

Abstract
The N-terminal six-transmembrane domain (TM) bundle of lactose permease of Escherichia coli is uniformly inverted when assembled in membranes lacking phosphatidylethanolamine (PE). Inversion is dependent on the net charge of cytoplasmically exposed protein domains containing positive and negative residues, net charge of the membrane surface, and low hydrophobicity of TM VII acting as a molecular hinge between the two halves of lactose permease (Bogdanov, M., Xie, J., Heacock, P., and Dowhan, W. (2008) J. Cell Biol. 182, 925–935). Net neutral lipids suppress the membrane translocation potential of negatively charged amino acids, thus increasing the cytoplasmic retention potential of positively charged amino acids. Herein, TM organization of sucrose permease (CscB) and phenylalanine permease (PheP) as a function of membrane lipid composition was investigated to extend these principles to other proteins. For CscB, topological dependence on PE only becomes evident after a significant increase in the net negative charge of the cytoplasmic surface of the N-terminal TM bundle. High negative charge is required to overcome the thermodynamic block to inversion due to the high hydrophobicity of TM VII. Increasing the positive charge of the cytoplasmic surface of the N-terminal TM hairpin of PheP, which is misoriented in PE-lacking cells, favors native orientation in the absence of PE. PheP and CscB also display co-existing dual topologies dependent on changes in the charge balance between protein domains and the membrane lipids. Therefore, the topology of both permeases is dependent on PE. However, CscB topology is governed by thermodynamic balance between opposing lipid-dependent electrostatic and hydrophobic interactions.
Keywords: Membrane Biogenesis, Membrane Lipids, Membrane Proteins, Phospholipid, Sugar Transport, Phosphatidylethanolamine, Protein Synthesis, Sucrose Permease, Transmembrane Domain
Introduction
The proper folding and final topology of a polytopic membrane protein is determined by an intimate interplay between topogenic sequences of the protein itself (1–4), the specialized proteinaceous machinery (translocon) (5–7), and the lipid bilayer (1, 8, 9). Total hydrophobicity (10, 11) of a transmembrane domain (TM)3 is a primary driving force for bona fide membrane integration. However, TM orientation appears to be primarily determined by the net charge of flanking extramembrane domains. Membrane protein topological organization can in most cases be described by the statistically derived and experimentally confirmed positive inside rule (12). The positive inside rule is based on the predominance of positively charged protein domains exposed to the cytoplasm with negative or net neutral domains exposed to the opposite side of the membrane. A TM segment may adopt an initial topology determined by direct charge interactions between the translocon and the protein (5, 6). However, the contribution of the translocon to making a topological decision is limited by time, size of a newly synthesized TM, and the effective size of the translocon pore (7). After release from the translocon into the surrounding lipid bilayer, final TM orientation and late folding events are driven by the thermodynamics of interactions of the protein with the lipid bilayer (1, 9) and itself (3) that may further decode the topogenic signals within nascent polypeptides.
Considerable evidence exists that the membrane lipid environment is a factor that influences the topological organization of some membrane proteins in a co-translational and postinsertional manner (13–17). So far, three solute transporters have been shown to respond to a change in lipid environment (Fig. 1). Each protein adopts a partially mirrored topological organization in the absence of the zwitterionic lipid phosphatidylethanolamine (PE) relative to their normal organization in the presence of PE (1, 16, 17); lack of PE results in a large increase in the negative charge density of the membrane surface due to the remaining anionic lipids phosphatidylglycerol and cardiolipin. Lactose permease (LacY), phenylalanine permease (PheP), and γ-aminobutyric acid permease (GabP) have conserved acidic residues located within cytoplasmic domains that also contain basic residues deemed important for cytoplasmic localization (supplemental Fig. S1). However, only LacY cytoplasmic domains strictly follow the positive inside rule. In the first half of PheP and GabP, some cytoplasmic domains carry a net zero or even net negative charge, and thus these permeases do not strictly obey the positive inside rule (Fig. 1). Although a net positive charge dictates cytoplasmic disposition, negatively charged residues can be topogenic and attenuate the effect of positively charged residues, thus favoring periplasmic location if they lie within a critical distance (up to 6 residues) from a TM (18). Acidic residues located within the normally cytoplasmic domain of the above permeases were postulated to act as lipid-sensitive topogenic signals (1, 14, 19). An increase in the net positive charge of the cytoplasmic surface of LacY, either by eliminating negatively charged residues or introducing positively charged residues in a position-independent manner, resulted in the N-terminal half of LacY adopting its native orientation even in PE-lacking membranes (1). However, eliminating both a negative and positive charge from the cytoplasmic surface of the protein still resulted in inversion in PE-lacking cells. Significantly increasing the negative charge density of the cytoplasmic face of this bundle resulted in topological inversion even in normal PE-containing membranes.
FIGURE 1.

Topological organization of secondary transporters LacY, PheP, and CscB as a function of membrane lipid composition. Topologies of LacY (1) and PheP (16) as reported previously initially assembled in PE-containing (A, LacY; C, PheP) or PE-lacking (B, LacY; D, PheP) cells are shown. The predicted topology of CscB in PE-containing cells is depicted in E. Cytoplasm is at the top of each panel, and rectangles indicate TMs numbered sequentially in Roman numerals from the N-terminal (NT) to the C-terminal (CT) domain. The extramembrane domains connecting the TMs are numbered sequentially, and their topological disposition in PE-containing cells is indicated by the prefix C for cytoplasmic or P for periplasmic. The approximate positions of acidic (●, negative charge) and basic (○, positive charge) amino acids are indicated, with the net charge of each extramembrane domain noted below the domain names. The amino acids involved in salt bridges between TMs VII, X, and XI in LacY are indicated as well as amino acids involved in the putative salt bridge between TMs VII and X in CscB. Note that this protein has only one acidic residue in its TM VII. B shows the resulting topological inversion of TMs I–VI and exposure of TM VII to the periplasm with the loss of salt bridges to TM X and TM XI when LacY is assembled in PE-lacking cells. D shows the resulting topological inversion of the N-terminal hairpin and the formation of a miniloop by TM III, when PheP is assembled in PE-lacking cells. Note the high content of aromatic amino acids in TM III.
The net neutral lipids monoglucosyl diacylglycerol (20), diglucosyl diacylglycerol (21), and phosphatidylcholine (8, 15) are able to support native topology of LacY in the absence of PE. Therefore, it was concluded that interaction between the collective charge properties of the membrane surface and the protein cytoplasmically exposed extramembrane domains directs the final orientation of TMs (i.e. the effects of changes in net charge were the same whether resulting from the lipid or the protein) (14, 19). A role for lipids with no net charge appears to be a dampening of the translocation potential of negative residues that work in opposition to the positive inside rule. This dampening allows the functional presence of negative residues in cytoplasmic domains without affecting protein organization (1). Therefore, according to the net charge balance hypothesis of membrane protein assembly (1, 14, 19), the proper charge balance between translocation signals and retention signals required to achieve proper TM topology is maintained by the appropriate charge density of the membrane surface. In Escherichia coli, membrane surface charge is determined by the mol % of anionic (phosphatidylglycerol and cardiolipin) and zwitterionic (PE) phospholipids. Moreover, in order for the N- and C-terminal six TM helical bundles of LacY to respond to the lipid environment independent of each other, either during initial assembly or during a change in lipid environment postassembly, there must exist a flexible hinge region between the independently folding domains. The hydrophilic nature of TM VII appears to influence the operation of the molecular hinge within LacY. Electrostatic neutralization of the Asp240 by a D240I mutation within this hinge increases the hydrophobicity of TM VII. The increased hydrophobicity prevents its release into the periplasm in PE-deficient cells, which simultaneously blocks the inversion of the N-terminal bundle of the protein (1). It is not known whether increasing the translocation potential of neighboring extramembrane domains can overcome this thermodynamic barrier to inversion of topology.
What is the information encoded in these protein that makes them “sensitive” to lipids? Why are some proteins or protein domains dependent on membrane lipid composition for native topological orientation? What is necessary and sufficient for TM reorientation within a membrane in response to changes in membrane lipid composition? On the basis of the current properties of lipid-sensitive domains and “flip-flopping” proteins, some predictions can be made about domains within other proteins that might require PE or other net neutral lipids to display native topology. A primary feature would be the presence of cytoplasmic domains containing a mixture of negatively and positively charged residues flanking a TM. Such “frustrated” domains must be free of restrictions preventing different orientations relative to other parts of the protein. Extramembrane domains containing a mixture of charged residues are relatively easy to detect by proteomics. However, more global and long range interactions within a protein cannot be readily assessed by statistical methods and require direct experimental investigation.
