Pineal proteins interacting with the EMSA probe were shown to include both FRA-2 and CREB based on abrogation in the presence of antisera (Fig. 4C). Complete abrogation of the lower band with FRA-2 antibodies indicated that this band was wholly composed of FRA-2-containing complexes. In contrast, the diminution of both bands in the presence of CREB antisera indicated that CREB was a constituent of both shifted bands. Because FRA-2 is apparently restricted to the lower band, this indicates that there is a nocturnal increase in the binding of another protein(s) to the upper band. Comparison of the effects of the CREB antisera on day and night protein extracts (Fig. 4, D and E) indicated that CREB binding was similar at these two times. This is consistent with Western blot data (see below) and indicates constitutive CREB binding to this sequence. In contrast, whereas a p-CREB antibody did not significantly affect day binding activity, there was a significant effect on night binding activity (Fig. 4E). Hence, CREB protein binding to the Rgs4 promoter appears to be phosphorylated at night, which is entirely consistent with phosphorylation of CREB being a common (noradrenergic) response across pinealocytes (39).
Sequence specificity of the pineal DNA binding activity was investigated by comparing the effects of competition with unlabeled wild-type Rgs4 or mutant Rgs4 oligonucleotide probes (Fig. 4, F and G). Binding to the Rgs4 sequence appeared to be of relatively low affinity because it was displaced with only a 4-fold molar excess of unlabeled probe (Fig. 4F, p < 0.05 at 4.8- and 16-fold molar excess, ANOVA and Dunnett's test; F = 234.1, df = 11). However, much higher concentrations of unlabeled mutant probes were required to displace binding (Fig. 4F, p < 0.05, at only 16-fold molar excess, ANOVA and Dunnett's test; F = 68.92, df = 11). indicating that the binding was sequence-specific, and involved the tgcgtca sequence.
The EMSA analysis is consistent with and supported by Western blot analysis of protein content (Fig. 5). In these blots, which are representative of multiple similar blots, we show that the brain cortex contains minimal amounts of FRA-2 and p-CREB protein compared with the pineal gland, and also that there are minor differences in CREB-1 protein abundance between day and night pineal gland samples.
FIGURE 5.

Western blot analysis of pineal gland and brain cortex protein content showing nocturnal up-regulation of FRA-2 and p-CREB in pineal glands. Representative chemiluminescent images of Fra-2, CREB-1, p-CREB, and p-ATF-1 expression in pineal glands sampled at either 12.00 or 24.00 h (P12/P24) or brain cortex (Cx). The image labeled Fra-2++ is an extended film exposure of the same blot in the upper FRA-2 image, and the CREB-1 image is a re-probe of the same Western blot indicating relatively invariant CREB-1 expression (within the two sample groups) and consequently equal gel loading/transfer. A similar CREB-1 re-probe was obtained for the lower p-CREB/p-ATF-1 blot (not shown). Note that FRA-2 expression is increased at 24.00 versus 12.00 h and that FRA-2 expression in cortex is minimal. The amounts of protein extract loaded in each lane were as follows: 6 μg, pineal nuclear; 15 μg, cortex nuclear; 20 μg, pineal whole cell; 35 μg, cortex whole cell). Horizontal bars represent positions of molecular weight markers.
Mechanistic Basis of FRA-2 Down-regulation of Nocturnal Rgs4 Expression
Following our demonstration that transcriptional down-regulation of Rgs4 by FRA-2 involved a direct action at the proximal AP-1R element in the Rgs4 promoter, we next addressed the mechanistic basis of this down-regulation (Fig. 6). A possible mechanism is interference with co-activator function and consequent transcriptional down-regulation. A likely co-activator in this locus is CBP/p300 (40), and by using EMSA, we demonstrated CBP association with the Rgs4 promoter sequence (Fig. 6A). To address a CBP-linked mechanism of nocturnal Rgs4 down-regulation, we then went back to our in vivo transgenic model and measured nocturnal CBP occupancy at the Rgs4 proximal promoter using quantitative ChIP assays (Fig. 6B). We observed that relative CBP occupancy of Rgs4 at a 5′-proximal region was significantly higher in DN-FRA-2 transgenic compared with wild-type pineal glands (Fig. 6B), whereas total cellular CBP levels as determined by Western blot were not different between the two (Fig. 6, C and D). These results indicate that nocturnal down-regulation of Rgs4 transcription involves (at least partially) a FRA-2-mediated dismissal of CBP from the Rgs4 promoter.
FIGURE 6.

