

Abstract
Voltage-dependent Ca2+ (CaV1.2) channels are the primary Ca2+ influx pathway in arterial smooth muscle cells and are essential for contractility regulation by a variety of stimuli, including intravascular pressure. Arterial smooth muscle cell CaV1.2 mRNA is alternatively spliced at exon 1 (e1), generating e1b or e1c variants, with e1c exhibiting relatively smooth muscle-specific expression in the cardiovascular system. Here, we examined physiological functions of CaV1.2e1 variants and tested the hypothesis that targeting CaV1.2e1 modulates resistance size cerebral artery contractility. Custom antibodies that selectively recognize CaV1.2 channel proteins containing sequences encoded by either e1b (CaV1.2e1b) or e1c (CaV1.2e1c) both detected CaV1.2 in rat and human cerebral arteries. shRNA targeting e1b or e1c reduced expression of that CaV1.2 variant, induced compensatory up-regulation of the other variant, decreased total CaV1.2, and reduced intravascular pressure- and depolarization-induced vasoconstriction. CaV1.2e1b and CaV1.2e1c knockdown reduced whole cell CaV1.2 currents, with CaV1.2e1c knockdown most effectively reducing total CaV1.2 and inducing the largest vasodilation. Knockdown of α2δ-1, a CaV1.2 auxiliary subunit, reduced surface expression of both CaV1.2e1 variants, inhibiting CaV1.2e1c more than CaV1.2e1b. e1b or e1c overexpression reduced CaV1.2 surface expression and whole cell currents, leading to vasodilation, with e1c overexpression inducing the largest effect. In summary, data indicate that arterial smooth muscle cells express CaV1.2 channels containing e1b or e1c-encoded N termini that contribute to CaV1.2 surface expression, α2δ-1 preferentially traffics the CaV1.2e1c variant to the plasma membrane, and targeting of CaV1.2e1 message or the CaV1.2 channel proximal N terminus induces vasodilation.
Keywords: Calcium, Calcium Channels, Membrane, shRNA, Smooth Muscle, Myogenic Tone
Introduction
Voltage-dependent L-type Ca2+ (CaV1.2)2 channels are hetero-oligomeric protein complexes consisting of a pore-forming CaV1.2 α1 and regulatory α2δ, β, and γ subunits (1–3). CaV1.2 channels provide the major Ca2+ entry pathway in vascular smooth muscle cells (4–6). Ca2+ influx via Cav1.2 channels regulates a wide variety of different smooth muscle functions, including contractility and gene expression (7, 8). Cav1.2 channels are essential for intravascular pressure-induced vasoconstriction (myogenic tone) in small, resistance size arteries and arterioles that regulate blood pressure and organ blood flow (9, 10). Systemic hypertension is associated with an elevation in arterial CaV1.2 protein and larger smooth muscle cell L-type Ca2+ channel currents (11, 12). Voltage-dependent Ca2+ channel blockers inhibit vascular smooth cell CaV1.2 channels, leading to vasodilation. Therefore, CaV1.2 channel blockers are a commonly used antihypertensive therapy (13).
The CaV1.2 subunit gene (CACNA1C) is subject to alternative splicing, which generates structural and functional diversity in channel proteins (14–16). Of 55 exons present in the human CaV1.2 gene, 19 are subject to alternative splicing. This provides a large potential variance in channel amino acid sequences (17–19). Splicing of the CaV1.2 gene in arterial smooth muscle cells of resistance size arteries is more conservative and restricted to three mutually exclusive (exons 1b/c, 21/22, and 31/32) and two alternate (exons 9* and 33) exons (15, 16). Such splice variation can lead to modifications in recombinant channel voltage dependence and membrane insertion by auxiliary subunits (15, 16, 18, 20). Previous studies detected CaV1.2 exon 1b and 1c mRNA in cardiac and cerebral artery smooth muscle cells (15). CaV1.2 exon 1c was the primary CaV1.2 mRNA variant in cerebral artery smooth muscle cells, with exon 1a mRNA absent (15). Cell-specific CaV1.2 splice variation may provide a means by which to selectively target these channels to modify physiological functions. However, although CaV1.2 exon 1 mRNA splicing has been described and the effects on current properties have been studied using recombinant proteins, it is unclear whether CaV1.2 proteins encoded by these splice variants exist in native cells. Furthermore, physiological functions of CaV1.2 exon 1 variants are unclear. Given that arterial smooth muscle cell CaV1.2 channels are a major regulator of vascular contractility, and these channels are associated with vascular pathologies, it is important to investigate physiological functions of splice variants and determine whether targeting these variants modifies vascular contractility. Here, we examined the expression and physiological functions of exon 1-encoded CaV1.2 channel variants in smooth muscle cells of small (<250-μm diameter) cerebral arteries and determined the potential for exon 1 and CaV1.2 N-terminal targeting to modify arterial contractility.