A relationship between net charge of the cytoplasmically exposed protein domains, presence and absence of PE, availability of a molecular hinge region, and sensitivity of protein domain orientation to lipid composition has only been established for LacY. Therefore, additional polytopic membrane proteins (Fig. 1) were chosen to test the net charge balance hypothesis. LacY homologous (sucrose permease; CscB) and non-homologous (PheP) polytopic membrane proteins were expressed in cells lacking PE. These proteins were also re-engineered to force acquisition of either normal or aberrant TM orientation as a function of membrane lipid composition. Inspection of the charge distribution of cytoplasmic domains of sugar permeases closely related to LacY (supplemental Fig. S1A) predicts that the topology of all, with the possible exception of CscB would be affected by the absence of PE (14). TM VII of CscB contains only one acidic residue and exhibits a higher hydrophobicity relative to TM VII of LacY. The higher hydrophobicity may prevent this domain from behaving as a molecular hinge, similar to the LacY D240I mutant (1). Although CscB transport activity was altered in the absence of PE, experiments aimed at lowering the hydrophobicity of CscB TM VII did not lead to CscB domain inversion. However, incremental decreases in the net positive charge of the cytoplasmic surface of the N-terminal TM bundle resulted in inversion of TMs first in PE-lacking cells and then in PE-containing cells. Furthermore, increasing the net positive charge of the cytoplasmic extramembrane domains of PheP prevented topological inversion in PE-lacking cells, similar to results found with LacY. These results extend to other proteins the importance of lipid-protein charge interactions in establishing membrane protein topological organization. Studies on CscB and further studies on LacY demonstrated that final topology is also determined by long range interactions within the proteins that thermodynamically balance the relative strength of hydrophobic forces and charge effects. Therefore, final topological organization is dependent on short range charge interactions between the nascent protein chains, and both the translocon and lipids as well as long range interactions within the protein. We also demonstrated that manipulation of protein domain charge and lipid composition can result in co-existence of multiple topological arrangements for the same protein. This observation provides molecular insight into how some proteins (22–24) exhibit multiple topological organizations within the same cell.
EXPERIMENTAL PROCEDURES
Materials
Chemicals were purchased from Sigma. 3-(N-Maleimido-propionyl biocytin) (MPB) and 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) were purchased from Molecular Probes-Invitrogen. Site-directed polyclonal antibodies directed against the C-terminal dodecapeptide of LacY and against the N-terminal domain of PheP were prepared by ProSci Inc. and by Zymed Laboratories Inc., respectively. Avidin-horseradish peroxidase, Protein A/G-agarose resin, spin columns, and Super Signal West Pico chemiluminescent substrate were from Pierce Thermo Fisher Scientific. Thesit was from Fluka. RbCl/CaCl2 transformation salts came from MP Biomedicals. Antiprotease CompleteTM EDTA-free was from Roche Applied Science. Nitrocellulose sheets (0.2 μm) for Western blotting were purchased from Schleicher & Schüll.
Plasmids
Ampicillin-resistant plasmids pT7-5/lacY (1) and pSP72/cscB (25) expressing LacY and CscB derivatives, respectively, under OPtac regulation, and pBR/pheP encoding derivatives expressing PheP (16) under its endogenous promoter were employed. Changes in amino acid residues and replacement of native residues with cysteine were accomplished by site-directed mutagenesis (1, 16) in derivatives of these proteins where native cysteines were replaced by serine (16, 25, 26). CscB derivatives were appended at the C terminus (CscB*) with a dodecapeptide derived from the C terminus of LacY so that LacY-specific antibody could be used for protein detection and immunoprecipitation. As determined by Western blot analysis, the amino acid replacements by cysteine did not affect steady state levels of LacY (1), PheP (16), or CscB (Fig. 2). Transformation of PE-deficient E. coli cells with plasmids was done according to a published protocol (27). The cells were made competent for transformation using RbCl/CaCl2 transformation salts, which is suitable for strains with normal as well as altered lipid compositions.
FIGURE 2.

Protein expression level of the various single-cysteine replacements in CscB* (A) and CscB*-SB (B) as a function of cellular PE content. The indicated cells were isolated by centrifugation, and inner membrane vesicles were prepared as described under “Experimental Procedures.” Equal amounts of membrane protein (10 μg) derived from the indicated cells were subjected to SDS-PAGE followed by Western blot analysis using LacY dodecapeptide-specific antibody as described under “Experimental Procedures.”
Bacterial Strains and Growth Conditions
Strain AL95 (pss93::kan lacY::Tn9) lacks the ability to synthesize PE unless it carries the pssA-containing plasmid pDD72GM (13). All growth media were supplemented with 50 mm MgCl2, which is required for viability of PE-lacking strains (28). Strain AL95 (grown at 37 °C) lacks PE, and strain AL95/pDD72GM (grown at 30 °C) retains the normal E. coli composition of phospholipids, including PE.
Unless otherwise indicated, cells carrying plasmids expressing LacY, PheP, or CscB were grown in LB medium containing ampicillin (100 μg/ml). Expression of plasmid-borne LacY and CscB was induced during growth in exponential phase by the addition of 1 mm isopropyl-β-d-thiogalactoside until cells reached an A600 of ∼0.6–0.7 unless otherwise indicated.
Determination of TM Orientation
Substituted cysteine accessibility method as applied to TM orientation (SCAMTM) is based on the controlled membrane permeability of a thiol-specific reagent (MPB, where maleimide is attached to biotin) (27, 29). This reagent was used to probe the TM topology of proteins under study, as described previously (1, 13, 27) using whole cells or cells broken by sonication. Reaction with MPB was carried out in buffer A (250 mm sucrose, 25 mm MgCl2, 0.1 mm KCl) at pH 7.4 (K-HEPES, 100 mm) or pH 9.1 (50 mm Tris-HCl) on whole cells (periplasmic exposed cysteines) or during sonication of cells (cytoplasmic and periplasmic exposed cysteines). Following inactivation of MPB, whole cells were sonicated as described previously (1). In order to address mixed topologies, a whole cell aliquot was incubated for 30 min at 25 °C with AMS at a final concentration of 5 mm to block periplasmic water-accessible cysteine residues from the outside of cells (13, 27). Then AMS was removed by two cycles of centrifugation and resuspension in 750 μl of buffer A (pH 7.4). Biotinylation of exposed cysteine residues was detected after isolation of LacY, CscB, or PheP using immunoprecipitation followed by SDS-PAGE, Western blotting using Avidin-horseradish peroxidase and chemiluminescent reagents, and visualization using a Fluor-STM MultiImager (Bio-Rad).
In all cases, equal amounts of cells were processed from samples before and after sonication, and the same amount of sample was applied to SDS gels. Figures show the presence or absence of signal in the Western blot region at the apparent molecular mass for LacY (35 kDa), CscB (33 kDa), or PheP (37 kDa). The results presented are representative of two or more determinations.
Immunoprecipitation and Western Blot Analysis
After MPB labeling, pellets derived from sonicated cells were suspended in 100 μl of buffer A (pH 7.4) by vigorous vortexing and then solubilized by the addition of an equal volume of 50 mm Tris-HCl buffer, pH 8.1, 2% SDS, and 1 mm EDTA followed by vigorous vortexing for 15 min at room temperature, incubation at 37 °C for 15 min, and an additional 15 min of vortexing at room temperature. Samples (200 μl) were diluted with 300 μl of cold 50 mm Tris-HCl buffer, pH 8.1, containing 0.15 m NaCl, 2% Thesit, 0.4% SDS, and 1 mm EDTA (buffer IP1). The samples were vortexed for 1 min and centrifuged for 10 min at 20,800 × gav and 4 °C. Supernatants were transferred to the cups of spin columns, permease-specific polyclonal antibodies were added, and supernatants were incubated overnight at 4 °C on a rocking platform. Antibody for PheP was described previously (16). Antibody directed at a C-terminal dodecapeptide of LacY (1) was used for immunoprecipitation of LacY and CscB. The antibody-antigen complex was isolated by the addition of 20 μl of a suspension of Protein A/G-agarose affinity resin followed by incubation at 4 °C on a rocking platform for 90 min. Washings and elution of the affinity resin were performed in the cup of spin columns at 4 °C with centrifugations at 20,800 × gav for 1 min. The antibody-antigen complex bound to agarose resin was washed once with buffer IP1; once with 50 mm Tris-HCl (pH 8.1), 1 m NaCl, 2% Thesit, 0.4% SDS; and once with 10 mm Tris-HCl, pH 8.1, by vortexing for 1 min followed by centrifugation. After the washing steps, the antigen and antibody were extracted from the resin by two rounds of vigorous vortexing with 25 μl of SDS sample buffer (10 mm Tris-HCl (pH 6.8), 5.6% SDS, 200 mm DTT, 10% glycerol, 0.01% bromphenol blue) followed by centrifugation. The solubilized permeases were subjected to SDS-PAGE. The samples were transferred from polyacrylamide gels to nitrocellulose membranes (0.2 μm) as described previously (27, 30). Avidin-horseradish peroxidase (1:10,000 dilution of 2 mg/ml stock solution) and Super Signal West Pico chemiluminescence substrate reagents were used to visualize biotinylated proteins according to the manufacturer's instructions.