Mechanistic basis for nocturnal Rgs4 down-regulation by FRA-2. A, CBP is associated with the Rgs4 promoter. Chemiluminescent image of a representative EMSAs showing bands of biotin-labeled oligonucleotide probe shifted in the presence of nuclear protein extracts (1.2 μg) from pineal gland (P). The shifted bands are partially abrogated in the presence of a CREB-1 antisera (sc-186) and completely abrogated in the presence of CBP antisera (A22). A rabbit IgG was used in the Control lane. B, ChIP analysis revealed increased nocturnal occupancy of CBP at the Rgs4 promoter in DN-FRA-2 transgenic (TG) rat pineal glands sampled at night (24.00 h) compared with wild-type (WT) controls. Data were quantified by QPCR, expressed as % of input DNA (mean ± S.E.) and summated in histograms (n = 4 assays from four individual groups of rats, p < 0.05, paired Student's t test). C, CBP protein levels are invariant between TG and WT rats. Representative chemiluminescent image of Western blot analysis of CBP protein levels in rat pineal glands sampled at night (24.00 h). Nuclear protein extracts (5 μg) were blotted, and CBP was detected using ECL. D, CBP protein levels were quantified by densitometry in multiple biological replicates and found to be similar in the two sample groups (n = 3 blots from three individual groups of rats). E, simplified summary of day-night changes in pineal gland noradrenergic (NE) activation, signaling molecule levels, and Rgs4 transcript in the rat pineal gland. F and G, schematic representations of possible transcription factor complex formations at the Rgs4 promoter. FRA-2 is associated with the Rgs4 promoter region, and binding activity increases at night. CREB binding activity does not change, but levels of p-CREB increase at night. An additional Rgs4 protein binding activity(s) also increases at night. These changes are associated with reduced CBP association predicting a decrease in transcriptional initiation associated with loss of RNA polymerase II (Pol II).
DISCUSSION
The genome of hormone-producing cells in the pineal gland, as in all rhythmically active systems, is confronted with daily fluxes in TF activity. We have recently catalogued the extent of these rhythmic changes, revealing a remarkable number and diversity of rhythmic factors (41). How melatonin-synthesizing pinealocytes respond to and coordinate this input provide the physiological context of this study. Our particular interest is in transcription factors linked to the key nocturnal regulator of pineal gene expression, namely cAMP (41). Although many pineal genes are up-regulated by cAMP, ∼30% are down-regulated (41), indicating a significant presence of transcriptional down-regulation that would be mediated (at least partially) by “negatively” acting TFs. Our work addresses the selective actions of the bZIP factor FRA-2 (14, 37) and investigates how the genome responds to changing levels of this factor with a selectively modified transcriptional output. We have also addressed how the actions of FRA-2 are integrated with the canonical regulator of cAMP-induced transcription, namely p-CREB, that is also nocturnally induced in the pineal gland (38).
In this study, we have defined a complement of the pineal transcriptome that is regulated by FRA-2. The FRA-2 regulon composes both positively and negatively regulated genes; our finding (supplemental Table S2) that 4-fold more genes are negatively regulated suggests that this mode of action is more common for FRA-2. We have also have gone on to show that for one transcript, Rgs4, FRA-2 regulation is direct, being mediated at a specific Rgs4 promoter site. The demonstration of a direct action is important because, together with our previous work (a direct action on Nr4a1 (18)), this argues that selective actions of Fra-2 are mediated at the level of cognate cis-acting sequence within our defined gene set. Genome-wide studies have shown that the number of loci exhibiting consensus AP-1 sequences appears to massively outweigh the number of AP-1-regulated genes (11), and our results would concur with this view because, although extensive, the FRA-2 regulon is relatively limited. The discordance between potential and actual gene targets is well illustrated by our findings that whereas one bZIP TF gene, Atf4, is regulated by FRA-2 (Table 1), many other bZIP genes that are expressed in the pineal gland, including Atf2, Fra-1, c-Fos, Jdp1, Jdp2, c-Jun, JunB, C/EBPγ, C/EBPβ, C/EBPδ, Dbp, and Maf1 (41), are independent of FRA-2 (this study). This is remarkable because with the single exception of c-Jun, all of these genes exhibit between 1 and 6 consensus AP-1F sites in their proximal (−2000 to +100) flanking sequence. These findings underline the importance of our functional approach to the understanding of rules that govern TF targeting of the genome and the value of the unique transgenic model used in this study.
