

Abstract
Inhibitors of AMPA-type glutamate ion channels are useful as biochemical probes for structure-function studies and as drug candidates for a number of neurological disorders and diseases. Here, we describe the identification of an RNA inhibitor or aptamer by an in vitro evolution approach and a characterization of its mechanism of inhibition on the sites of interaction by equilibrium binding and on the receptor channel opening rate by a laser-pulse photolysis technique. Our results show that the aptamer is a noncompetitive inhibitor that selectively inhibits the GluA2Qflip AMPA receptor subunit without any effect on other AMPA receptor subunits or kainate or NMDA receptors. On the GluA2 subunit, this aptamer preferentially inhibits the flip variant. Furthermore, the aptamer preferentially inhibits the closed-channel state of GluA2Qflip with a KI = 1.5 μm or by ∼15-fold over the open-channel state. The potency and selectivity of this aptamer rival those of small molecule inhibitors. Together, these properties make this aptamer a promising candidate for the development of water-soluble, highly potent, and GluA2 subunit-selective drugs.
Keywords: Glutamate Receptor Ionotropic (AMPA, NMDA); Ion Channels; Neurotransmitter Receptors; Protein-Nucleic Acid Interaction; RNA; RNA Binding Protein
Introduction
Developing inhibitors to selectively target a single subunit among a multisubunit protein or receptor family is a worthy effort for the following reasons. First, the role of the single subunit can be uniquely tested in a complex biological background, such as in vivo, leaving other subunits untouched. Such a test can be carried out at any particular time if the target function changes during development. In this scenario, the function of this subunit can be inhibited in a reversible, graded fashion in that the degree of inhibition of the protein function can be manipulated by the amount and the time of exposure when the inhibitor is applied, and such an inhibition can be reversibly relieved when the inhibitor is removed. Second, if the inhibitor is a drug candidate, selectivity is generally a desired property. A drug with higher selectivity may have a higher therapeutic effect when the excessive activity of a single protein subunit to which the drug molecule binds is linked to the pathogenesis of a disease. Third, development of an inhibitor to differentiate exclusively its binding to and inhibition of one subunit can provide valuable insights into the structural and functional differences of the subunit from all other subunits of the same family. As such, the most effective way to probe the structure of a particular subunit and to regulate the function of that subunit may be found. Guided by these reasons, we set out to find an inhibitor uniquely selective to a single subunit of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)3 receptors.
AMPA receptors are one of the three subtypes of glutamate ion channels that also include kainate and N-methyl-d-aspartate (NMDA) subtypes (1–4). AMPA receptors mediate most of fast synaptic neurotransmission in the mammalian central nervous system, and their function and regulation are critical for synaptic plasticity (4, 5). GluA1–4 (previously known as GluR1–4 or GluRA–D) encode four subunits of mammalian AMPA receptors. The primary molecular architecture of AMPA receptor subunits is most likely similar, given the fact that all subunits have ∼900 amino acids and share 70% homology of the encoding genes, although the genes are alternatively spliced and edited (1–3). AMPA receptor subunits are differentially expressed and developmentally regulated. For instance, in embryonic rat brain, GluA2 mRNA is ubiquitous (6). GluA1–3 are expressed in greater proportion in regions such as hippocampus (7), whereas GluA4 is mainly expressed early during development (8, 9). Although GluA1–4 can form homomeric channels individually (10, 11), each subunit has some distinct functional properties. For examples, in response to the binding of glutamate, each of the GluA2–4 homomeric receptors opens the channel with a kinetic rate constant about severalfold larger than GluA1 does, yet all AMPA receptors close their channels with roughly a similar rate (12, 13). Given these similarities and differences among various AMPA receptor subunits, it would be useful to develop subunit-selective inhibitors of AMPA receptors.
To develop subunit-selective inhibitors, we needed to choose an unconventional methodology. The main, conventional approach to find inhibitors is synthetic chemistry, which produces small molecule inhibitors, the most common type of molecular agents and drug candidates (14–16). In fact, a large number of small molecule inhibitors that target AMPA receptors have been synthesized (17–19). To date, however, no inhibitor is known to be capable of selectively inhibiting a single AMPA receptor subunit. Therefore, from the outset, we did not have a structural template of either a synthetic or a natural product inhibitor alike that might be most obvious to modify and/or improve upon to generate a subunit-selective inhibitor.
The approach we took was based on an in vitro evolution for isolating RNA inhibitors or aptamers from an RNA library (20, 21). This approach relies on RT-PCR to “breed” desired RNA molecules by exponential enrichment of their sequences over background, through multiple iteration cycles, against a specific target protein or receptor. The useful RNA molecules are selected because they are the best fits to the protein target based on geometrical complementarity. Therefore, this approach does not require prior knowledge of the structure of the protein target or the existence of any lead compounds (22).
For this study, we chose to identify aptamers that would selectively inhibit the GluA2 AMPA receptor subunit. We chose this subunit not just for a proof of principle but for a high utility of having a GluA2 subunit-selective inhibitor. GluA2 controls key functional properties of heteromeric AMPA receptors, such as Ca2+ permeability, single-channel conductance, and rectification (23, 24). These properties of GluA2 are attributed to Arg607, a residue at the glutamine/arginine (or Q/R) site introduced into the pore loop by RNA editing (23, 24). The Q/R editing is exclusive to GluA2 in AMPA receptors, and the editing is extremely efficient (i.e. >99% of GluA2 in adult brain is in the edited, R isoform) (25). Editing defect in GluA2, however, leads to generation of the highly Ca2+ permeable Q isoform through which excessive Ca2+ ions enter the cell. Consequently, intracellular calcium overload causes cell death, which underlines various neurological disorders such as stroke (26) and amyotrophic lateral sclerosis (27). Thus, GluA2 subunit-selective aptamers are potential drug candidates. These aptamers could be also used as structural probes.
To find GluA2-selective aptamers, we chose the closed-channel receptor conformation as the target of selection, rather than the open channel or a mixture of the closed- and the open-channel conformations. (For the purpose of this work, the closed-channel conformation is defined as the unliganded, resting form of the receptor; this is “A” as in a general mechanism of channel opening in the “Experimental Procedures.”) The choice of this conformation was based on our earlier hypothesis that the closed-channel conformation is more flexible or more modifiable in the context of inhibitor binding/inhibition (28). Therefore, the closed-channel conformation would be a better structural scaffold for geometrical complementarity selection (29). As such, an RNA aptamer, selected to uniquely recognize one conformation of a subunit, is not expected to bind avidly to either other receptor subunits or even other conformations of the same subunit due to incorrect or imperfect molecular recognition, thereby producing subunit discrimination and selectivity.
To maximize our chance of success in finding subunit-selective inhibitors, we did not select competitive inhibitor type, despite the fact that there is abundant, structural information available for competitive inhibitors and the agonist binding sites (30–34). Instead, we selected noncompetitive inhibitors (or noncompetitive aptamers). Because noncompetitive inhibitors bind to regulatory sites, distinct to the site agonist binds, they are generally considered more selective or less promiscuous in differentiating isoforms.