Image Acquisition and Processing
Western blots were imaged using a Fluor-S Max MultiImager equipped with a CCD camera and a Nikon 50-mm 1:1.4 AD (F = 1.4) at the ultrasensitive chemiluminescence setting as described previously (1). Quantity One versions 4.6.5.094 and 4.4.1 (Bio-Rad) were used to collect and store the images as JPEG files, which were later imported into Adobe Photoshop CS to construct the figures. Images were expanded or reduced so that the horizontal strip containing the targeted protein on all images within the same figure was approximately the same size. Images were then masked to only show the protein strips of interest, which were then aligned and labeled. The only valid comparison in intensity is between whole cell and sonicated sets (images treated identically) run on the same gel. Pairs of images from different gels or from discontinuous regions of the same gel are separated by white spaces in the figures. Final figures were saved at 300 dpi as TIFF files.
Sugar Transport Assay
For uphill transport, PE-lacking and PE-containing strains were washed once with 100 mm HEPES (pH 7.5), 50 mm MgCl2 and adjusted to an A600 of 10 (∼0.7 mg of protein/ml). Transport was initiated by the addition of 10 μl of [14C(U)]sucrose (0.5 μCi/ml, 0.4 mm final concentration) to 1 ml of cells. Aliquots of 100 μl were removed at various times, quenched with 3 ml of ice-cold 100 mm HEPES (pH 7.5), 100 mm LiCl, 50 mm MgCl2, immediately filtered through GF/F filters, and washed using 10 ml of the same buffer. Filters were dried and counted by liquid scintillation. Uptake values were corrected for sucrose uptake by cells carrying the pSP72 vector only. De-energized cells were obtained by pretreating the cells with a 50 μm concentration of the protonophore carbonyl cyanide-p-trifluoromethoxyphenylhydrazone for 5 min prior to assessing transport, as described above.
Preparation of Inner Membrane Vesicles
The pellets of cells grown in exponential phase in LB medium were resuspended to a concentration of A600 = 15 in 100 mm HEPES (pH 7.5), 25 mm MgCl2, 1 mm DTT, and anti-protease. Total cell lysates were prepared by subjecting the cell suspensions to a single passage through a motor-driven French pressure cell at 10,000 p.s.i. to form inner membrane vesicles. Undisrupted cells and outer membrane vesicles were removed from the lysates by centrifugation at 30,000 × gav for 20 min at 4 °C. The inner membrane fraction was collected by centrifugation at 150,000 × gav for 1.5 h at 4 °C and resuspended in the original buffer with 250 mm sucrose to a protein concentration of 15–20 mg/ml. Aliquots were stored at −80 °C until further use.
Hydrophobicity Calculations
Amino acid sequences were taken from the transport classification data base Web site (31) and used to calculate hydrophobicities of TMs and extramembrane domains using the Goldman-Engelman-Steitz scale (32) The hydrophobicity calculations using this scale are estimates of free energies of transfer from water to a hydrocarbon and are considered a good predictor of free energy changes for membrane integration of hydrophobic protein segments (33, 34), especially those containing polar residues (35).
RESULTS
Expression of CscB and Transport Activity
Plasmid pSP72/cscB* (gene for Cys-less CscB with a LacY C terminus subcloned into pSP72) was transformed into PE-containing strain AL95/pDD72GM and PE-lacking strain AL95. CscB* activity was measured by the ability to actively accumulate sucrose against a concentration gradient (proton-coupled uphill transport). Both the initial rate and steady-state level of sucrose transport were severely inhibited in PE-lacking cells compared with PE-containing cells (Fig. 3). Transport was also sensitive to the addition of a protonophore, verifying that uphill transport was occurring in both cell types. However, the CscB* expression levels in the membranes of PE-lacking cells, as measured by Western blotting with anti-LacY dodecapeptide, were nearly 3 times higher than in PE-containing cells (Fig. 3, inset) as determined by densitometry; because strain AL95 (lacY::Tn9) lacks LacY, anti-LacY can be utilized to quantify the expression level of CscB* in both types of cells. Therefore, PE-lacking E. coli membranes contain more CscB* but with impaired sucrose uphill transport function.
FIGURE 3.
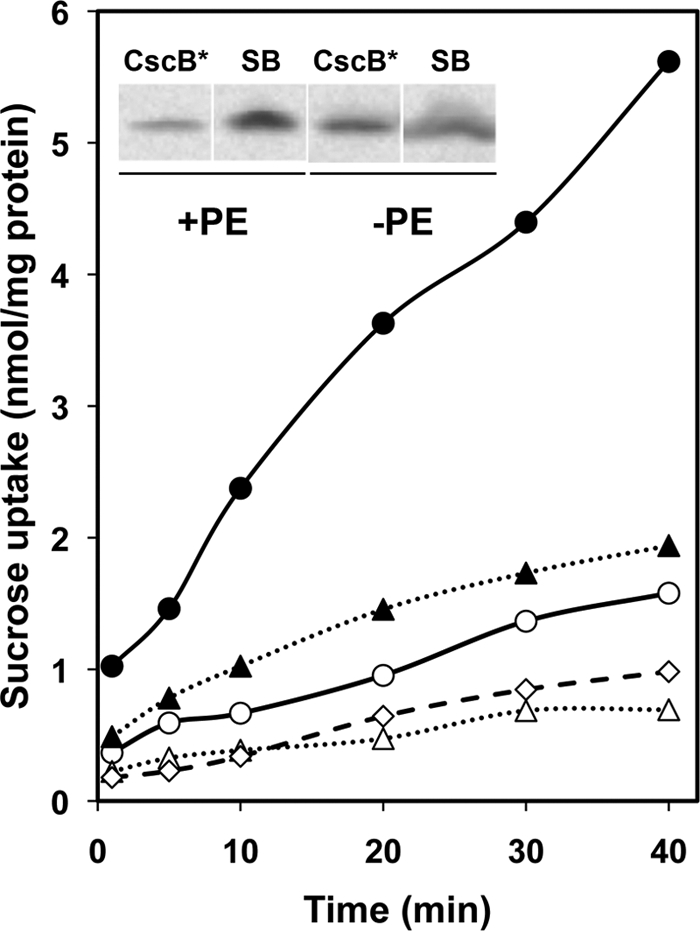
Uphill transport of sucrose by PE-containing and PE-lacking cells. Shown is uptake of sucrose normalized to total cell protein determined as a function of time in either strain AL95/pDD72GM carrying plasmid pSP72cscB* (+PE, ●) or pSP72cscB*-SB (+PE, ○) or strain AL95 carrying plasmid pSP72cscB* (−PE, ▴) or pSP72cscB*-SB (−PE, ▵), as described under “Experimental Procedures.” The addition of a protonophore eliminated any detectable accumulation of sucrose (+PE and −PE, ♢). Inset, expression levels of CscB* and CscB*-SB (SB) in PE-containing and PE-lacking cells. The inner membrane fraction was isolated as described under “Experimental Procedures.” Equal amounts of membrane protein (10 μg) from the above cells (lacY::Tn9) were subjected to SDS-PAGE, followed by Western blot analysis using LacY dodecapeptide-specific antibody. An increased level (3 times) of CscB* was detected in PE-lacking cells relative to the level of CscB* in PE-containing cells. An increased level (8 times) of CscB*-SB was detected in both PE-containing and PE-lacking cells relative to the CscB* level in PE-containing cells. (Quantification of protein expression was obtained by densitometric analysis using a Fluor-S Max MultiImager with ImageJ software).
Engineering a new charge pair mimicking the LacY counterpart between TM VII and TM XI (see Fig. 1 and supplemental Fig. S1A) was carried out later to see if this had any effect on the lipid-dependent topological organization of CscB. The salt bridge mutant (CscB*-SB) was constructed by introducing an Asp codon (GAC) at position Asn234 (TM VII) and a Lys codon (AAG) at position Ser356 (TM XI) in CscB* (plasmid pSP72/cscB*-SB). In the presence of an additional salt bridge, the uphill transport of sucrose was significantly decreased in PE-containing cells, whereas almost no transport was observed in PE-lacking cells (Fig. 3). Although very low, the transport observed in PE-containing cell was energy-dependent because it was sensitive to dissipation of the proton gradient by protonophore. Western blotting indicated that CscB*-SB expression levels in the membranes of PE-containing and PE-lacking cells were nearly the same (Fig. 3, inset) and 8 times higher than the level of CscB* in PE-containing cells. Therefore, the differences in sucrose transport activity do not stem from differences in the amount of CscB in membrane. The dependence on PE for transport activity by CscB provided the rationale to investigate whether lipid composition affects CscB topology.