In rat, we have now validated FRA-2 regulation for nine genes: Fra-2 itself, Cd24, DII (14), Nr4a1 (NGFI-B (18), ATF-4, Cox6a2, Mt1, Opn1sw, and Rgs4 (present study). One of these targets (DII) has also been validated with a Fra-2 RNAi strategy (42). In addition, we have also validated genes that are independent of FRA-2: Aanat (14) ICER, Id-1, Dbp, E4bp4, and Per2 (present study). This study has therefore provided a data set of differentially regulated genes that can be used to question the mechanisms of selective gene targeting by FRA-2.
cis-Sequence Logic of FRA-2 Gene Regulation
Using our sets of validated FRA-2-regulated and nonregulated genes, we employed a number of different bioinformatic analyses to search for common regulatory sequences. Previous studies have shown that FRA-2 may interact with a variety of different AP-1/CRE sequences (10, 43–45), and therefore we adopted largely unbiased approaches to the identification of common sequences. Although one algorithm (ModuleMiner (27)) showed that AP-1 sequences are enriched in the extended 5′-flanking sequences of the Fra-2-repressed subset, analysis of more proximal sequence in both sets of validated genes using a different bioinformatic approach failed to identify a common AP-1/CRE-based sequence or module organization that could underlie the differential regulation. This outcome may be interpreted to indicate that, in general, AP-1 sites distal to the TSS have a greater regulatory significance compared with more proximal sites; this interpretation would accord, for example, with recent studies of estrogen receptor genomic targeting (46). However, caution must be exercised in making this interpretation. First, the ModuleMiner algorithm is restricted to extended 5′-flanking sequence and consequently cannot be used to address proximal sequence in a parallel and independent manner. Second, larger sets of validated genes may be required to provide a clear outcome. Third, inherent limitations in the in silico identification of functional AP-1 sites may exclude a valid outcome. This point is illustrated in this study by our finding that the (core) consensus AP-1 sequence at −415 of rat Rgs4 (AP-1F, tgactca) was not functional, possibly due to sequence divergence in non-core sequence (MatInspector analysis (47)). Finally, the absence of a clear outcome may simply reflect a diversity of cis-acting mechanisms that are used to effect FRA-2-regulated transcription of these genes. In respect of this, our bioinformatic analysis did highlight a regulatory sequence motif that, although not entirely common, was found in the proximal sequence (−2000 to +100) of 8 or nine FRA-2 regulated genes. This motif is a member of the AP-1R position weight matrix (PWM) group and included, in some genes, the core sequence tgcgtca, which we have previously identified as a FRA-2 target sequence (18). Consequently, AP-1R sequences may form one group of cis-acting elements that mediate the actions of FRA-2.
cis-Regulatory Role of AP-1R Sequences
The AP-1R regulatory sequence (tgcgtca) identified here in Rgs4 and other FRA-2-regulated genes is classified in the MatInspector matrix library within the subfamily of AP-1R PWMs termed BACH2.01 (BACH2 bound TRE), i.e. a 12-O-tetradecanoylphorbol-13-acetate-response element that binds the bZIP factor BACH2 (48). However, in numerous genomic contexts this tgcgtca core sequence has been shown to interact with other factors including JUND (18) and CREB (49, 50).
These sites can therefore integrate the activity of AP-1, CREB and possibly other factors; this can involve replacement of CREB (at the CRE half-site) by FRA-2 when these proteins become differentially expressed in the ovary, for example (50). However, the situation is different in the context of the pineal gland because both factors (p-CREB and FRA-2) are increased at night in this gland. Therefore, the pineal gland may represent a distinct physiological example where FRA-2 appears to override CREB function in the context of the Rgs4 locus. We addressed this hypothesis in detailed studies of the Rgs4 AP-1R element.
Role of the Proximal AP-1R Sequence in Rgs4 Regulation
The proximal Rgs4 tgcgtca element is highly conserved across mammalian and higher vertebrate species, which may argue for functional selection (supplemental Fig. S2). Recent ChIP-Seq analysis of five vertebrate genomes (51) has shown, at least for two transcription factors (CEBPA and HNF4A), that “aligned binding events” are rare. The possibility that a positionally conserved functional Rgs4 tgcgtca element may represent an “ultrashared binding event” as defined previously (51) must await further functional analysis of this element in other species.
Our current studies using ChIP, transfection, and EMSA assays have provided clear evidence of an action of FRA-2 at the −155 AP-1R element in rat Rgs4. Gene sequence-specific FRA-2 binding activity was found to increase at night in a quantitatively similar manner to that observed previously for consensus AP-1 probes (52). However, FRA-2 binding to the Rgs4 AP-1R element was clearly retained during the day, which accords with maintenance of FRA-2 protein in the daytime pineal gland (this study and Refs. 37, 52). Nevertheless, this finding is consistent with the hypothesis that an increase in FRA-2 loading at the Rgs4 AP-1R site at night could displace other factors leading to transcriptional down-regulation. Surprisingly, this hypothesized displacement does not include CREB because we found that CREB binding to the AP-1R element was fully maintained at night, and additionally, Rgs4-bound CREB was nocturnally phosphorylated. Our findings are therefore consistent with a model (Fig. 5) in which FRA-2 loading of the Rgs4 AP-1R element is increased at night while CREB binding is maintained.