By applying these mechanism-based design principles, together with the use of an in vitro evolution approach, we have successfully isolated, and report here, an aptamer that is not only potent but also exclusively selective to the GluA2 AMPA receptor subunit.
EXPERIMENTAL PROCEDURES
Cell Culture and Receptor Expression
The cDNAs encoding various subunits of glutamate ion channels were used for transient receptor expression in HEK-293S cells (35). The cell culture, transfection, and preparation of membrane lipids containing functional, intact GluA2Qflip receptors for aptamer selection were carried out according to the protocols reported previously (35).
Aptamer Selection
The preparation of the RNA library with ∼1015 random sequences and the running of in vitro selection of aptamers against GluA2Qflip in membrane lipids were described previously (35) (see also supplemental Fig. S1). For binding at each selection cycle, the RNA library was dissolved in the extracellular buffer, which contained:145 mm NaCl, 3 mm KCl, 1 mm CaCl2, 2 mm MgCl2, and 10 mm HEPES (pH 7.4). The final concentration of membrane-bound receptor for binding was 8 nm, determined by [3H]AMPA binding (35). The binding mixture was incubated at 22 °C for 50 min in the presence of 0.3 units/μl RNase inhibitor. For elution, 1 mm (final concentration) of (−)1-(4-aminophenyl)-3-methylcarbamoyl-4-methyl-7,8-methylenedioxy-3,4-dihydro-5H-2,3-benzodiazepine, which we designated as BDZ-f, was used. The eluted RNAs were subject to RT-PCR (supplemental Fig. S1). To suppress enrichment of nonspecific RNAs bound to any unwanted “targets,” such as lipids, we also ran three negative selections at rounds 4, 8, and 13 in which plain HEK-293 cell membrane fragments lacking only GluA2Qflip receptors were used to absorb nonspecific RNAs. For identifying consensus sequences, the DNA pools from rounds 12 and 14 (i.e. 14 was the final round) were cloned separately into the pGEM-T easy vector (Invitrogen) and sequenced.
Homologous Competitive Binding Assay
The 5′-end 32P-labeled aptamers, AF44 and AF42, were prepared as described (35). An aliquot of 10 nm of 32P-labeled (hot) aptamer was mixed with a series of concentrations (i.e. 0–400 nm, final concentration) of the unlabeled (cold) aptamer. The final concentration of the receptor and hot aptamer was 0.4 and 0.1 nm. The same concentrations of the receptor and the hot aptamer were used when kainate (1 mm, final concentration) was included to assess aptamer binding to the receptor in the presence of kainate. The receptor/library mixture was incubated at 22 °C for 1 h for binding, followed by filtering and centrifugation (35). The radioactivity on the filter was quantified with the use of a scintillation counter (Beckman Coulter LS6500). The nonspecific binding was estimated by Equation 1 (36). The specific binding was calculated as the difference between the total binding in cpm and the estimated nonspecific cpm. The specific binding was normalized to the percentage without the unlabeled AF44/AF42. Assuming a one-site binding model, the Kd of an aptamer was estimated by Equation 1 (36),
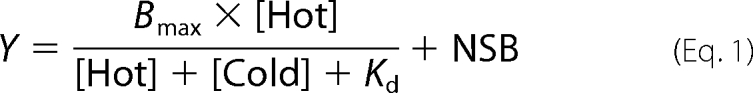 |
where [Hot]/[Cold] are the concentrations of unbound, hot aptamer/cold aptamer, respectively; NSB represents nonspecific binding. Bmax is the maximal number of binding sites.
Displacement of Aptamer Binding by NBQX
6-Nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX) was mixed (200 μm, final concentration) with hot aptamer/GluA2Qflip to assess whether NBQX bound to the same site with AF44 or AF42 as well as AF1422. Dimethyl sulfoxide (DMSO) was used for initially dissolving NBQX. Hot AF44 or AF42 of 0.1 nm (final concentration) mixed with the receptor was incubated with DMSO alone (as a blank control) or NBQX/DMSO at 22 °C for 1 h. The binding in the absence and presence of DMSO and NBQX/DMSO was quantified the same way as in homologous competitive binding.
Whole-cell Current Recording
The procedure for whole-cell current recording to assay putative aptamers was described previously (35). All recordings were at −60 mV and 22 °C. The recording electrode was filled with the buffer: 110 mm CsF, 30 mm CsCl, 4 mm NaCl, 0.5 mm CaCl2, 5 mm EGTA, and 10 mm HEPES (pH 7.4 adjusted by CsOH). The extracellular buffer composition was provided under “Aptamer Selection.” All other buffers used were described previously (35). The whole-cell current was recorded using an Axopatch 200B amplifier at a cutoff frequency of 2–20 kHz by a built-in, four-pole Bessel filter and digitized at 5–50 kHz sampling frequency using a Digidata 1322A (Molecular Devices). pClamp 8 (Molecular Devices) was used for data acquisition.
Laser-pulse Photolysis Measurements
The laser-pulse photolysis technique was used to characterize the mechanism of inhibition by measuring the effect of an aptamer on the channel-opening kinetics (28). Briefly, γ-O-(α-carboxy-2-nitrobenzyl)glutamate (caged glutamate) (37) (Invitrogen) with or without aptamer dissolved in the extracellular buffer was applied to a cell using a flow device (28, 35). A single, 355-nm laser pulse with a pulse length of 8 ns and pulse energy of 200–800 μJ, generated from a pulsed Q-switched Nd:YAG laser (Continuum), was used for photolyzing the caged glutamate. To calibrate the concentration of released glutamate, we applied two solutions of free glutamate with known concentrations to the same cell using the same flow device before and after a laser flash (38). The current amplitudes obtained from this calibration were compared with the amplitude from the laser measurement with reference to the dose-response relationship for GluA2Qflip (39).
Kinetic Data Analysis, Mechanism of Channel Opening
The opening of the GluA2Qflip channel in response to glutamate binding was described kinetically in a general mechanism (Scheme 1; 39).
SCHEME 1.
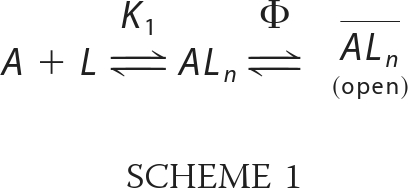
A represents the closed, unliganded form of the receptor, L represents the ligand, ALn represents the closed-channel forms bound with ligands, ALn represents the open-channel state, K1 represents the intrinsic dissociation constant of activating ligand, and Φ represents the channel opening equilibrium constant; n is the number of the ligand molecules that bind to the receptor to open the channel (i.e. n = 1–4). Based on this mechanism and also the assumption that the ligand binding rate was fast as compared with the channel-opening rate, the observed rate constant of channel opening (kobs) was written as Equation 2.