Orientation of CscB Assembled in PE-containing and PE-lacking Cells
The topology of putative hydrophilic domains connecting TMs of CscB was determined based on the accessibility to sulfhydryl reagents of single cysteine replacements in these domains in whole and broken cells using SCAMTM. MPB is a biotinylated sulfhydryl-specific probe that readily passes through the pores of the outer membrane but is relatively impermeable to the inner membrane (29, 36, 37). Therefore, cysteines located on the periplasmic side of the inner membrane should be derivatized by MPB, whereas cysteines located on cytoplasmic side of the inner membrane should be protected (13, 29, 38), unless the membrane is disrupted before or during labeling. Single cysteine replacement derivatives of CscB* and CscB*-SB were expressed from plasmids in a PE-containing (with pDD72GM) or a PE-lacking (without pDD72GM) strain (AL95). The replacements were in putative extramembrane domains connecting TMs (Fig. 4A), as predicted from the CscB secondary structure (25). Western blot analysis showed that all of these derivatives were present in the membrane fraction with some variability but easily within detectable levels (Fig. 2).
FIGURE 4.

Mapping of CscB* and CscB*-SB topology in PE-containing and PE-lacking cells. A, TM orientation of CscB* (cysteineless) and CscB*-SB with positions of single-cysteine replacements noted. CscB* only has a salt bridge between Asp237 and Lys317, whereas CscB*-SB has an additional salt bridge between Asp243 and Lys356. B, strain AL95 either with (+PE) or without (−PE) plasmid pDD72GM was used as the host for expression of single cysteine replacements in CscB*. C, strain AL95 either with (+PE) or without (−PE) plasmid pDD72GM was used as the host for expression of single cysteine replacements in CscB*-SB. Domains containing cysteine replacements are indicated. Cys-less denotes CscB* and CscB*-SB with no cysteine replacements. Whole cells were labeled with MPB either before (−) or during (+) cell disruption by sonication, and the membrane extracts were subjected to immunoprecipitation, SDS-PAGE, and Western blotting as described under “Experimental Procedures.”
The predicted biotinylation patterns were observed for single cysteine derivatives of CscB* (Fig. 4B) expressed and probed in PE-containing cells, which were the same as that observed in PE-lacking cells. Derivatives with single cysteines in periplasmic (P) domains were labeled without and with cell disruption. The derivatives with single cysteines in the cytoplasmic (C) domains were only labeled after cell disintegration by sonication. No labeling of CscB was detected when the host strain (either PE-containing or PE-lacking cells) expressing cysteine-less CscB (i.e. CscB*) was probed (Fig. 4B). Western blot analysis showed that the protein was present (Fig. 2A). Therefore, although almost no transport activity is observed in PE-lacking cells, CscB still exhibits a proper topology.
The lack of inversion in PE-lacking cells might be explained by the higher hydrophobicity of CscB TM VII compared with LacY TM VII or the presence of only a single charged residue in TM VII of CscB. However, mapping the TM orientation in CscB*-SB led to the same observation as in CscB*. Whether assembled in PE-containing or PE-lacking strains, CscB*-SB displays a wild type topology (Fig. 4C).
Introduction of N234D only reduced the overall hydrophobicity of TM VII from −25.1 to −20.7 kcal/mol. This hydrophobicity is still much higher than the hydrophobicity of TM VII in LacY normally containing two Asp residues (−5.2 kcal/mol) and higher than the value of the D240I replacement in TM VII of LacY (−17.7 kcal/mol). Therefore, the barrier to lipid-dependent inversion of CscB appears to be the high hydrophobicity of TM VII, similar to the D240I replacement in TM VII of LacY (1). Attempts to further lower the hydrophobicity by replacing the CscB TM VII with the LacY TM VII were not possible due to lack of stable expression of the chimeric protein.
Testing the Lipid-Protein Charge Balance Hypothesis with CscB
Sequence comparison within related sugar permeases (supplemental Fig. S1A), including CscB and LacY, reveals a high content of negatively charged residues in the cytoplasmic face of the N-terminal six TM bundles. It was demonstrated (1) that modifying the net charge of the cytoplasmic face of this bundle in LacY by adding or removing positively charged residues in the cytoplasmic domains C2, C4, and C6 can either prevent its inversion in PE-lacking cells or force its inversion in PE-containing cells, respectively. A sequence comparison for the cytoplasmic domains C2–C6 between CscB and LacY shows that domain content of charged residues (Fig. 1, A and E) is identical in domain C2 (net charge of +2) but quite different in domain C4 (net charge of +2 for LacY versus 0 for CscB) and domain C6 (net charge of +2 for LacY versus +1 for CscB). Although the net charge of the CscB C6 domain is lower than the same domain in LacY (+1 versus +2, respectively), the CscB C6 domain contains more charged residues than found in LacY (7 basic and 6 acidic versus 4 basic and 2 acidic, respectively), making CscB more hydrophilic (+26.5 kcal/mol for LacY versus +98.1 kcal/mol for CscB). In order to differentiate between charge distribution and high hydrophobicity of TM VII in CscB as a barrier to lipid sensitivity of topology, we investigated the effects of increasing the negative charge of the cytoplasmic surface of the N-terminal TM bundle of CscB.
First a series of mutants carrying diagnostic S197C in C6 and the salt bridge mutations (N234D/S356K) were constructed, in which positive charges were replaced by neutral residues (see Table 1 for a complete list of the studied mutants). Topology of these various constructs was assessed in PE-containing and PE-lacking cells. A change in net charge (Fig. 5A and Table 2) from +2 to −1 or −2 for C6 separately (+2/0/−1 or +2/0/−2 template) or in combination with a 0 to −1 or −2 change in C4 (+2/−1/−2 or +2/−2/−2 template) had no effect on C6 orientation in both PE-containing and PE-lacking cells. Similarly, combining the changes in C6 with a change in C2 from +2 to 0 (0/0/−2 template) had no effect on orientation of C6. Interestingly, simultaneously combining changes in C2, C4, and C6 (0/−2/−2 and −2/0/−2 template) had no effect on CscB orientation in PE-containing cells but resulted in a small level of periplasmic exposure of S197C (in C6) in PE-lacking cells before sonication. The latter result suggests a possible mixture of topologies where molecules with the correct topology dominated. Combining the changes in C2, C4 and C6 (−2/−2/−2 template) resulted in a full periplasmic exposure of S197C in PE-lacking cells, suggesting the complete flipping of the C6 domain for the whole population of CscB. However, a correct topology was still observed in PE-containing cells. Therefore, a more extensive manipulation of the charge content of the CscB cytoplasmic domains C2–C6 resulted a lipid-dependent sensitivity for CscB topology. However, removing the artificial salt bridge between TMs VII and XI (reversing the mutation at residue 234) had no effect on CscB orientation in either PE-containing cells (which exhibited a correct C6 orientation) or PE-lacking cells (which exhibited C6 exposure to the periplasm) (Fig. 5A, bottom row).
TABLE 1.
Charge mutants used in CscB topology studies
The various mutations in C2, C4, and/or C6 domains are listed. Name and number of residues changed are indicated. The net charge of the resulting template is indicated in the left column.
| Net charge of C2/C4/C6 | C2 domain mutation(s) | C4 domain mutation(s) | C6 domain mutation(s) |
|---|---|---|---|
| +2/0/+1 | |||
| +2/0/−1 | K206Q/K207Q | ||
| +2/0/−2 | K206Q/K207Q/K215Q | ||
| +2/−1/−2 | K134Q | K206Q/K207Q/K215Q | |
| 0/0/−2 | K76Q/K77Q | K206Q/K207Q/K215Q | |
| 0/−2/−2 | K76Q/K77Q | K134Q/R137N | K206Q/K207Q/K215Q |
| −2/0/−2 | K72Q/L75E | K206Q/K207Q/K215Q | |
| K76Q/K77Q | |||
| +2/−2/−2 | K134Q/R137N | K206Q/K207Q/K215Q | |
| −2/−2/−2 | K72Q/L75E | K134Q/R137N | K206Q/K207Q/K215Q |
| K76Q/K77Q | |||
| −2/−2/−3 | K72Q/L75E | K134Q/R137N | K206Q/K207Q |
| K76Q/K77Q | K215Q/R217N | ||
| −2/−2/−5 | K72Q/L75E | K134Q/R137N | R189N/K191Q/K193Q |
| K76Q/K77Q | K206Q/K207Q/K215Q | ||
| R189N/K191Q/K193Q | |||
| −2/−2/−6 | K72Q/L75E | K134Q/R137N | K206Q/K207Q |
| K76Q/K77Q | K215Q/R217N |
FIGURE 5.