The results of our EMSAs also indicated the recruitment of additional protein(s) at the Rgs4 AP-1R element at night (see Fig. 5). The identity of these protein(s) is unknown, but these may also contribute to transcriptional down-regulation. A potential role for an additional regulatory factor is indicated by the finding that DN-FRA-2 does not completely alleviate the nocturnal reduction in Rgs4 expression despite fully competing FRA-2 DNA binding activity (Fig. 1) (14). It is unlikely that the protein(s) include the other major FOS/JUN binding activity (JUND) because this is a large and relatively invariant component of pineal AP-1 binding activity (14, 52). An AP-1R-associated ETSF site (see Fig. 1) is conserved across mouse and human promoters and could provide for an interaction with ETS-domain proteins (53). Other factors that potentially bind the EMSA probe sequence are a winged helix TF (− strand, ctgACGCatgg) and Glial cells missing homolog 1/chorion-specific transcription factor GCMa (+ strand, atCCCCcatgc). However, our microarray analysis (41) did not reveal evidence of these factors in the rat pineal gland. Clearly, additional screens are required to identify the unknown AP-1R element-binding proteins.
Our studies have provided the most extensive analysis of AP-1-Rgs4 interactions to date but must be considered in the context of previous studies of Rgs4 gene regulation. A potential AP-1 binding sequence has previously been recognized in the mouse Rgs4 gene and suggested (based on sequence data alone) to mediate cocaine-induced changes in Rgs4 expression (54). However, this tgactca sequence at −7600 in the mouse 5′-flanking region is not conserved in either the rat or human gene sequences. Other factors known to regulate Rgs4 expression include Phox2b (55) and the NF-κB subunit p65 (56). Recently, a study of the human Rgs4 promoter (57) has provided evidence of a repressive activity at a Bcl6 site at −253 of hRgs4. However, one-half of this site is not conserved in rat (hRgs4 Bcl6, ccttttctagaa; rRgs4 Bcl6, ccttttctgttg) and consequently is not detected in a MatInspector binding site (Genomatix) search. In the study of Yang et al. (57) the authors consider the (hRgs4) tgcgtca sequence as a CRE site and show that removal of the “CRE”-containing region (from −195 to −83 relative to TSS) increases human promoter activity. This is in agreement with our mutation analysis in the rat. However, mutation of the CRE site in the context of a −435-bp human promoter construct resulted in a relative decrease in promoter activity (57). This apparent discrepancy with our finding is probably explained by relative differences in the role of the human CRE site in the different sequence, and cellular (confluent PC6 cells) contexts where a repressive action of Bcl6 appears to dominate.
Inverse Regulation of Rgs4 and Rgs2
Additional insight into the selective genomic targeting of Rgs4 can be gained from comparison with the related gene, Rgs2, which is independent of FRA-2 (present study) and is, in fact, up-regulated, nocturnally in the pineal (41), and by cAMP in PC12 cells (30). Our bioinformatic analysis does not indicate clearly why Rgs2 is not a FRA-2 target; there are 9 AP-1F sequences and 9 AP-1R sequences including one BACH2.01 element proximal to Rgs2 (−2000 to +100; MatInspector analysis). However, the single Rgs2 BACH2.01 element has the core sequence tgagtca rather than tgcgtca and therefore may not form a target for FRA-2 on this basis. Rather, CRE sites proximal to Rgs2 (8 CREB sites within −2000 to +100) may act to directly up-regulate Rgs2 via a cAMP-CREB pathway (34). The differential regulation of Rgs4 and Rgs2 observed in this pineal is interesting because these genes are also differentially regulated in the striatum by dopamine receptor activation (58). Taken together, these findings indicate a divergence of cis-regulatory control between these two genes that may underlie a functional requirement for reciprocal expression of these factors.
Mechanism of Nocturnal Down-regulation of Rgs4 Transcription
Our demonstration of an inverse relationship between FRA-2 loading and CBP association with the Rgs4 promoter has provided a mechanistic basis for the transcriptional down-regulation of Rgs4 in the nocturnal pineal gland (Fig. 6, F and G). CBP is a ubiquitous, but limiting, transcriptional co-activator and signaling integrator that is required by many nuclear trans-acting complexes, including AP-1, and acts to facilitate RNA polymerase II function (40). Consequently, a FRA-2-associated loss of CBP recruitment (44) would be anticipated to result in nocturnal Rgs4 down-regulation.