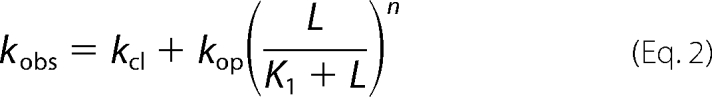 |
In Equation 2, kcl and kop are the channel-closing and channel-opening rate constants, respectively. Furthermore, kobs was calculated from Equation 3, where Imax is the maximum current amplitude and It is the current amplitude at time t. Our previous studies of AMPA receptors, including a mutant AMPA receptor, for their channel-opening kinetic mechanisms led us to conclude that binding of two glutamate molecules per receptor (i.e. n = 2) was sufficient to open the channel (40). Using the laser-pulse photolysis technique, we previously determined the kop of (8.0 ± 0.49) × 104 s−1 and the kcl of (2.6 ± 0.20) × 103 s−1, respectively, for the channel-opening kinetic constants of the GluA2Qflip receptor (39).
Kinetic Data Analysis, Mechanism of Inhibition
The noncompetitive mechanism of inhibition was established by measuring the effect of AF44/AF42 on the channel-opening rate constants (41, 42). By this mechanism (see Scheme 2 below), an inhibitor binds to both the closed- and open-channel states through a regulatory site, and the binding results in inhibition of kobs as in Equation 4, where I is the molar concentration of the inhibitor (other symbols have been defined earlier).
SCHEME 2.
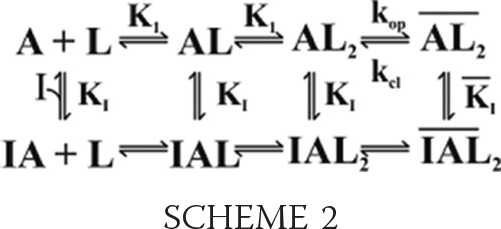
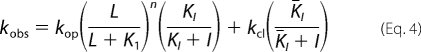 |
In deriving Equation 4, one inhibitory site was assumed. At low concentrations of glutamate (L ≪ K1), kobs reflected kcl because the contribution of the kop portion in Equation 4 to the overall rate, kobs, was negligible. Thus, Equation 4 was reduced into Equation 5, and the effect of the inhibitor on kcl could be assessed (28) by using Equation 5.
 |
When kobs > kcl, the effect of AF44/AF42 on kop was measured at a series of high glutamate concentrations. By noncompetitive inhibition, AF44/AF42 would affect kop (Equation 6), as was observed (Fig. 5D, right panel).
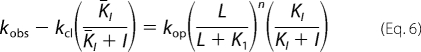 |
Because the magnitude of kcl reflects the lifetime (τ) of the open channel (i.e. τ = 1/kcl), the effect of an inhibitor on kcl reveals whether or not it inhibits the open-channel conformation or state (28). In contrast, kop reflects the closed-channel state, the effect on kop, therefore, reveals whether the inhibitor is effective on the closed-channel state (28). Experimentally, we established that for GluA2Qflip, at 100 μm photolytically released glutamate, which we set as the low glutamate concentration, kcl was measured (28, 43). In other words, under this condition or L ≪ K1, Equation 2 is reduced to kobs ≈ kcl (28, 43). In contrast, kop could be determined at a glutamate concentration of about 300 μm (28, 43). Correspondingly, the effect of an aptamer on kcl and kop could be characterized separately.
FIGURE 5.

Characterization of the mechanism of inhibition of AF44/AF42 by binding and kinetic measurements. A, by homologous competition binding assay of AF44 (cold and hot) to GluA2Qflip receptor, the binding constant, Kd, was calculated based on triplicate data sets, using Equation 1, to be 44 ± 18 nm for the unliganded, closed-channel conformation (left panel) and 48 ± 13 nm for the open-channel conformation (right panel) of GluA2Qflip, respectively. B, likewise, Kd for AF42 was found to be 57 ± 21 nm for the closed-channel conformation (left panel) and 44 ± 11 nm for the open-channel (right panel) conformation of GluA2Qflip, respectively. C, percentage of displacement of the binding of each aptamer in the presence of NBQX. Binding of hot AF44 or hot AF42 without DMSO and NBQX was set to be 100% (dashed line). As a control, we tested the effect of 8% DMSO on the binding of AF44 and AF42 to GluA2Qflip; this was the same amount of DMSO used to dissolve NBQX. Triplicate data sets for binding were collected, and the average value is displayed. D, the laser-pulse photolysis measurement of the effect of AF44/AF42 on the channel-opening rate of GluA2Qflip. At 300 μm photolytically released glutamate concentration (left panel), kobs was 4975 ± 173 s−1 for the control (open circles), but was 4273 ± 146 s−1 in the presence of 1 μm AF44/AF42 (open diamonds). The first order rate constants were calculated as the best fit (see the solid line in both traces) by Equation 3. The current amplitude in the absence and presence of aptamer was 0.70 and 0.51 nA, respectively. At 100 μm photolytically released glutamate concentration, kobs, which reflected kcl, was similarly estimated to be 2297 ± 36 s−1, and the current amplitude was 0.23 nA in the absence of AF44/AF42 (open circles). In the presence of 1 μm AF44/AF42 (open diamonds), kobs was found to be 2223 ± 25 s−1, and the current amplitude was 0.15 nA. The initial spikes prior to the current rise were discharge signals from the laser flash.
Amplitude Data Analysis
The ratio of the whole-cell current amplitude in the absence and presence of a putative aptamer or A/A(I) as a function of aptamer concentration was used to independently measure inhibition constants (28), as illustrated in Equations 7 and 8 (28). These equations were derived based on the same general mechanism of channel opening described earlier. KI,app is the apparent inhibition constant for the inhibitor; other terms have been defined previously.
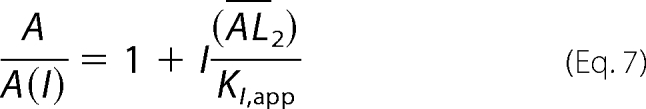 |
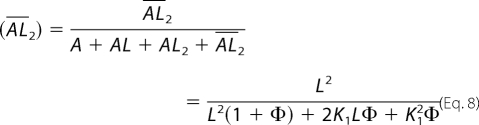 |
According to Equation 7, at low glutamate concentrations (i.e. L ≪ K1), the majority of the receptors were in a closed-channel population. Thus, the inhibition constant for the closed-channel conformation or state was determined from A/A(I) versus inhibitor concentration by Equations 7 and 8. Likewise, at a saturating ligand concentration (i.e. L ≫ K1), the majority of the receptors were in the open-channel state. Thus, the inhibition constant for the open-channel state was determined. In the case of GluA2Qflip, because the EC50 of GluA2Qflip with glutamate is 1.3 mm and the channel-opening probability of GluA2Qflip is near unity (39), 100 μm glutamate concentration corresponds to the majority of the channels (i.e. ∼96%) being in the closed-channel state (39). At 3 mm glutamate concentration, which is a saturating concentration (39), almost all of the channels are in the open state (39). Thus, the KI value of an aptamer for the open- and closed-channel states of GluA2Qflip was determined using these two glutamate concentrations.