Effect of changes in the net charge of the N-terminal bundle cytoplasmic domains on TM orientation of CscB* in PE-containing and PE-lacking cells. A, CscB*-SB derivatives containing a single diagnostic cysteine at Ser197 (C6) with various charge replacements in the C2, C4, and C6 domains were expressed in PE-containing (+PE) and PE-lacking (−PE) cells and subjected to SCAMTM analysis (see Table 1 for specific changes made). N234 in the last lane indicates removal of the additional salt bridge with regeneration of CscB*. B, sequence alignment of LacY and CscB C6 domains showing negatively (lowercase type) and positively (boldface type) charged residues as well as the total net charge of the domains (on the left). C, effect of changes in the C6 domain charge clusters on CscB*-SB topology in PE-containing cells. CscB*-SB derivatives containing a single diagnostic cysteine at Ser197 (C6) with additional charge replacements in the C6 domain, as described in the sequence alignment above the blots, were expressed in PE-containing cells. The total net charge of the C6 domain is indicated to the left of the sequences. SCAMTM analysis with (+) or without (−) sonication is shown for CscB*-SB containing the indicated charge changes in Cluster 1 and Cluster 2 in the left panels. SCAMTM analysis for CscB* with (SB) and without (N234) the salt bridge and containing changes in Clusters 1 and 2 is shown in the right panels.
TABLE 2.
Dependence of CscB domain C6 and NT topology on domain charge nature and membrane phospholipid composition
| C2 charge naturea | C4 charge naturea | C6 charge naturea | +PE topology C6/NTb | −PE topology C6/NTb |
|---|---|---|---|---|
| +2 (2) | 0 (3) | +1 (7) | N/ND | N/ND |
| +2 (2) | 0 (3) | −1 (5) | N/ND | N/ND |
| +2 (2) | 0 (3) | −2 (4) | N/ND | N/ND |
| +2 (2) | −1 (2) | −2 (4) | N/ND | N/ND |
| +2 (2) | −2 (1) | −2 (4) | N/ND | N/ND |
| 0 (0) | 0 (3) | −2 (4) | N/ND | N/ND |
| 0 (0) | −2 (1) | −2 (4) | N/ND | M/ND |
| −2 (0) | 0 (3) | −2 (4) | N/M | M/M |
| −2 (0) | −2 (1) | −2 (4) | N/I | I/I |
| −2 (0) | −2 (1) | −5 (1) | N/ND | ND |
| −2 (0) | −2 (1) | −3 (2) | N/ND | ND |
| −2 (0) | −2 (1) | −6 (0) | I/ND | ND |
a Net charge of domain with number of positive charges shown in parentheses.
b N, normal; ND, not determined; M, mixed; I, inverted.
The difference in the response of CscB charge content changes in PE-containing and PE-lacking cells validated the importance of PE as a topological determinant for cytoplasmic domains carrying a high number of negatively charged residues. To further investigate this hypothesis, we decided to assess the conditions necessary to observe an inversion of CscB topology in the presence of PE. One of the main differences between CscB and LacY lies in their C6 domain sequence. CscB displays a shorter yet more hydrophilic sequence containing clusters of charged amino acids in the middle (already made more negative) and at both ends (Fig. 5B). The importance of the end clusters in determining topology was investigated by making the following three mutants. Each mutant carried diagnostic S197C in C6, the salt bridge mutations (N234D/S356K), positive charge neutralization (K206Q/K207Q) in the middle of C6, and alterations in the positive charge content of the end clusters in the C6 domain (see Table 1 and Fig. 5C for a complete list of the studied mutants). Positively charged residues were eliminated in Cluster 1 (RFKDKD, at the N terminus of TM VI), in Cluster 2 (KDR, at the C terminus of TM VI), or in both at the same time (Cluster 1 + 2). These mutants exhibit a net charge in C2, C4, and C6 domains of −2/−2/−5, −2/−2/−3, and −2/−2/−6, respectively.
Elimination of positive charges in either cluster led to little or no labeling of the C6 domain without cell disruption. However, the elimination of all C6 domain-positive charges resulted in the complete inversion of this domain as shown by the same extent of labeling with and without cell disruption (Fig. 5C and Table 2). The presence (SB) or absence (N234) of the additional salt bridge had no affect on the topology (Fig. 5C). Therefore, in PE-containing cells, the presence of three positive charges in the cytoplasmic face of the N-terminal bundle was sufficient for C6 to display a correct topology, whereas up to seven positive charges were required in PE-lacking cells (Table 2). These results clearly emphasize the crucial role played by PE in enhancing the retention potential of positive charges, particularly in the presence of negative charges, and are consistent with previous results with LacY. Therefore, positive residues are dominant over negative residues as topological determinants in membranes with normal phospholipid composition, but there is an attenuation of this dominance in the absence of PE (1). Changing the net charge of C6 from +1 to −6 also reduced hydrophilicity from +98.1 to +58.9 kcal/mol. This is still well above the value of +26.5 kcal/mol for LacY C6, indicating that the barrier to inversion for CscB is the hydrophobicity of TM VII and not the hydrophilicity of C6.
Topological Behavior of CscB*-SB Upstream or Downstream of C6
A diagnostic cysteine in C6 does not provide topological information on the rest of the protein. We investigated the behavior of the remainder of CscB*-SB downstream of the C6 domain in mutants, where C6 inversion was observed in PE-containing (−2/−2/−6) and PE-lacking (−2/−2/−2) cells. As demonstrated by the same biotinylation patterns in PE-containing and PE-lacking cells (Fig. 6A), CscB*-SB exhibits a normal topological disposition from C8 to the C-terminal domain in these mutants (i.e. the same as CscB* and CscB*-SB) (Fig. 4B). In this regard, CscB and LacY behave similarly because their C-terminal bundle topologies do not change when the net negative charge content in N-terminal cytoplasmic domains is increased.
FIGURE 6.

Effect of net charge changes in the N-terminal bundle on TM orientation of the CscB*-SB C-terminal bundle and NT in PE-containing and PE-lacking cells. A, CscB*-SB derivatives with charge replacements affecting domain C6 orientation and containing a single diagnostic cysteine in the indicated C or P domain of the C-terminal bundle were expressed in PE-containing (SB with C2/C4/C6, −2/−2/−6) and PE-lacking (SB with C2/C4/C6, −2/−2/−2) cells. B, CscB*-SB derivatives containing a single diagnostic cysteine at Ala10 (NT) and charge replacements in C2, C4, or C6 domains (−2/−2/−2 or −2/0/−2) were expressed in PE-containing and PE-lacking cells. SCAMTM analysis at the indicated pH with (+) or without (−) sonication is shown for the different CscB*-SB charge mutants studied. C, CscB*-SB derivatives containing a single diagnostic cysteine at Ser197 (C6) or Ala10 (NT) and charge replacements in the C2, C4, and C6 domains (−2/−2/−2) were expressed in PE-containing cells. SCAMTM analysis at the pH 9.1 with (+) or without (−) sonication is shown for the different CscB*-SB charge mutants studied. D, CscB*-SB derivatives containing a single diagnostic cysteine at Ser197 (C6) and charge replacements in C2, C4, and C6 domains (−2/0/−2 and 0/−2/−2) were expressed in PE-lacking cells. SCAMTM analysis at pH 9.1 with (+) or without (−) sonication and with (+) or without (−) preincubation with AMS is shown for the different CscB*-SB charge mutants studied.
In order to definitively establish the orientation of the whole N-terminal helical bundle, CscB*-SB containing a diagnostic A10C (NT) replacement, which flanks the N-terminal helical bundle from the end opposite to C6, was analyzed (Fig. 6B and Table 2). In PE-lacking cells, a change in C2, C4, and C6 net charge from +2/0/+1 to −2/−2/−2 for CscB*-SB resulted in a slight labeling of a cysteine located in the N-terminal domain without sonication at pH 7.4 (Fig. 5A). Such a labeling pattern can be due to a mixed orientation of the domain or reduced reactivity of the diagnostic cysteine. However, increasing the labeling pH from 7.4 to 9.1 led to a full labeling of this cysteine (Fig. 6B). It was previously shown that cell integrity is maintained at pH 9.1 and that cysteines within cytoplasmic domains are not labeled at pH 9.1 unless cells are sonicated (27); see also Fig. 6C for additional control for lack of exposure of C6 at pH 9.1. This result indicates that the weak labeling at pH 7.4 without sonication is due to a lack of accessibility of a periplasmically exposed cysteine at neutral pH, which becomes exposed at higher pH (39). Therefore, in the absence of PE, the periplasmic exposure of C6 and NT is consistent with complete inversion of the N-terminal six-TM bundle of CscB*-SB. In PE-containing cells, although almost no labeling of NT in CscB*-SB was observed at pH 7.4 (Fig. 6B, −2/−2/−2 template), a strong labeling was seen with and without cell disruption at pH 9.1 (Figs. 6B and 5C, −2/−2/−2 template). This result indicates a periplasmic localization of CscB N terminus within the −2/−2/−2 template. However, labeling of the C6 domain at pH 9.1 only occurred after sonication (Fig. 6C), consistent with a cytoplasmic location in PE-containing cells. The fact that the N-terminal bundle in PE-containing cells does not behave as a single TM bundle as observed during initial insertion and folding for LacY suggests that the two ends of the bundle display different sensitivities to the lipid environment. Therefore, it is also possible that the N terminus of CscB may be inverted in PE-lacking cells in some of the mutants where C6 displays a normal orientation. Differential sensitivity to lipid composition suggests a second flexible domain within the N-terminal bundle, which was also observed for LacY after its reorientation due to introduction of PE following assembly in the absence of PE (1).