Alternative Mechanisms of FRA-2 Regulation at Other Genomic Loci
Our analysis of the Rgs4 promoter has been important in detailing the mechanism of one direct action of FRA-2 at a target gene promoter. However, as discussed above, our bioinformatic analysis of FRA-2 target genes is consistent with the presence of multiple alternative mechanisms. These alternatives may involve either distinct cis-based mechanisms, for example involving remote AP-1-CRE interactions (59), or possibly higher order mechanisms involving changes in chromatin organization (60, 61).
Regulation of Rgs4 in the Context of Physiological and Pathological Roles
RGS4 is one member of an extensive family of related proteins that regulate the duration of G-protein-coupled receptor-linked signaling through enhancement of Gα GTPase activity (62). Rgs4 is highly expressed in the brain neocortex (63) and has been implicated in many neuro-diseases, including schizophrenia (64). Although the status of Rgs4 as a candidate schizophrenia vulnerability gene is now questioned (65), there is now a body of evidence showing that Rgs4 expression is reduced in the brains of schizophrenic patients (66). Our studies have now positioned FRA-2 as a candidate regulator of brain Rgs4 expression in disease-related states. Other work in our laboratory has also provided evidence for a potential functional association between FRA-2 and Rgs4 in a covert pathology associated with brain lesions (67). The possibility that disease-related SNPs in the hRgs4 promoter (68, 69) may involve the APIR sequence is not currently supported (UCSC genome browser analysis). One SNP (dbSNP id. Rs12402634) is close, at position −365 of TSS, but does not appear to be functional (69).
The role of RGS4 in the pineal gland is currently unknown and is not addressed in this study. The relatively high level (∼38% of frontal cortex levels (70)) and rhythmicity of expression suggests a role within the circadian signaling pathways, possibly gating melatonin production through enhanced adrenergic function at night (71–73). However, our pineal transcriptome analysis (41) has revealed that other Rgs transcripts exhibit either minor or no rhythm in expression (Rgs3, -5, -8, -9, -12, and -14) or are up-regulated nocturnally (Rgs2, Rgs17 (predicted)). Therefore, the apparently unique nocturnal down-regulation of Rgs4 (versus other Rgs genes) is inconsistent with a general enhancement of G-protein activity; this implies a specific biochemical role for pineal RGS4.
Implications for Future Studies
We have identified a novel DNA element in the Rgs4 gene that binds FRA-2 and mediates a repressive action on Rgs4 transcription as identified in our transgenic rat model. Further analysis of this interaction requires in vivo mutation of the Rgs4 AP-1R sequence. The functional regulatory element identified here would not and has not been identified in conventional promoter inspections (26). Such discoveries are consistent with the emerging view of complexity and diversity within protein-DNA interactions (74, 75). The latter study has confirmed the widespread existence of alternative or “secondary” DNA-binding motifs among transcription factors; potentially providing a hierarchy of sites with different affinities for TFs (76). In vivo functional screens for TF targets as demonstrated here in our transgenic model have important implications for the identification and analysis of regulatory single nucleotide polymorphisms that may be associated with human disease (77).
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Intramural Research Program (to D. C. K.). This work was also supported by the Wellcome Trust (to J. D. and D. A. C.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2 and Tables S1 and S2.
- bZIP
- basic region-leucine zipper
- AP-1R
- AP-1-related sequence
- AP-1F
- AP-1 family sequence
- CBP
- cAMP-response element-binding protein-binding protein
- CRE
- cAMP response element
- DN-FRA-2
- dominant negative FRA-2
- TF
- transcription factor
- ANOVA
- analysis of variance
- PWM
- position weight matrix
- TG
- transgenic
- QPCR
- quantitative PCR
- TSS
- transcriptional start site
- df
- degrees of freedom.