Unless noted otherwise, each data point was an average of at least three measurements. Each of the whole-cell recording data point was collected from at least three cells. Uncertainties reported refer to S.D. Origin software (version 7) was used for data analysis and plotting.
RESULTS
Isolation of an RNA Aptamer That Inhibits GluA2Qflip AMPA Receptor Subunit
To ensure that the aptamers to be identified would recognize the functional GluA2Qflip AMPA receptors, we transiently expressed the intact GluA2Qflip receptors in HEK-293 cells and used the cellular membrane harboring the receptor for aptamer selection (35). After 14 selection cycles (supplemental Fig. S1), we cloned the DNA libraries from rounds 12 and 14. Overall, 92 clones were randomly chosen and sequenced. Four enriched sequences were identified. An enriched sequence was defined as one that appeared at least twice in the entire sequence pool (Fig. 1A).
FIGURE 1.

Selection of aptamers against the closed-channel conformation of the GluA2Qflip AMPA receptor subunit. A, the four enriched RNA sequences were identified from 92 clones randomly chosen from rounds 12 and 14. Their names are shown on the left; on the right are their copy numbers (or the number of appearance of the same sequence in the 92 sequences). The variable region that contained 50 nucleotides (or N50) is marked in black letters, whereas the constant regions are indicated in color and are displayed at the bottom. It should be noted that AF1422 had a point mutation from GGC to GCC at the 5′-end constant region next to the variable region, and AF1437 had seven consecutive nucleotides missing (four nucleotides within variable region and three nucleotides within the 3′-end constant region), from the evolution process. B, representative traces of the whole-cell current response of GluA2Qflip to 100 μm glutamate in the absence (left panel) and the presence (right panel) of 1 μm aptamer AF1422. The current was recorded at −60 mV, (pH 7.4) and 22 °C with the same HEK-293 cells expressing GluA2Qflip. C, all four selected RNA sequences were assayed using whole-cell current recording with GluA2Qflip. The result was represented by the ratio of the current amplitude in the absence and presence, or A/A(I), of 1 μm aptamer and 100 μm glutamate. The third round of the library or “pool 3” at 1 μm was used as a control. The blue asterisk indicates p ≤ 0.05 from the two-tailed Student's t test (H0, μ = μ0 = 1, 1 being the theoretical value of no inhibition, marked as the red dashed line).
Using whole-cell recording, we tested the putative inhibitory property for each sequence with HEK-293 cells expressing GluA2Qflip receptors, the selection target. By the ratio of the whole-cell current amplitude in the absence and presence of an aptamer or A/A(I) (Fig. 1B), we found that AF1422, the most enriched sequence (i.e. 75% appearance frequency), was an inhibitor, whereas the other sequences were not (Fig. 1C).
Identification of Minimal, Functional Aptamer Sequence, AF44 and AF42 Inhibitor Pair
Guided by Mfold, an RNA secondary structure prediction program (44), we constructed a number of shorter versions of AF1422 to identify the minimal, yet functional RNA sequence (Fig. 2A). We initially tested, by monitoring the A/A(I) value, the inhibitory function of single, but shorter RNA pieces one at a time (Fig. 2A), but found that none worked alone as an inhibitor (Fig. 2B). Eventually, we established that the shortest, yet functional version of AF1422 was a pair, i.e. AF44 and AF42 (AF44 and AF42 represent a 44-nucleotide and a 42-nucleotide RNA molecules, respectively). In other words, the use of either AF44 or AF42 alone did not render any inhibition; yet, an equal molar mixture of AF44 and AF42 reproduced an inhibition as full as AF1422 (Fig. 2B).
FIGURE 2.

The minimal, functional sequence of AF1422, AF44/AF42 inhibitor pair. A, in truncating the full-length sequence of AF1422 to identify the minimal, functional sequence, only the most stable secondary structure of each sequence, as predicted by the Mfold program, was displayed and constructed. The free energy is listed below these structures. The 5′- and 3′-end constant regions are shown in red and blue. B, a truncated aptamer was tested either alone or together as shown, such as AF44/AF42 functional pair, by measuring the whole-cell current response of GluA2Qflip with and without a shortened RNA piece(s). The blue asterisk indicates p ≤ 0.05 from the two-tailed Student's t test (H0, μ = μ0 = 1, 1 being the theoretical value of no inhibition, marked as the red dashed line). Additional information for sequence reduction is provided in supplemental Fig. S2.
To create shorter pieces of RNAs (Fig. 2A), we preserved the predicted secondary structures that were thought to be important. For example, AF78 was constructed to test whether the stem-loop structure (shown in red of the predicted structure of AF1422, Fig. 2A), comprised of the initial 20 or so nucleotides at the 5′ constant region, played any functional role. The fact that AF78 turned out to be noninhibitory suggested that this region was essential for inhibition. However, AF20, which contained only this stem loop (Fig. 2A), was not sufficient to act either alone as an inhibitor or together with AF42 as an inhibitory pair (Fig. 2B). In contrast, AF44, which carries this stem-loop module, was capable of acting with AF42 as an inhibitory pair (Fig. 2B). Furthermore, based on the functional combination of AF44 and AF42, a removal of the terminal loop from the AF42 resulting AF38 led to a total loss of function, suggesting that this terminal loop was essential as well.
To create AF44, we modified the projected secondary structure of AF1422 (Fig. 2A) by the following way. First, we significantly shortened the stem formed from U19/G99 to U31/G86 by retaining only three-GC base pairs, i.e. C20/G98, G27/C90, and G30/C87 (see supplemental Fig. S2A). Second, to maintain the two central loops as in AF1422 (Fig. 2), we replaced nucleotide G22 with A22 (see supplemental Fig. S2B) (note that G22 is numbered as the nucleotide position in the truncated sequence as in supplemental Fig. S2B; in the wild type AF1422 sequence, it corresponds to G32 in supplemental Fig. S2A). Without this replacement, the shorter sequence carrying G22 (i.e. AF44 wildtype) showed no activity at all with GluA2 receptor channels when it combined with AF42 (data not shown). This was because the wild type sequence in AF44(G22) could no longer fold into the same secondary structure as in the central loop of AF1422 (Fig. 2). Yet, AF44(A22) was projected to fold into a structure resembling the central loop of AF1422 with a shortened stem (Fig. 2 and supplemental Fig. S2B). Taken together, AF44 and AF42 as a whole entity were considered a minimal, functional inhibitor entity and were used for all the subsequent studies below. (For convenience, AF44(A22) was simply termed AF44 in this work.)
Characterization of Inhibition Constant of AF44/AF42 with GluA2Qflip
Using Equations 7 and 8, we characterized the inhibition constant of AF44/AF42 with GluA2Qflip, the selection target. A KI of 1.5 ± 0.1 μm (the solid line in Fig. 3A) was determined for the closed-channel conformation from a series of aptamer concentrations and at 100 μm glutamate concentration (see “Experimental Procedures” for choosing this concentration for the assay of the closed-channel conformation). Yet, with the same range of aptamer concentration, AF44/AF42 was ineffective in inhibiting the open-channel conformation (open symbol in Fig. 3A). The inhibition by AF44/AF42 became detectable only when its concentration was raised much higher. For example, the whole-cell current amplitude at 3 mm glutamate was reduced by ∼20% at 6.25 μm AF44/AF42 (Fig. 3B). As such, the inhibition constant of AF44/AF42 for the open-channel state was estimated to be ∼23 μm (using Equations 7 and 8). Based on the ratio of KI values, AF44/AF42 was selective toward the closed-channel conformation of GluA2Qflip channels by ∼15-fold.