Further investigation of CscB*-SB NT domain topology was done using the −2/0/−2 mutant, which displayed a mixed topology for the C6 domain in PE-lacking cells (Fig. 5A). In this case, topology assessment at pH 7.4 and pH 9.1 showed partial labeling of the cysteine without cell disruption (Fig. 6B) for the NT domain in both PE-containing and PE-lacking cells. Therefore, the whole N-terminal bundle in PE-lacking cells shows a mixture of topologies, whereas only the NT domain shows a mixture of topologies in PE-containing cells. This conclusion was further confirmed using topology assessment at pH 9.1 for −2/0/−2 and 0/−2/−2 C6 mutants in PE-lacking cells, which showed an apparent mixed topology for domain C6 at pH 7.4 (Fig. 5A). A mixed topology was still displayed for the C6 domain when assessed at pH 9.1 (Fig. 6D) for both mutants. Preincubation of CscB*-SB with AMS prior to MPB treatment (Fig. 6D) blocked the small amount of C6 labeling in intact PE-lacking cells, further verifying the existence of a subpopulation of CscB with domain C6 exposed to the periplasm in PE-lacking cells.
Relationship between Interfacial Lipid-Protein Charge Balance and Availability of a Hinge Region
We postulated that the requirement for the higher negative charge for cytoplasmic domains to effect a topological rearrangement of CscB in both PE-lacking and PE-containing cells may be due to the higher hydrophobicity of TM VII in CscB versus LacY. This may indeed be the case because introduction of an additional Asp into the CscB TM VII did not decrease hydrophobicity (−20.7 kcal/mol) to the level of the LacY TM VII (−5.6 kcal/mol). TM flipping of LacY appears to rely on the intrinsic structural flexibility and low hydrophobicity provided by TM VII as a molecular hinge. This hinge is necessary and sufficient for TM rearrangement in response to changes in lipid environment (1). Increasing the overall hydrophobicity of TM VII to −17.7 kcal/mol by the D240I replacement prevented TM VII from being released into the periplasm in PE-lacking cells and blocked the inversion of the N-terminal bundle of wild type LacY (C2/C4/C6, +2/+2/+2) (1). Increasing the net charge of these cytoplasmic domains to −2/−2/−2 resulted in topological inversion of the bundle in PE-containing cells with Asp240 in TM VII. However, inversion was prevented in the D240I replacement. If the translocation potential of negative residues is increased in the absence of PE, then the D240I replacement may not be able to prevent inversion of the −2/−2/−2 mutant in PE-lacking cells. This was tested using a diagnostic cysteine in the middle of cytoplasmic domain C6 (H205C) within the −2/−2/−2 LacY template carrying a D240I substitution. H205C was equally derivatized by MPB before and after membrane disruption and thus fully exposed to the periplasm of PE-lacking cells (Fig. 7, lanes 3 and 4). The same diagnostic cysteine residue in the +2/+2/+2 LacY template carrying Asp240 was also labeled by MPB whether or not PE-lacking cells were disrupted (Fig. 7, lanes 1 and 2) as expected. Therefore, increasing TM VII hydrophobicity (D240I substitution) of the −2/−2/−2 mutant was sufficient to prevent the inversion in the presence of PE (1) but not in the absence of PE. Thus, the higher translocation potential of negative residues in the absence of PE overrides the thermodynamic disadvantage of presumably exposing a more hydrophobic TM VII of LacY to the periplasm allowing inversion.
FIGURE 7.
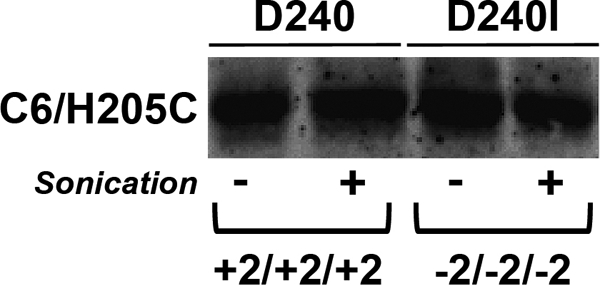
Relationship between hydrophobicity of LacY TM VII and net negative charge of the cytoplasmic surface of the N-terminal bundle of LacY expressed in PE-lacking cells. The LacY multiple charge mutant (C2/C4/C6, −2/−2/−2) with a D240I substitution in TM VII or the wild type LacY (C2/C4/C6, +2/+2/+2) with Asp240 in TM VII each with a diagnostic cysteine substitution in C6 (H205C) was expressed in PE-lacking cells. Cells were labeled with MPB either before (−) or during sonication (+) and analyzed as described under “Experimental Procedures.”
Testing the Lipid-dependent Charge Balance Hypothesis with PheP
We suggested that polytopic membrane proteins containing competing opposite charges on one side of the membrane might share a common mechanism for topogenesis (1, 14, 19). To further test this hypothesis, we extended our studies to PheP as another model protein with little homology to the sugar permeases. Sequence alignment of PheP with its four most similar homologues of the amino acid/polyamine/organocation family (supplemental Fig. S1B) reveals conservation of negative and positive residues within the first three cytoplasmic domains but with a significant departure from the positive inside rule (see Fig. 1C for PheP). In the NT domain, there are 4 positively charged residues and 3 negatively charged residues (net charge +1); in domain C2, there are 3 negatively charged residues and 2 positively charged residues (net charge is −1); in domain C4, there are 2 negatively charged residues and 2 positively charged residues (net charge is 0); and in domain C6, there are 4 negatively charged and 3 positively charged residues (net charge is −1).
Using plasmids carrying a Cys-less PheP with a single diagnostic Q50C replacement in the short periplasmic P1 domain, we made several net charge changes in NT and C2, followed by expression in PE-lacking cells and SCAMTM analysis (Fig. 8 and Table 3). As expected for the wild type PheP with respect to NT and C2 domains (Fig. 8, lanes +1/−1), a cysteine in P1 was derivatized only after sonication, consistent with previous results that established aberrant inversion of the hairpin in PE-lacking cells (17). However, neutralizing two acidic residues in NT (changing net charge from +1 to +3, lane +3/−1) or one acidic residue in C2 (changing net charge from −1 to 0, lane +1/0) resulted in mixed topology for the P1 domain with 60% in the native orientation.
FIGURE 8.

Relationship between TM orientation and net negative charge of the N-terminal hairpin of PheP expressed in PE-lacking cells. SCAMTM analysis is shown for wild type PheP (+1/−1) or PheP with a change in net charge from +1 to +3 for NT (+3/−1) or from −1 to 0 for C2 alone (+1/0) or combined changes in NT and C2 (+3/0). NT/C2 indicates the net charge of each domain, respectively. All PheP mutants contained a single cysteine replacement at position Gln50 (P1) of PheP. The approximate proportion of periplasmic exposure (%P) of each protein band was determined by the intensity of the signal minus sonication divided by the intensity of the signal after sonication with quantification using software for a Personal Molecular ImagerTM FX.
TABLE 3.
Dependence of PheP N-terminal hairpin topology on domain net charge and membrane phospholipid composition
| NT net charge | C2 net charge | +PE cell topology | −PE cell topology |
|---|---|---|---|
| +1 | −1 | Normal | Inverted |
| +3 | −1 | Normal | 60/40a |
| +1 | 0 | Normal | 60/40a |
| +3 | 0 | Normal | Normal |
a Percentage distribution between normal and inverted topology.
Failure to completely prevent misorientation may be due to the negative charge of C2. Thus, a simultaneous increase in net positive charge of NT by neutralization of two acidic residues in NT (changing net charge from +1 to +3) and one acidic residue in C2 (changing net charge from −1 to 0) resulted in almost 100% properly oriented PheP molecules (Fig. 8, lane +3/0). It is not surprising that a mixed topology was obtained, given the departure of the NT and C2 domains from the positive inside rule. However, it is clear that decreasing the negative charge density (increasing the positive charge effect) of these domains favors retention in the absence of PE, as predicted by the charge balance hypothesis.