REFERENCES
- 1. Vinson C., Myakishev M., Acharya A., Mir A. A., Moll J. R., Bonovich M. (2002) Mol. Cell. Biol. 22, 6321–6335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jochum W., Passegué E., Wagner E. F. (2001) Oncogene 20, 2401–2412 [DOI] [PubMed] [Google Scholar]
- 3. Wagner E. F. (2010) Ann. Rheum. Dis. 69, Suppl. 1, i86–i88 [DOI] [PubMed] [Google Scholar]
- 4. Bozec A., Bakiri L., Hoebertz A., Eferl R., Schilling A. F., Komnenovic V., Scheuch H., Priemel M., Stewart C. L., Amling M., Wagner E. F. (2008) Nature 454, 221–225 [DOI] [PubMed] [Google Scholar]
- 5. Engel L., Gupta B. B., Lorenzkowski V., Heinrich B., Schwerdtle I., Gerhold S., Holthues H., Vollrath L., Spessert R. (2005) Neuroscience 132, 511–518 [DOI] [PubMed] [Google Scholar]
- 6. Carro M. S., Lim W. K., Alvarez M. J., Bollo R. J., Zhao X., Snyder E. Y., Sulman E. P., Anne S. L., Doetsch F., Colman H., Lasorella A., Aldape K., Califano A., Iavarone A. (2010) Nature 463, 318–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eferl R., Hasselblatt P., Rath M., Popper H., Zenz R., Komnenovic V., Idarraga M. H., Kenner L., Wagner E. F. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 10525–10530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hai T., Curran T. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 3720–3724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ryseck R. P., Bravo R. (1991) Oncogene 6, 533–542 [PubMed] [Google Scholar]
- 10. Bakiri L., Matsuo K., Wisniewska M., Wagner E. F., Yaniv M. (2002) Mol. Cell. Biol. 22, 4952–4964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhou H., Zarubin T., Ji Z., Min Z., Zhu W., Downey J. S., Lin S., Han J. (2005) DNA Res. 12, 139–150 [DOI] [PubMed] [Google Scholar]
- 12. Suzuki T., Okuno H., Yoshida T., Endo T., Nishina H., Iba H. (1991) Nucleic Acids Res. 19, 5537–5542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kerppola T. K., Curran T. (1993) Mol. Cell. Biol. 13, 5479–5489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Smith M., Burke Z., Humphries A., Wells T., Klein D., Carter D., Baler R. (2001) Mol. Cell. Biol. 21, 3704–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baler R., Covington S., Klein D. C. (1999) Biol. Cell 91, 699–705 [PubMed] [Google Scholar]
- 16. Burke Z., Wells T., Carter D., Klein D., Baler R. (1999) J. Neurochem. 73, 1343–1349 [DOI] [PubMed] [Google Scholar]
- 17. Zenz R., Wagner E. F. (2006) Int. J. Biochem. Cell Biol. 38, 1043–1049 [DOI] [PubMed] [Google Scholar]
- 18. Humphries A., Weller J., Klein D., Baler R., Carter D. A. (2004) J. Neurochem. 91, 946–955 [DOI] [PubMed] [Google Scholar]
- 19. Carter D. A. (1992) Brain Res. Mol. Brain Res. 16, 111–118 [DOI] [PubMed] [Google Scholar]
- 20. Gaildrat P., Møller M., Mukda S., Humphries A., Carter D. A., Ganapathy V., Klein D. C. (2005) J. Biol. Chem. 280, 16851–16860 [DOI] [PubMed] [Google Scholar]
- 21. Cartharius K., Frech K., Grote K., Klocke B., Haltmeier M., Klingenhoff A., Frisch M., Bayerlein M., Werner T. (2005) Bioinformatics 21, 2933–2942 [DOI] [PubMed] [Google Scholar]
- 22. Blanchette M., Kent W. J., Riemer C., Elnitski L., Smit A. F., Roskin K. M., Baertsch R., Rosenbloom K., Clawson H., Green E. D., Haussler D., Miller W. (2004) Genome Res. 14, 708–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Siepel A., Bejerano G., Pedersen J. S., Hinrichs A. S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L. W., Richards S., Weinstock G. M., Wilson R. K., Gibbs R. A., Kent W. J., Miller W., Haussler D. (2005) Genome Res. 15, 1034–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suzuki Y., Yamashita R., Nakai K., Sugano S. (2002) Nucleic Acids Res. 30, 328–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun H., Palaniswamy S. K., Pohar T. T., Jin V. X., Huang T. H., Davuluri R. V. (2006) Nucleic Acids Res. 34, D98–D103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ding L., Mychaleckyj J. C., Hegde A. N. (2007) Gene 401, 46–60 [DOI] [PubMed] [Google Scholar]
- 27. Van Loo P., Aerts S., Thienpont B., De Moor B., Moreau Y., Marynen P. (2008) Genome Biol. 9, R66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsankova N. M., Kumar A., Nestler E. J. (2004) J. Neurosci. 24, 5603–5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Krumins A. M., Barker S. A., Huang C., Sunahara R. K., Yu K., Wilkie T. M., Gold S. J., Mumby S. M. (2004) J. Biol. Chem. 279, 2593–2599 [DOI] [PubMed] [Google Scholar]