Our data indicate that cerebral artery smooth muscle cell CaV1.2 channels contain either exon 1b (CaV1.2e1b) or exon 1c (CaV1.2e1c) encoded N termini. Both of these endogenous CaV1.2 channel N-terminal variants are located primarily in the plasma membrane. shRNA targeting exon 1b or 1c selectively reduced the expression of each variant, leading to vasodilation. Overexpression of exon 1b or 1c peptide sequences did not alter total CaV1.2 channel expression but inhibited CaV1.2 surface trafficking, leading to vasodilation. Our data also indicate that α2δ-1 inserts both CaV1.2 N-terminal variants into the arterial smooth muscle cell plasma membrane but preferentially inserts CaV1.2e1c channels. These data identify physiological functions of native arterial smooth muscle cell CaV1.2e1 variants and indicate that exon 1 targeting induces cerebral artery vasodilation.
EXPERIMENTAL PROCEDURES
Antibody Generation and Dot Blots
Antibodies were raised to the previously described peptide sequences encoded by exon 1b (MVNENTRMYVPEENHQG, anti-e1b) or exon 1c (MLCCALDCAC, anti-e1c) (15) in guinea pig (Genmed Synthesis Inc., San Francisco CA) and chicken (Covance, Denver, PA), respectively. Chicken IgY protein was further purified from egg yolks in our laboratory using an Eggstract IgY purification system (Promega, Madison, WI). Commercially available rabbit anti-α2δ-1 (Aviva Systems Biology) and rabbit anti-CaV1.2, raised to a conserved sequence (TTKINMDDLQPSENEDKS) in the intracellular loop between repeats II and III (Alomone Labs), were also used.
Antigenic peptides that were used to raise anti-e1b and anti-e1c antibodies were spotted at 0.01, 0.05, and 0.1 μg onto nitrocellulose membranes and probed with anti-e1b or anti-e1c antibodies. Reactivity was determined by chemiluminescence (Pierce) from HRP-conjugated secondary antibodies.
Cell Isolation and Tissue Preparation
Animal protocols used were reviewed and approved by the Animal Care and Use Committee at the University of Tennessee Health Science Center. Male Sprague-Dawley rats (∼250 g) were euthanized by intraperitoneal injection of sodium pentobarbital solution (150 mg/kg). The brain was removed, and resistance size cerebral arteries (posterior cerebral, cerebellar, and middle cerebral, <250 μm) were harvested in physiological saline solution containing 5 mm KCl, 112 mm NaCl2, 24 mm NaHCO3, 1.2 mm MgSO4, 1.2 mm KH2PO4, 1.8 mm CaCl2, and 10 mm glucose.
Cell Culture and Transient Transfection
HEK293 cells (ATCC) were transfected with full-length pIRES-CaV1.2e1b-hrGFPII or pIRES-CaV1.2e1c-hrGFPII and pcDNA3.1-α2δ-1 and pGW-β1b subunits using the Ca2+ phosphate method. The cells were maintained in DMEM (Cellgro, Manassas, VA) supplemented with 10% FBS and 1% penicillin-streptomycin in culture (21% O2, 5% CO2 at 37 °C) and used for biochemical experiments 32–72 h after transfection. Transfected cell lysates were obtained in radioimmune precipitation assay buffer (Sigma) prior to analysis by Western blotting. pcDNA3.1-α2δ-1 was kindly provided by Dr. Diane Lipscomb (Brown University) and pGW-β1b by Dr. Henry Colecraft (John Hopkins University School of Medicine).