DISCUSSION
Thus far, the dependence on membrane lipid composition (the balance between zwitterionic and anionic phospholipids) of the topological organization of three E. coli polytopic membrane proteins (LacY, PheP, and GabP) has been determined. These proteins are all solute permeases of the secondary transporter type. However, LacY and the amino acid permeases belong to different subfamilies and display very different topological patterns in the inverted state when compared with LacY. Although all cytoplasmic domains of LacY follow the positive inside rule, the presence of acidic amino acids in the cytoplasmic domains of the N-terminal six TM bundle is responsible for its topological inversion in membranes lacking PE (1). The results with LacY strongly indicate that in the absence of conformational restraints, there is a competition between the domain retention potential of positive residues and the domain translocation potential of negative residues for extramembrane domains flanking TMs. The effect of zwitterionic and neutral lipids, which dilute the surface negative charge density of the anionic lipids, is to make positive residues dominant over negative residues as topological determinants (1). An additional factor in determining lipid sensitivity of protein domains is the property of TMs that can act permissively as molecular hinges between inverted and non-inverted domains (TM VII in LacY and probably TM III in PheP and GabP) (14). TMs can also act restrictively as a thermodynamic barrier to topological inversion (TM VII of CscB as now established or TM VII of LacY containing D240I). Thus far, the interplay between the charge nature of extramembrane domains, the charge properties of the membrane lipid bilayer surface, and the effect of long range interactions as determinants of polytopic membrane protein topology has only been tested with LacY. In order to extend these principles to other proteins, the role of extramembrane domain charge distribution in determining lipid-dependent topological organization was investigated for CscB and PheP.
Although CscB uphill transport is greatly reduced in PE-lacking cells, CscB topological organization is not changed, although the cytoplasmic domains of the N-terminal six-TM bundle contain a mixture of acidic and basic amino acids. Progressively making the charge of the cytoplasmic domains less positive (i.e. more negative) resulted in an inversion of TMs first in PE-lacking and then in PE-containing cells (Table 2). Eliminating three positive charges in the C6 domain or in addition removing two positive charges from C4 was not enough to induce inversion of TMs in either cell type. However, decreasing C2 and C6 net positive charge (−2/0/−2) resulted in a mixture of topologies for domains NT and C6 in PE-lacking cells, suggesting that the whole N-terminal six-TM bundle exists with a mixture of topologies. A reduction in net positive charge in all three domains (0/−2/−2) also led to mixed topologies for domain C6 (domain NT was not probed). Further increases in the net negative charge (−2/−2/−2) resulted in complete inversion of the NT and C6 domains in PE-lacking cells. These results strongly suggest that the whole N-terminal bundle was inverted. TM inversion also was observed in PE-containing cells but in all cases only with a higher net negative charge for the cytoplasmic domains. The −2/0/−2 template showed a mixture of topologies for NT but not C6, whereas the −2/−2/−2 template showed a completely inverted NT domain with wild type orientation of the C6 domain. In order to observe inversion of C6, a higher increase in net negative charge (from −6 to −10, −2/−2/−6 template) with complete removal of positively charged residues in the C6 domain was required. Although NT orientation was not probed in this template, the fact that NT was inverted in the −2/−2/−2 template suggests that the whole N-terminal bundle was inverted in the most negative template. Interestingly, in PE-containing cells, the presence of three positive charges in the cytoplasmic face of the N-terminal bundle is sufficient for C6 to display a correct topology, whereas up to seven positive charges were required in PE-lacking cells. These results support the previous conclusion (1) that PE enhances the retention potential of positively charged residues and reduces the translocation potential of negative residues. These results provide a molecular basis for the operation of the positive inside rule for domains containing a mixture of positive and negative residues, particularly when the latter are in excess.
The fact that PheP and GabP are also sensitive to lipid composition (16, 17) and have acidic residues within cytoplasmic domains that also contain positive residues suggests a molecular basis for the requirement for PE similar to that of LacY in establishing proper topology. Cytoplasmic domains NT and C2, which are misoriented in PheP assembled in PE-lacking cells, have a net charge of +1 (with three negative residues) and −1 (with three positive residues), respectively. Thus, reversal of aberrant topology required a significant decrease in the number of negative charges in the absence of PE. Changing the net charge density to +3 in NT or 0 in C2 resulted in a mixed topology rather than a complete prevention of inversion for the N-terminal hairpin of PheP in PE-lacking cells (Fig. 8 and Table 3). Simultaneously changing the charge density of NT to +3 and C2 to 0 resulted in wild type topology of the N-terminal hairpin of PheP in PE-lacking cells. Therefore, the role of PE in suppressing the translocation of negative residues has been extended to PheP and CscB. Broader establishment of this principle provides a molecular explanation for how net negative cytoplasmic domains remain cytoplasmic and why positive charge is dominant over negative charge as a topological determinant.
Inversion of the N-terminal bundle of LacY in PE-lacking and PE-containing cells is dependent on exposure of the marginally hydrophobic TM VII to the periplasm; increasing the hydrophobicity of TM VII by a D240I mutation prevents inversion (1). TM VII of CscB does not appear to provide such a switch, probably due to its higher hydrophobicity even with an added acidic residue. In fact, its high hydrophobicity appears to desensitize CscB topology to membrane lipid composition. However, for LacY, the block to topological inversion in PE-lacking cells caused by an increase in the hydrophobicity of TM VII (D240I change in TM VII) could be suppressed in PE-lacking cells by further increasing the net negative charge of the cytoplasmic surface of the N-terminal bundle from +6 to −6 (Fig. 7). This is consistent with the progressive increase in the net negative charge of C6 resulting in a complete inversion of C6 in CscB first in PE-lacking cells and then in PE-containing cells required due to the high hydrophobicity of TM VII. Applying the above principle of thermodynamic balance to PheP also explains why TM III (miniloop that does not traverse the membrane; Fig. 1D) provides the flexible hinge in PE-lacking cells. The net charge of NT/C2/C4/C6 (+1/−1/0/0) does not follow the positive inside rule except for NT. The hydrophobicities of TMs II, III, IV, and V range from −27 to −40 kcal/mol, which should prevent any TM from acting as a hinge domain in PE-lacking cells given the near neutral charge of the flanking extramembrane domains. However, TM III is rich in aromatic residues (Fig. 1D and supplemental Fig. S1B), which are most stable in TMs when localized near the membrane-solvent interface (9). This is consistent with this domain being a miniloop that does not traverse the membrane in PE-lacking cells and thereby provides the hinge domain.
The above results with LacY, CscB, and PheP illustrate how long range interactions between local electrostatic and hydrophobic forces can override each other in the context of a given membrane protein sequence and given lipid composition. It should also be noted that the high hydrophobicity of TM VII in CscB prevents periplasmic orientation of domains that carry a net negative charge in opposition to the positive inside rule in both PE-containing and PE-lacking cells (Table 2). The importance of long range interactions in determining final topology further indicates that membrane proteins during translation exist in an uncommitted organizational state until partially or fully synthesized. Final organization is therefore determined during late folding events after exit from the translocon (1, 3, 14).
Altering the charge nature of extramembrane domains or the membrane surface charge resulted in CscB and PheP being expressed in several different topological arrangements. We observed domains completely inverted relative to other domains in normal orientation or with parts of the protein in an inverted orientation and other parts that co-existed as a mixture of two different orientations for the same domain. Some model proteins have been shown to exhibit mixed topology when protein domain charge distribution is altered (40–42), but mixed orientation has not been previously attributed to membrane lipid composition.
There are differences in membrane protein assembly between prokaryotes and eukaryotes. In eukaryotes, translation rates are slower, which might provide more time for topological reorganization. Glycosylation of extramembrane domains also occurs, which might prevent postassembly topological reorganization. These differences may thermodynamically lock proteins in organizations that prevent postassembly changes in structure as proteins move through the changing lipid composition of organelles along vesicle-mediated intracellular trafficking pathways. However, examples exist in eukaryotes where lipid-protein interactions may play a role in defining or changing topological organization. Ductin (a component of vacuolar ATPase and connexon channel subunit) (22) and epoxide hydrolase/bile acid transporter (24) are two proteins that exhibit dual topologies upon initial assembly in the endoplasmic reticulum. After each form moves to its respective target organelle, they exhibit stable unique but different topologies and functions. Results reported here provide a molecular basis for the co-existence of two topological forms of a protein dependent on protein structure and lipid environment. In yeast with severely reduced levels of PE and/or phosphatidylserine, transport of tryptophan (43), cadmium (44), arginine (45), and ferrichrome (46) is also greatly reduced. In the latter two cases, the respective permeases were trapped in the Golgi along their normal vesicular movement to the plasma membrane. A change in lipid environment in moving from the endoplasmic reticulum to the Golgi may induce misorganization of these transporters, thus affecting the availability of signals required for exit from the Golgi.