- 30. Pepperl D. J., Shah-Basu S., VanLeeuwen D., Granneman J. G., MacKenzie R. G. (1998) Biochem. Biophys. Res. Commun. 243, 52–55 [DOI] [PubMed] [Google Scholar]
- 31. Stehle J. H., Foulkes N. S., Molina C. A., Simonneaux V., Pévet P., Sassone-Corsi P. (1993) Nature 365, 314–320 [DOI] [PubMed] [Google Scholar]
- 32. Flammer J. R., Popova K. N., Pflum M. K. (2006) Biochemistry 45, 9615–9623 [DOI] [PubMed] [Google Scholar]
- 33. Conkright M. D., Guzmán E., Flechner L., Su A. I., Hogenesch J. B., Montminy M. (2003) Mol. Cell 11, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 34. Zhang X., Odom D. T., Koo S. H., Conkright M. D., Canettieri G., Best J., Chen H., Jenner R., Herbolsheimer E., Jacobsen E., Kadam S., Ecker J. R., Emerson B., Hogenesch J. B., Unterman T., Young R. A., Montminy M. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 4459–4464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eriksson M., Taskinen M., Leppä S. (2007) J. Cell. Physiol. 210, 538–548 [DOI] [PubMed] [Google Scholar]
- 36. Iwamoto T., Mamiya N., Masushige S., Kida S. (2005) Cytotechnology 47, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baler R., Klein D. C. (1995) J. Biol. Chem. 270, 27319–27325 [DOI] [PubMed] [Google Scholar]
- 38. Maronde E., Pfeffer M., Olcese J., Molina C. A., Schlotter F., Dehghani F., Korf H. W., Stehle J. H. (1999) J. Neurosci. 19, 3326–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tamotsu S., Schomerus C., Stehle J. H., Roseboom P. H., Korf H. W. (1995) Cell Tissue Res. 282, 219–226 [DOI] [PubMed] [Google Scholar]
- 40. Kamei Y., Xu L., Heinzel T., Torchia J., Kurokawa R., Gloss B., Lin S. C., Heyman R. A., Rose D. W., Glass C. K., Rosenfeld M. G. (1996) Cell 85, 403–414 [DOI] [PubMed] [Google Scholar]
- 41. Bailey M. J., Coon S. L., Carter D. A., Humphries A., Kim J. S., Shi Q., Gaildrat P., Morin F., Ganguly S., Hogenesch J. B., Weller J. L., Rath M. F., Møller M., Baler R., Sugden D., Rangel Z. G., Munson P. J., Klein D. C. (2009) J. Biol. Chem. 284, 7606–7622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chik C. L., Wloka M. T., Price D. M., Ho A. K. (2007) Endocrinology 148, 3523–3531 [DOI] [PubMed] [Google Scholar]
- 43. Benkoussa M., Brand C., Delmotte M. H., Formstecher P., Lefebvre P. (2002) Mol. Cell. Biol. 22, 4522–4534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Boss V., Roback J. D., Young A. N., Roback L. J., Weisenhorn D. M., Medina-Flores R., Wainer B. H. (2001) J. Neurosci. 21, 18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Seldeen K. L., McDonald C. B., Deegan B. J., Farooq A. (2009) Biochemistry 48, 1975–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carroll J. S., Liu X. S., Brodsky A. S., Li W., Meyer C. A., Szary A. J., Eeckhoute J., Shao W., Hestermann E. V., Geistlinger T. R., Fox E. A., Silver P. A., Brown M. (2005) Cell 122, 33–43 [DOI] [PubMed] [Google Scholar]
- 47. Siddharthan R. (2010) PLoS One 5, e9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oyake T., Itoh K., Motohashi H., Hayashi N., Hoshino H., Nishizawa M., Yamamoto M., Igarashi K. (1996) Mol. Cell. Biol. 16, 6083–6095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Konradi C., Kobierski L. A., Nguyen T. V., Heckers S., Hyman S. E. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 7005–7009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yivgi-Ohana N., Sher N., Melamed-Book N., Eimerl S., Koler M., Manna P. R., Stocco D. M., Orly J. (2009) Endocrinology 150, 977–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schmidt D., Wilson M. D., Ballester B., Schwalie P. C., Brown G. D., Marshall A., Kutter C., Watt S., Martinez-Jimenez C. P., Mackay S., Talianidis I., Flicek P., Odom D. T. (2010) Science 328, 1036–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guillaumond F., Sage D., Deprez P., Bosler O., Becquet D., François-Bellan A. M. (2000) J. Neurochem. 75, 1398–1407 [DOI] [PubMed] [Google Scholar]
- 53. Wasylyk B., Wasylyk C., Flores P., Begue A., Leprince D., Stehelin D. (1990) Nature 346, 191–193 [DOI] [PubMed] [Google Scholar]