FIGURE 3.

Characterization of inhibition constant and selectivity of AF44/AF42. A, by whole-cell current recording assay, AF44/AF42 inhibited the closed-channel, but not the open-channel, conformation of GluA2Qflip within the aptamer concentration shown. The KI for AF44/AF42 was estimated to be 1.5 ± 0.1 μm for the closed-channel conformation of GluA2Qflip (i.e. the upper solid line) by Equations 7 and 8. B, the whole-cell current response to 3 mm glutamate in the absence (left panel) and presence of 6.25 μm AF44/AF42. The reduction of current amplitude at this aptamer concentration is ∼20%. C, similarly, KI for BDZ-f was determined to be 3.8 ± 0.4 μm for the closed-channel conformation and 5.4 ± 0.8 μm for the open-channel confirmation, respectively. D, by whole-cell current recording assay, AF44/AF42 selectively inhibited the closed-channel conformation of GluA2Qflip but did not affect any other channels as shown. For this assay, the glutamate concentration was chosen to be equivalent to ∼4 and ∼95% fraction of the open channels. Specifically, the glutamate concentration was 0.04 mm (for the closed-channel conformation) and 3 mm (for the open-channel conformation) of GluA1flip; 0.1 and 3 mm for GluA2Qflip, GluA3flip, and GluA4flip; and 0.04/3 mm for GluK1 and GluK2. For NMDA channel, glutamate concentrations of 0.02 and 0.05 mm were used. AF44/AF42 was kept at 1 μm. Each data point was based on at least three measurements collected from at least three cells. The single asterisk indicates p ≤ 0.05 from the two-tailed Student's t test (H0, μ = μ0 = 1, 1 being the theoretical value of no inhibition), and the double asterisks indicate p ≤ 0.01 from the two-sample Student's t test, as a comparison between closed and open forms (H0, μ1 = μ2). E, the selectivity of BDZ-f was determined similarly as with AF44/AF42. The BDZ-f concentration used was 20 μm, and the glutamate concentrations used are described in D.
It is worth noting that BDZ-f, a 2,3-benzodiazepine compound and the one we used as the selective pressure for the identification of AF1422 (supplemental Fig. S1), the predecessor of AF44/AF42, exhibited a KI of 3.8 μm for the closed-channel conformation and 5.4 μm for the open-channel conformation of GluA2Qflip, respectively (Fig. 3C). Thus, the selectivity of BDZ-f for the closed-channel state over the open-channel state is merely 1.4-fold. In contrast, AF44/AF42 showed 15-fold higher selectivity toward the closed-channel state of GluA2Qflip while maintaining a high potency (in fact, AF44/AF42 showed a 2.5-fold improvement in potency, i.e. KI of 1.5 μm for AF44/AF42 versus 3.8 μm for BDZ-f).
Subunit Selectivity of AF44/AF42
The subunit selectivity of AF44/AF42 was assessed with individual subunits of AMPA, kainate, and NMDA receptors expressed in HEK-293 cells and was represented by the A/A(I) value collected from whole-cell recording (Fig. 3D). The selectivity was further determined at two glutamate concentrations, representing the closed-channel and open-channel conformations (see the solid and open columns in Fig. 3D). We found that AF44/AF42 did not affect the rest of AMPA receptor subunits, i.e. GluA1, -3, and -4. Furthermore, AF44/AF42 did not affect either kainate channels (i.e. GluK1 and GluK2) or NMDA channels (i.e. GluN1a/2A and GluN1a/2B) (Fig. 3D). It should be mentioned that GluN1a/2A and GluN1a/2B are two dominant NMDA receptor complexes in vivo (45) and neither GluN1a nor GluN2A or GluN2B can form a functional channel by itself (46). These results thus demonstrated that AF44/AF42 is an inhibitor pair that possesses a unique selectivity toward GluA2 but without any unwanted activity on any other subunits of the glutamate ion channel receptor family. These properties are expected because of our design strategy. In contrast, BDZ-f, the chemical compound that we used in the selection of AF1422 inhibited not only GluA2Qflip but also GluA1flip (Fig. 3E). Thus, AF44/AF42 is a better inhibitor in terms of subunit selectivity.
AF44/AF42 Is More Potent and Selective to the Flip Than to the Flop Isoform of GluA2Q
Alternative splicing in AMPA receptors generates two variants, i.e. flip and flop (47). The flip/flop sequence cassette is part of the extracellular ligand binding domain, and the C terminus of this sequence cassette precedes the last transmembrane domain. The flip and flop variants of an AMPA receptor subunit generally have different kinetic properties. The flop variants of AMPA receptor subunits, with the exception of GluA1, have similar kop, yet different kcl values (12, 48); they desensitize at least three times faster but recover more slowly from desensitization than the flip counterparts (10, 12, 48, 49).
Because of the difference in these properties for the flip and flop variants of GluA2Q, we tested whether AF44/AF42 differentially inhibited the flip and flop. Indeed, AF44/AF42 preferred to inhibit the closed-channel state of GluA2Qflop, but with a significantly weaker potency (Fig. 4, A and B). The comparison of this result (Fig. 4) with that of the same experiment but with the flip variant (Fig. 3, A and B) suggested that AF44/AF42 preferentially inhibited flip over the flop variant of GluA2Q.
FIGURE 4.
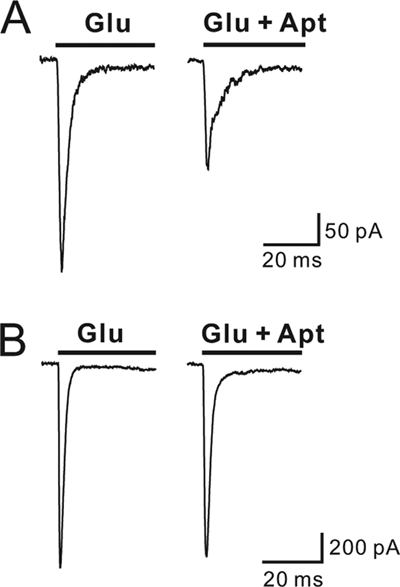
Inhibition of the flop isoform of GluA2Q or GluA2Qflop by AF44/AF42. A, AF44/AF42 inhibited the closed-channel conformation of Glu2AQflop expressed in HEK-293 cells. The left panel is the whole-cell current response to 100 μm glutamate, whereas the right panel is the whole-cell current to the same glutamate concentration but in the presence of 6.25 μm AF44/AF42. B, the inhibition of the open-channel conformation of GluA2Qflop by AF44/AF42 was assayed at 3 mm glutamate. Shown are two representative whole-cell current traces in the absence (left panel) and presence (right panel) of 6.25 μm AF44/AF42.