Protein Analysis and Biochemistry
Experiments were performed using HEK293 cell lysate, human cerebral artery lysate, or rat cerebral artery lysate. A live human cerebral cortex sample was obtained through National Disease Research Interchange (Philadelphia, PA) using an institutional review board-exempt protocol. Human cerebral arteries were manually dissected from cortex using forceps. Rat cerebral arteries were obtained from 1–2 animals/sample. Human and rat arteries were homogenized in 1× Laemmli buffer containing 2% β-mercaptoethanol to obtain whole arterial lysate. HEK293 cell lysate was obtained using radioimmune precipitation assay buffer. Laemmli buffer containing 2% β-mercaptoethanol was added to a final concentration of 1×. Cellular debris was removed by centrifugation. Protein concentrations were determined using the method of Henkel and Bieger (21). The proteins were separated using SDS-PAGE and analyzed by Western blotting. Antibodies used were anti α2δ-1 (Aviva Systems Biology), CaV1.2 (Alomone), anti-e1b, anti-e1c, and actin (Millipore). The bands were visualized on a Kodak Image F-Pro system using a West Pico Chemiluminescence kit (Pierce). For quantification, the protein band intensities were normalized to actin and then to control samples.
Knockdown Using shRNA
shRNA sequences designed to CaV1.2 exon 1b (GGATGTACGTTCCAGAGGAAA) or CaV1.2 exon1c (CTCTGCTGTGCTCTGGACTGT) were inserted into the pRNA6.1/Neo vector (Genscript, Piscataway, NJ). shRNA sequences designed to recognize α2δ-1 were the same as those described previously (22). A control vector encoding a scrambled sequence (scrmV, TGCGCGTCCGTTGGTCTCTTA) was also used. Vectors encoding shRNA sequences were inserted into rat cerebral arteries using reverse permeabilization (23). Arteries were then placed into DMEM/F-12 supplemented with 1% penicillin-streptomycin in a sterile culture incubator (21% O2, 5% CO2 at 37 °C) for 4 days, as described previously (24).
Surface Biotinylation
The cellular distribution of α2δ-1 and CaV1.2 subunit proteins in intact cerebral arteries was determined using arterial surface biotinylation. Arteries were incubated for 1 h in a 1 mg/ml mixture each of EZ-Link Sulfo-NHS-LC-LC-Biotin and EZ-Link Maleimide-PEG2-Biotin reagents (Pierce) in PBS (Invitrogen). Unbound biotin was removed by quenching using 100 mm glycine and washing with PBS.
For protein determination, biotinylated arteries were homogenized in radioimmune precipitation assay buffer (Sigma), and cellular debris removed by centrifugation. The supernatant comprised the total protein lysate. Total protein concentration was then determined using the method of Henkel and Bieger (21) to allow normalization for avidin (monomeric avidin; Pierce) pull-down of biotinylated surface proteins. Following pulldown, the supernatant comprised the nonbiotinylated (cytosolic) protein fraction, while surface proteins remained bound to the avidin beads. Proteins were eluted from beads by boiling in 1× Laemmli buffer containing 2-mercaptoethanol (2%). Total, surface, and cytosolic proteins were analyzed using Western blotting. Band intensity was determined using Quantity One software (Bio-Rad).
Patch Clamp Electrophysiology
Smooth muscle cells were enzymatically dissociated from cerebral arteries, as described previously (25). Whole cell patch clamp recordings were acquired at room temperature using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA) and pCLAMP 8.2. Borosilicate glass electrodes of resistance 3–6 MΩ were filled with pipette solution containing 135 mmol/liter CsMeSO4, 5 mmol/liter CsCl, 5 mmol/liter EGTA, 4 mmol/liter MgATP, 0.25 mmol/liter NaGTP, 10 mmol/liter HEPES, and 10 mmol/liter glucose (pH 7.2, with CsOH). The extracellular bath solution contained 140 mmol/liter N-methyl-d-glucamine, 1 mmol/liter MgCl2, 10 mmol/liter HEPES, 20 mmol/liter BaCl2, and 10 mmol/liter glucose (pH 7.4, adjusted with aspartate). Isolated cells not visibly attached to neighboring cells were used for patch clamp. Cell capacitance was measured by applying a 5-mV test pulse and correcting transients with series resistance compensation. For measurement of current-voltage (I-V) relationships, the cells were clamped at −80 mV, and whole cell currents were evoked every 5 s by 300-ms step depolarizations to between −80 and +40 mV in 10-mV increments. Whole cell currents were filtered at 1 kHz and digitized at 5 kHz. P/4 protocols were used to subtract leak and capacitive transients.