In conclusion, TM orientation depends on integration of responses to at least three parameters: net charge of the cytoplasmically exposed protein surface, charge character of the membrane surface, and availability of an unrestricted hinge region between domains with different responses to the lipid composition (14). This report extends to additional proteins the role net neutral charged lipids play in suppressing negative amino acids as topological determinants in favor of positive residues. These results provide a molecular basis for the positive inside rule and the dominance of positive residues over negative residues. In addition, the importance of long range interactions in establishing final topological organization of polytopic membrane proteins was more fully illustrated with CscB and LacY. Finally, the co-existence of dual topological organization was shown to be dependent on protein sequence and lipid environment. Thus far, only solute permeases in E. coli have been demonstrated to require PE for proper topology and function. However, PE-lacking cells show defects in cell division, display filamentous growth, require high concentrations of divalent cations in the growth medium, and have increased permeability of the outer membrane (47). Therefore, other proteins may require PE for native structure and function, suggesting that the presence of lipid-responsive topogenic elements is a widespread phenomenon.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant GM R37 20478 (to W. D.). This work was also supported by a grant from the John Dunn Research Foundation (to W. D.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- TM
- transmembrane domain
- PE
- phosphatidylethanolamine
- LacY
- lactose permease
- PheP
- phenylalanine permease
- CscB
- sucrose permease
- GabP
- γ-aminobutyric acid permease
- MPB
- 3-(N-maleimidylpropionyl) biocytin
- AMS
- 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid
- SCAMTM
- substituted cysteine accessibility method applied to TM orientation
- CscB*
- sucrose permease lacking native cysteine residues and containing a C-terminal dodecapeptide derived from LacY
- CscB*-SB
- CscB* with an engineered salt bridge.
REFERENCES
- 1. Bogdanov M., Xie J., Heacock P., Dowhan W. (2008) J. Cell Biol. 182, 925–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Monné M., Hessa T., Thissen L., von Heijne G. (2005) FEBS J. 272, 28–36 [DOI] [PubMed] [Google Scholar]
- 3. Seppälä S., Slusky J. S., Lloris-Garcerá P., Rapp M., von Heijne G. (2010) Science 328, 1698–1700 [DOI] [PubMed] [Google Scholar]
- 4. von Heijne G., Gavel Y. (1988) Eur. J. Biochem. 174, 671–678 [DOI] [PubMed] [Google Scholar]
- 5. Goder V., Junne T., Spiess M. (2004) Mol. Biol. Cell. 15, 1470–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Junne T., Kocik L., Spiess M. (2010) Mol. Biol. Cell. 21, 1662–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Skach W. R. (2009) Nat. Struct. Mol. Biol. 16, 606–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bogdanov M., Heacock P., Guan Z., Dowhan W. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 15057–15062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. White S. H., von Heijne G. (2005) Biochem. Soc. Trans. 33, 1012–1015 [DOI] [PubMed] [Google Scholar]
- 10. Granseth E., von Heijne G., Elofsson A. (2005) J. Mol. Biol. 346, 377–385 [DOI] [PubMed] [Google Scholar]
- 11. Lee E., Manoil C. (1994) J. Biol. Chem. 269, 28822–28828 [PubMed] [Google Scholar]
- 12. von Heijne G. (1989) Nature 341, 456–458 [DOI] [PubMed] [Google Scholar]
- 13. Bogdanov M., Heacock P. N., Dowhan W. (2002) EMBO J. 21, 2107–2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dowhan W., Bogdanov M. (2009) Annu. Rev. Biochem. 78, 515–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang X., Bogdanov M., Dowhan W. (2002) EMBO J. 21, 5673–5681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang W., Bogdanov M., Pi J., Pittard A. J., Dowhan W. (2003) J. Biol. Chem. 278, 50128–50135 [DOI] [PubMed] [Google Scholar]
- 17. Zhang W., Campbell H. A., King S. C., Dowhan W. (2005) J. Biol. Chem. 280, 26032–26038 [DOI] [PubMed] [Google Scholar]
- 18. Rutz C., Rosenthal W., Schülein R. (1999) J. Biol. Chem. 274, 33757–33763 [DOI] [PubMed] [Google Scholar]
- 19. Bogdanov M., Xie J., Dowhan W. (2009) J. Biol. Chem. 284, 9637–9641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wikström M., Xie J., Bogdanov M., Mileykovskaya E., Heacock P., Wieslander A., Dowhan W. (2004) J. Biol. Chem. 279, 10484–10493 [DOI] [PubMed] [Google Scholar]
- 21. Wikström M., Kelly A. A., Georgiev A., Eriksson H. M., Klement M. R., Bogdanov M., Dowhan W., Wieslander A. (2009) J. Biol. Chem. 284, 954–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dunlop J., Jones P. C., Finbow M. E. (1995) EMBO J. 14, 3609–3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. von Heijne G. (2006) Nat. Rev. Mol. Cell Biol. 7, 909–918 [DOI] [PubMed] [Google Scholar]
- 24. Zhu Q., von Dippe P., Xing W., Levy D. (1999) J. Biol. Chem. 274, 27898–27904 [DOI] [PubMed] [Google Scholar]
- 25. Sahin-Tóth M., Frillingos S., Lawrence M. C., Kaback H. R. (2000) Biochemistry 39, 6164–6169 [DOI] [PubMed] [Google Scholar]
- 26. Frillingos S., Sahin-Tóth M., Wu J., Kaback H. R. (1998) FASEB J. 12, 1281–1299 [DOI] [PubMed] [Google Scholar]
- 27. Bogdanov M., Heacock P. N., Dowhan W. (2010) Methods Mol. Biol. 619, 79–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DeChavigny A., Heacock P. N., Dowhan W. (1991) J. Biol. Chem. 266, 5323–5332 [PubMed] [Google Scholar]
- 29. Bogdanov M., Zhang W., Xie J., Dowhan W. (2005) Methods 36, 148–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bogdanov M., Sun J., Kaback H. R., Dowhan W. (1996) J. Biol. Chem. 271, 11615–11618 [DOI] [PubMed] [Google Scholar]
- 31. Saier M. H., Jr., Tran C. V., Barabote R. D. (2006) Nucleic Acids Res. 34, D181–D186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Engelman D. M., Steitz T. A., Goldman A. (1986) Annu. Rev. Biophys. Biophys. Chem. 15, 321–353 [DOI] [PubMed] [Google Scholar]
- 33. Guo D., Liu J., Motlagh A., Jewell J., Miller K. W. (1996) J. Biol. Chem. 271, 30829–30834 [DOI] [PubMed] [Google Scholar]
- 34. Soekarjo M., Eisenhawer M., Kuhn A., Vogel H. (1996) Biochemistry 35, 1232–1241 [DOI] [PubMed] [Google Scholar]
- 35. Steitz T. A., Goldman A., Engelman D. M. (1982) Biophys. J. 37, 124–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Long J. C., Wang S., Vik S. B. (1998) J. Biol. Chem. 273, 16235–16240 [DOI] [PubMed] [Google Scholar]
- 37. Loo T. W., Clarke D. M. (1995) J. Biol. Chem. 270, 843–848 [DOI] [PubMed] [Google Scholar]
- 38. Wada T., Long J. C., Zhang D., Vik S. B. (1999) J. Biol. Chem. 274, 17353–17357 [DOI] [PubMed] [Google Scholar]
- 39. Xie J., Bogdanov M., Heacock P., Dowhan W. (2006) J. Biol. Chem. 281, 19172–19178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Beltzer J. P., Fiedler K., Fuhrer C., Geffen I., Handschin C., Wessels H. P., Spiess M. (1991) J. Biol. Chem. 266, 973–978 [PubMed] [Google Scholar]
- 41. Nilsson I., von Heijne G. (1990) Cell 62, 1135–1141 [DOI] [PubMed] [Google Scholar]
- 42. van Klompenburg W., Nilsson I., von Heijne G., de Kruijff B. (1997) EMBO J. 16, 4261–4266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nakamura H., Miura K., Fukuda Y., Shibuya I., Ohta A., Takagi M. (2000) Biosci. Biotechnol. Biochem. 64, 167–172 [DOI] [PubMed] [Google Scholar]
- 44. Gulshan K., Shahi P., Moye-Rowley W. S. (2010) Mol. Biol. Cell. 21, 443–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Opekarová M., Robl I., Tanner W. (2002) Biochim. Biophys. Acta. 1564, 9–13 [DOI] [PubMed] [Google Scholar]
- 46. Guo Y., Au W. C., Shakoury-Elizeh M., Protchenko O., Basrai M., Prinz W. A., Philpott C. C. (2010) J. Biol. Chem. 285, 39564–39573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dowhan W. (2009) J. Lipid Res. 50, S305–S310 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.