Studying Mechanism of Inhibition of AF44/AF42 by Homologous Competition Binding Experiment
The fact that AF44/AF42 inhibited both the closed-channel and the open-channel conformations of GluA2Qflip observed in the amplitude measurements (Fig. 3, A and B), although the inhibition of the latter was considerably less potent, was consistent with a noncompetitive mechanism. If this mechanism was indeed operative, AF44/AF42 would be expected to bind to a noncompetitive site, and such a site would be distinct from the glutamate binding site and would be accessible through both the closed-channel and the open-channel conformations. In fact, in a homologous competition binding experiment (36) in which the nonradiolabeled aptamer (or cold aptamer) was used to compete against the same, but radiolabeled, aptamer (or hot aptamer), AF44 bound to not only the closed-channel conformation (i.e. the unliganded, closed-channel receptor form) but also the open-channel conformation (Fig. 5A). Specifically, AF44 bound to the two different receptor conformations with equal affinity, i.e. Kd = 44 ± 18 nm (Fig. 5A, left panel, closed-channel conformation) and Kd = 48 ± 13 nm (Fig. 5A, right panel, open-channel conformation), respectively. Likewise, AF42 was found to bind to both the closed-channel conformation (Kd = 57 ± 21 nm; Fig. 5B, left panel) and the open-channel conformation (Kd = 44 ± 11 nm; Fig. 5B, right panel), respectively. The binding results were consistent with noncompetitive sites because these sites were supposed to be accessible through both the closed- and the open-channel conformations. The fact that AF44 or AF42 alone did not exert any inhibition and yet each could bind the receptor further suggested that AF44 and AF42 bound to two different sites (Fig. 5, A and B). These sites were noncompetitive type because binding to the receptor by either AF44 or AF42 (Fig. 5C) or the full-length AF1422 (supplemental Fig. S3) was unaffected in the presence of NBQX, a classic competitive inhibitor (50).
Studying Mechanism of Action of AF44/AF42 by a Laser-pulse Photolysis Measurement of Effect of AF44/AF42 on Channel-opening Rate of GluA2Qflip
Using a laser-pulse photolysis technique with ∼60-ms time resolution (37), we further characterized the mechanism of inhibition of AF44/AF42 by measuring its effect on both kcl and kop of GluA2Qflip (Fig. 5D) (28, 43). Specifically, kcl and kop of GluA2Qflip were determined separately at 100 and 300 μm glutamate photolytically released by the laser-pulse photolysis of caged glutamate (see rate equations and quantitative treatment of the kinetic data under “Experimental Procedures”). The laser-pulse photolysis experiment also permitted us to follow simultaneously both the rate of channel opening and the current amplitude at a given glutamate concentration prior to channel desensitization (Fig. 5D) (28).
At 1 μm aptamer concentration and at 300 μm glutamate concentration, AF44/AF42 inhibited kop as compared with the control (Fig. 5D, left panel). This was expected because kop reflected the closed-channel conformation, and thus, the appearance of an inhibitory effect by the aptamer was consistent with the notion that AF44/AF42 inhibited the closed-channel state.
However, at the same aptamer concentration (i.e. AF44/AF42 of 1 μm) but at 100 μm photolytically released glutamate where kcl was measured (28, 43), AF44/AF42 did not show an inhibition of kcl, although it inhibited the current amplitude (Fig. 5D, right panel). Only at a higher concentration of the aptamer was the inhibition of kcl observed, along with further reduction of amplitude (supplemental Fig. S4). These results demonstrated that AF44/AF42 had a stronger inhibition toward the close-channel conformation than the open-channel conformation. This conclusion was again consistent with the results described earlier. It should be noted that at a low glutamate concentration (i.e. 100 μm photolytically released glutamate) where kcl was measured (28, 43), that AF44/AF42 inhibited the current amplitude at a low aptamer concentration but without inhibiting kcl was plausible because the macroscopic amplitude observed at a low glutamate concentration (i.e. 100 μm photolytically released glutamate) reflected ensemble receptors mostly from the closed-channel receptor population, yet kcl reflected the open-channel conformation (see Equations 5–7 under “Experimental Procedures”).
The results from the binding site assessment (Fig. 5, A–C) and the kinetic characterization of the effect of AF44/AF42 on both kcl and kop (Fig. 5D; supplemental Fig. S4) as well as the amplitude measurement (Figs. 3, A and B, and 4) are all consistent with the conclusion that AF44/AF42 is a noncompetitive inhibitor selective to the GluA2 closed-channel conformation. Conversely, either a competitive or an uncompetitive mode of action is inconsistent with our data because AF44/AF42 would bind to only the closed-channel conformation as a competitive inhibitor or only to the open-channel conformation as an uncompetitive inhibitor. The A/A(I) would also increase as the increase of glutamate concentration for an uncompetitive model, which contradicts to the observation of the effect of AF44/AF42 on the amplitude (Figs. 3A and 4).
DISCUSSION
In this study, we have described a de novo design, isolation, and mechanistic characterization of a novel aptamer pair (i.e. AF44/AF42) that acts as a potent, noncompetitive inhibitor. AF44/AF42 is exclusively selective to the GluA2 subunit and is without any activity even on the rest of the AMPA receptor subunits, although they are the closest to GluA2 in terms of sequence homology and presumed protein structure. Between different isoforms of the GluA2 subunit, AF44/AF42 is strongly selective toward the flip isoform or GluA2Qflip. For GluA2Qflip, AF44/AF42 is selective to the closed-channel over the open-channel conformation by ∼15-fold. To our knowledge, no inhibitor that shows a single AMPA receptor subunit selectivity has been documented thus far.
The unique GluA2 subunit selectivity of AF44/AF42 is achieved through the interaction of the aptamer with the GluA2Qflip closed-channel conformation, which we used as the selection target. The choice of using exclusively the closed-channel conformation as the target of aptamer selection was based on our hypothesis that the closed-channel conformation is more flexible and modifiable than the open-channel conformation (28). A more flexible protein target (with a presumed subset of conformational repertory) is more amenable to find a precise fit from a vast number of guest molecules (i.e. a vast number of RNA folds in our library). In other words, the best fit binds best. It is thus not surprising that AF44/AF42 prefers to inhibit the closed-channel conformation of the flip isoform of GluA2Q (Figs. 3, A and B, and 4).
Additional evidence that the closed-channel conformation is more amenable to the isolation of subunit-selective inhibitor came from another experiment we performed previously (51). In that study, we used the open-channel conformation of the GluA2Qflip, the same receptor we used here, to identify the open-channel conformation-selective aptamers. (We prepared the long-lasting open-channel conformation for the selection by using kainate, which opens the GluA2Qflip channel but does not desensitize it.) Indeed, an aptamer selective to the open-channel conformation has been successfully isolated (51). However, the same aptamer (51) is almost equally effective, as an inhibitor, on the rest of AMPA receptor subunits, i.e. GluA1, -3, and -4, despite the fact that none of these subunits were ever subjected to geometrical complementarity selection. Taken together, these results suggest that the open-channel conformations of all four AMPA receptors are more similar, whereas their closed-channel conformations are more unique. We therefore predict that developing subunit-selective inhibitors, i.e. chemical or nucleic acid inhibitors alike, is more feasible by targeting the closed-channel conformation of AMPA receptors.