- 54. Zhang D., Zhang L., Tang Y., Zhang Q., Lou D., Sharp F. R., Zhang J., Xu M. (2005) Neuropsychopharmacology 30, 1443–1454 [DOI] [PubMed] [Google Scholar]
- 55. Grillet N., Dubreuil V., Dufour H. D., Brunet J. F. (2003) J. Neurosci. 23, 10613–10621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hu W., Li F., Mahavadi S., Murthy K. S. (2008) Biochem. J. 412, 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang J., Huang J., Chatterjee T. K., Twait E., Fisher R. A. (2010) J. Biol. Chem. 285, 29760–29769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Taymans J. M., Leysen J. E., Langlois X. (2003) J. Neurochem. 84, 1118–1127 [DOI] [PubMed] [Google Scholar]
- 59. Guberman A. S., Scassa M. E., Giono L. E., Varone C. L., Cánepa E. T. (2003) J. Biol. Chem. 278, 2317–2326 [DOI] [PubMed] [Google Scholar]
- 60. Cha-Molstad H., Keller D. M., Yochum G. S., Impey S., Goodman R. H. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 13572–13577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ho A. K., Price D. M., Dukewich W. G., Steinberg N., Arnason T. G., Chik C. L. (2007) Endocrinology 148, 4592–4600 [DOI] [PubMed] [Google Scholar]
- 62. Willars G. B. (2006) Semin. Cell Dev. Biol. 17, 363–376 [DOI] [PubMed] [Google Scholar]
- 63. Ebert P. J., Campbell D. B., Levitt P. (2006) Neuroscience 142, 1145–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Levitt P., Ebert P., Mirnics K., Nimgaonkar V. L., Lewis D. A. (2006) Biol. Psychiatry 60, 534–537 [DOI] [PubMed] [Google Scholar]
- 65. Sanders A. R., Duan J., Levinson D. F., Shi J., He D., Hou C., Burrell G. J., Rice J. P., Nertney D. A., Olincy A., Rozic P., Vinogradov S., Buccola N. G., Mowry B. J., Freedman R., Amin F., Black D. W., Silverman J. M., Byerley W. F., Crowe R. R., Cloninger C. R., Martinez M., Gejman P. V. (2008) Am. J. Psychiatry 165, 497–506 [DOI] [PubMed] [Google Scholar]
- 66. Mirnics K., Middleton F. A., Stanwood G. D., Lewis D. A., Levitt P. (2001) Mol. Psychiatry 6, 293–301 [DOI] [PubMed] [Google Scholar]
- 67. Poirier G. L., Shires K. L., Sugden D., Amin E., Thomas K. L., Carter D. A., Aggleton J. P. (2008) Thalamus Relat. Syst. 4, 59–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chowdari K. V., Mirnics K., Semwal P., Wood J., Lawrence E., Bhatia T., Deshpande S. N., Thelma B. K., Ferrell R. E., Middleton F. A., Devlin B., Levitt P., Lewis D. A., Nimgaonkar V. L. (2002) Hum. Mol. Genet. 11, 1373–1380 [DOI] [PubMed] [Google Scholar]
- 69. Chowdari K. V., Bamne M., Wood J., Talkowski M. E., Mirnics K., Levitt P., Lewis D. A., Nimgaonkar V. L. (2008) Schizophr. Bull. 34, 118–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Su A. I., Wiltshire T., Batalov S., Lapp H., Ching K. A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G., Cooke M. P., Walker J. R., Hogenesch J. B. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 6062–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Klein D. C. (2007) J. Biol. Chem. 282, 4233–4237 [DOI] [PubMed] [Google Scholar]
- 72. Klein D. C., Coon S. L., Roseboom P. H., Weller J. L., Bernard M., Gastel J. A., Zatz M., Iuvone P. M., Rodriguez I. R., Bégay V., Falcón J., Cahill G. M., Cassone V. M., Baler R. (1997) Recent Prog. Horm. Res. 52, 307–358 [PubMed] [Google Scholar]
- 73. Liu W., Yuen E. Y., Allen P. B., Feng J., Greengard P., Yan Z. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 18338–18343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lapidot M., Michal L., Mizrahi-Man O., Pilpel Y. (2008) PLoS Genet. 4, e1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Badis G., Berger M. F., Philippakis A. A., Talukder S., Gehrke A. R., Jaeger S. A., Chan E. T., Metzler G., Vedenko A., Chen X., Kuznetsov H., Wang C. F., Coburn D., Newburger D. E., Morris Q., Hughes T. R., Bulyk M. L. (2009) Science 324, 1720–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bruce A. W., López-Contreras A. J., Flicek P., Down T. A., Dhami P., Dillon S. C., Koch C. M., Langford C. F., Dunham I., Andrews R. M., Vetrie D. (2009) Genome Res. 19, 994–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chorley B. N., Wang X., Campbell M. R., Pittman G. S., Noureddine M. A., Bell D. A. (2008) Mutat. Res. 659, 147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.