The identification and characterization of nucleic acid inhibitors have yielded additional structural and functional insights into the GluA2Qflip. The unique selectivity of AF44/AF42 (and its predecessor aptamer AF1422) to the closed-channel conformation is in stark contrast to its chemical selective agent or the selective pressure (29), BDZ-f (see Fig. 3). BDZ-f, a small organic compound, inhibits both the open- and the closed-channel conformations, although it prefers, albeit only marginally (i.e. by 1.4-fold), the closed-channel conformation (Fig. 3C). Yet, the AF44/AF42 pair shows a dominant preference of the closed-channel conformation over the open-channel one (i.e. by ∼15-fold) (Fig. 3, A and B). We speculate that one of the main reasons for this much “improved” selectivity by AF1422, the actual aptamer that was evolved, was due to a more exquisite molecular recognition at the level of geometrical complementarity between the AF1422 and the receptor. AF1422 is predicted to resemble a “dumbbell” molecule, which is replaced at both ends by a pair of smaller, functional RNA folds, i.e. AF44/AF42 (see the structural similarities in Fig. 2A). The AF44/AF42 pair, for instance, occupies a much larger surface area than BDZ-f. By estimating atomic volume of AF44 and AF42 using the NucProt program (52), we find that the radius of AF44 and AF42 is 14.8 and 14.6 Å, respectively. Yet the radius of BDZ-f is only ∼5Å (estimated by the use of Chem3D Ultra 10.0). A molecule with a larger surface area generally exhibits more of multivalent binding and interaction to the target molecule. Multivalent binding increases the binding strength of a molecule to a single target, when the target displays potentially multiple, discrete sites for binding (53).
Based on this dumbbell model, we can estimate that the relative distance between the two ends of the dumbbell in AF1422 is 60–80 Å. This estimate is based on the secondary structure prediction from Mfold in that AF1422 contains five base-pairing helixes (Fig. 2A) with 23 base pairs in the middle, and there are five base pairs on each end (Fig. 2A). An average of 2.8 Å per base pair for the A form RNA helix without stacking is further assumed (54). Therefore, the two ends of the dumbbell-shaped AF1422 most likely represent the two modular sites for AF1422 or AF44/AF42. As such, the 60–80 Å range is really an estimate of the distance separation between the two subsites, although the precise location of these two sites is unknown.
Currently, there is no structural information on the noncompetitive sites of 2,3-benzodiazepine inhibitors, including the one we used here for aptamer selection, on any of the AMPA receptors. Mapping the location of these sites on the receptor will be a significant subject of study in the future. The RNA aptamers we present here, i.e. the AF44/AF42 pair, can be readily developed into labeling probes for mapping the sites of binding to achieve a greater understanding of the receptor structure and regulation of receptor function.
Using a high concentration of either aptamer AF44/AF42 (supplemental Fig. S5A) or chemical compound BDZ-f (supplemental Fig. S5B), we noticed that the initial, fast desensitizing phase of the GluA2Qflip receptor current could be virtually inhibited, but the nondesensitizing phase remained or seemed insensitive to inhibitors. At this point, the nature of this residual activity is unclear, but it is not due to the properties of AF44/AF42 we have found. Nevertheless, the existence of residual activity that cannot be inhibited by these noncompetitive inhibitors suggests that (a) other types of inhibitors should be used and/or (b) new aptamers should be developed in the future specifically against this nondesensitizing receptor state for the purpose of a complete control of the receptor activity.
How are the properties of AF44/AF42 compared with those of small molecule inhibitors? First, the exclusive selectivity of AF44/AF42 toward GluA2 AMPA receptor subunit is unique. Second, although AF44/AF42 has similar potency with BDZ-f, which we used for isolating this aptamer, AF44/AF42 is completely water-soluble. In contrast, BDZ-f and other 2,3-benzodiazepine compounds (19) have a serious water solubility problem, which limits their use in vivo. Thus AF44/AF42 represents a water-soluble, highly potent, and GluA2 subunit-selective inhibitor. The unique selectivity of AF44/AF42 toward the GluA2 subunit without any effect on other glutamate receptor subunits offers a possibility of controlling the in vivo function of the GluA2 subunit more precisely and quantitatively with minimal or none off-target activity.
Supplementary Material
Acknowledgments
We thank Mohammad Qneibi and Andrew Wu for some data collection.
This work was supported in part by National Institutes of Health Grant R01 NS060812. This work was also supported by the Department of Defense (Grants W81XWH-04-1-0106 and GW080163) and the Muscular Dystrophy Association.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5.
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- NMDA
- N-methyl-d-aspartate
- BDZ-f
- 1-(4-aminophenyl)-3-methylcarbamoyl-4-methyl-7,8-methylenedioxy-3,4-dihydro-5H-2,3-benzodiazepine
- NBQX
- 6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione
- Caged glutamate
- γ-O-(α-carboxy-2-nitrobenzyl)glutamate
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Hollmann M., Heinemann S. (1994) Annu. Rev. Neurosci. 17, 31–108 [DOI] [PubMed] [Google Scholar]
- 2. Seeburg P. H. (1993) Trends Neurosci. 16, 359–365 [DOI] [PubMed] [Google Scholar]
- 3. Dingledine R., Borges K., Bowie D., Traynelis S. F. (1999) Pharmacol. Rev. 51, 7–61 [PubMed] [Google Scholar]
- 4. Palmer C. L., Cotton L., Henley J. M. (2005) Pharmacol. Rev. 57, 253–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Malinow R., Malenka R. C. (2002) Annu. Rev. Neurosci. 25, 103–126 [DOI] [PubMed] [Google Scholar]
- 6. Monyer H., Seeburg P. H., Wisden W. (1991) Neuron 6, 799–810 [DOI] [PubMed] [Google Scholar]
- 7. Keinänen K., Wisden W., Sommer B., Werner P., Herb A., Verdoorn T. A., Sakmann B., Seeburg P. H. (1990) Science 249, 556–560 [DOI] [PubMed] [Google Scholar]
- 8. Kawahara Y., Ito K., Sun H., Aizawa H., Kanazawa I., Kwak S. (2004) Nature 427, 801. [DOI] [PubMed] [Google Scholar]
- 9. Zhu J. J., Esteban J. A., Hayashi Y., Malinow R. (2000) Nat. Neurosci. 3, 1098–1106 [DOI] [PubMed] [Google Scholar]
- 10. Mosbacher J., Schoepfer R., Monyer H., Burnashev N., Seeburg P. H., Ruppersberg J. P. (1994) Science 266, 1059–1062 [DOI] [PubMed] [Google Scholar]
- 11. Boulter J., Hollmann M., O'Shea-Greenfield A., Hartley M., Deneris E., Maron C., Heinemann S. (1990) Science 249, 1033–1037 [DOI] [PubMed] [Google Scholar]
- 12. Pei W., Huang Z., Wang C., Han Y., Park J. S., Niu L. (2009) Biochemistry 48, 3767–3777 [DOI] [PubMed] [Google Scholar]
- 13. Li G., Sheng Z., Huang Z., Niu L. (2005) Biochemistry 44, 5835–5841 [DOI] [PubMed] [Google Scholar]
- 14. Dixon S. J., Stockwell B. R. (2009) Curr Opin. Chem. Biol. 13, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marcaurelle L. A., Johannes C. W. (2008) Prog. Drug Res. 66, 187, 189–216 [DOI] [PubMed] [Google Scholar]
- 16. Hruby V. J. (2009) J. Org. Chem. 74, 9245–9264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zappalà M., Grasso S., Micale N., Polimeni S., De Micheli C. (2001) Mini. Rev. Med. Chem. 1, 243–253 [DOI] [PubMed] [Google Scholar]
- 18. Sólyom S., Tarnawa I. (2002) Curr. Pharm. Des. 8, 913–939 [DOI] [PubMed] [Google Scholar]
- 19. Weiser T. (2005) Curr. Drug Targets CNS Neurol. Disord. 4, 153–159 [DOI] [PubMed] [Google Scholar]
- 20. Tuerk C., Gold L. (1990) Science 249, 505–510 [DOI] [PubMed] [Google Scholar]
- 21. Ellington A. D., Szostak J. W. (1990) Nature 346, 818–822 [DOI] [PubMed] [Google Scholar]
- 22. Nimjee S. M., Rusconi C. P., Sullenger B. A. (2005) Annu. Rev. Med. 56, 555–583 [DOI] [PubMed] [Google Scholar]
- 23. Lomeli H., Mosbacher J., Melcher T., Höger T., Geiger J. R., Kuner T., Monyer H., Higuchi M., Bach A., Seeburg P. H. (1994) Science 266, 1709–1713 [DOI] [PubMed] [Google Scholar]
- 24. Kuner T., Beck C., Sakmann B., Seeburg P. H. (2001) J. Neurosci. 21, 4162–4172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sommer B., Köhler M., Sprengel R., Seeburg P. H. (1991) Cell 67, 11–19 [DOI] [PubMed] [Google Scholar]
- 26. Oguro K., Oguro N., Kojima T., Grooms S. Y., Calderone A., Zheng X., Bennett M. V., Zukin R. S. (1999) J. Neurosci. 19, 9218–9227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kwak S., Kawahara Y. (2005) J. Mol. Med. 83, 110–120 [DOI] [PubMed] [Google Scholar]
- 28. Ritz M., Micale N., Grasso S., Niu L. (2008) Biochemistry 47, 1061–1069 [DOI] [PubMed] [Google Scholar]
- 29. Wilson D. S., Szostak J. W. (1999) Annu. Rev. Biochem. 68, 611–647 [DOI] [PubMed] [Google Scholar]
- 30. Ahmed A. H., Thompson M. D., Fenwick M. K., Romero B., Loh A. P., Jane D. E., Sondermann H., Oswald R. E. (2009) Biochemistry 48, 3894–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ahmed A. H., Wang Q., Sondermann H., Oswald R. E. (2009) Proteins 75, 628–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lunn M. L., Hogner A., Stensbøl T. B., Gouaux E., Egebjerg J., Kastrup J. S. (2003) J. Med. Chem. 46, 872–875 [DOI] [PubMed] [Google Scholar]
- 33. Jin R., Banke T. G., Mayer M. L., Traynelis S. F., Gouaux E. (2003) Nat. Neurosci. 6, 803–810 [DOI] [PubMed] [Google Scholar]
- 34. Jin R., Horning M., Mayer M. L., Gouaux E. (2002) Biochemistry 41, 15635–15643 [DOI] [PubMed] [Google Scholar]
- 35. Huang Z., Pei W., Jayaseelan S., Shi H., Niu L. (2007) Biochemistry 46, 12648–12655 [DOI] [PubMed] [Google Scholar]
- 36. Swillens S. (1995) Mol. Pharmacol. 47, 1197–1203 [PubMed] [Google Scholar]
- 37. Wieboldt R., Gee K. R., Niu L., Ramesh D., Carpenter B. K., Hess G. P. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 8752–8756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li G., Niu L. (2004) J. Biol. Chem. 279, 3990–3997 [DOI] [PubMed] [Google Scholar]
- 39. Li G., Pei W., Niu L. (2003) Biochemistry 42, 12358–12366 [DOI] [PubMed] [Google Scholar]
- 40. Pei W., Ritz M., McCarthy M., Huang Z., Niu L. (2007) J. Biol. Chem. 282, 22731–22736 [DOI] [PubMed] [Google Scholar]
- 41. Niu L., Hess G. P. (1993) Biochemistry 32, 3831–3835 [DOI] [PubMed] [Google Scholar]
- 42. Niu L., Abood L. G., Hess G. P. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 12008–12012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li G., Oswald R. E., Niu L. (2003) Biochemistry 42, 12367–12375 [DOI] [PubMed] [Google Scholar]
- 44. Zuker M. (2003) Nucleic Acids Res. 31, 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wenthold R. J., Petralia R. S., Blahos J., II, Niedzielski A. S. (1996) J. Neurosci. 16, 1982–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Erreger K., Dravid S. M., Banke T. G., Wyllie D. J., Traynelis S. F. (2005) J. Physiol. 563, 345–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sommer B., Keinänen K., Verdoorn T. A., Wisden W., Burnashev N., Herb A., Köhler M., Takagi T., Sakmann B., Seeburg P. H. (1990) Science 249, 1580–1585 [DOI] [PubMed] [Google Scholar]
- 48. Pei W., Huang Z., Niu L. (2007) Biochemistry 46, 2027–2036 [DOI] [PubMed] [Google Scholar]
- 49. Koike M., Tsukada S., Tsuzuki K., Kijima H., Ozawa S. (2000) J. Neurosci. 20, 2166–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Honoré T., Davies S. N., Drejer J., Fletcher E. J., Jacobsen P., Lodge D., Nielsen F. E. (1988) Science 241, 701–703 [DOI] [PubMed] [Google Scholar]
- 51. Huang Z., Han Y., Wang C., Niu L. (2010) Biochemistry 49, 5790–5798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Voss N. R., Gerstein M. (2005) J. Mol. Biol. 346, 477–492 [DOI] [PubMed] [Google Scholar]
- 53. Goodsell D. S., Olson A. J. (2000) Annu. Rev. Biophys Biomol. Struct. 29, 105–153 [DOI] [PubMed] [Google Scholar]
- 54. Tanaka Y., Fujii S., Hiroaki H., Sakata T., Tanaka T., Uesugi S., Tomita K., Kyogoku Y. (1999) Nucleic Acids Res. 27, 949–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.