Pressurized Artery Myography
Where applicable, the endothelium was denuded, as described previously (26). The diameter of pressurized cerebral arteries was measured by edge detection myography (26). Myogenic tone (%) was calculated as 100 × (1 − active diameter/passive diameter).
Confocal Imaging
Nucleotide sequences encoding exon 1b or 1c were inserted downstream of EGFP in the pEGFP-N3 vector (Loftstrand Labs, Gaithersburg, MD). EGFP-tagged exon 1b (e1b-EGFP) and exon 1c (e1c-EGFP) constructs were inserted into cerebral arteries using reverse permeabilization (23). Control arteries were mock permeabilized or permeabilized with empty pEGFP-N3 vector as indicated. The arteries were placed into DMEM/F-12 supplemented with 1% penicillin-streptomycin in a sterile culture incubator (21% O2, 5% CO2 at 37 °C). After 2–4 days, the arteries were mounted on rectangular glass cannulae (220 × 40 μm). EGFP was excited at 488 nm, and emission was collected at 510 nm using a Zeiss LSM 510 Pascal laser-scanning confocal microscope.
Statistical Analysis
Summary data are presented as the means ± S.E. Significance was determined using analysis of variance followed by Student-Newmann-Keul's post hoc test for multiple groups or paired or unpaired t tests with Welsh correction. p < 0.05 was considered significant. Where p > 0.05, power analysis was performed to verify that sample size gave a value >0.8.
RESULTS
CaV1.2e1b and CaV1.2e1c Proteins Are Expressed in Rat and Human Resistance Size Cerebral Arteries
Rat cerebral artery smooth muscle cells express two different CaV1.2 mRNA variants spliced at exon 1, termed CaV1.2e1b and CaV1.2e1c (15, 16). Whether CaV1.2e1b and CaV1.2e1c proteins are present in arterial smooth muscle cells is unclear. Custom antibodies (anti-e1b and anti-e1c) were raised to each CaV1.2 exon 1-encoded N-terminal amino acid sequence. The specificity of each antibody was confirmed using a dot blot of antigenic peptides and Western blot analysis of lysates from HEK293 cells transfected with cDNA encoding either CaV1.2e1b or CaV1.2e1c together with α2δ-1 and β1b subunits. Anti-e1b and anti-e1c antibodies selectively detected their respective antigenic peptide (Fig. 1A). Anti-e1b and anti-e1c antibodies also detected recombinant proteins expressed in HEK293 cells with molecular masses of ∼240 and 190 kDa, consistent with long and short CaV1.2 (Fig. 1B). Anti-e1b and anti-e1c antibodies selectively detected CaV1.2e1b and CaV1.2e1c subunit proteins, respectively, and did not detect the other splice variant (Fig. 1B). Anti-e1b and anti-e1c antibodies each detected ∼240- and 190-kDa bands in lysate from resistance size (<250-μm diameter) cerebral arteries (Fig. 1C). A commercially available antibody raised against an amino sequence encoded by exon 18, a site that does not undergo splicing, detected proteins of identical molecular mass in cerebral artery lysate (Fig. 1C). In summary, these data indicate that CaV1.2e1b and CaV1.2e1c proteins are expressed in rat cerebral arteries.
FIGURE 1.

Rat and human cerebral arteries express CaV1.2e1b and CaV1.2e1c proteins. A, antibodies to exon 1b and 1c only recognized their respective antigenic peptides spotted onto nitrocellulose membrane. The amount of peptide spotted is indicated to the left. B, Western blot showing detection of CaV1.2e1b and CaV1.2e1c subunit protein in lysates from transfected HEK293 cells. Mock (left lane) showed no reactivity to anti-CaV1.2, anti-e1b, or anti-e1c antibodies. The blot is representative of four experiments. C, Western blot illustrating detection of CaV1.2e1b and CaV1.2e1c subunit protein in lysates from cerebral arteries. The blot is representative of five experiments. D, bands corresponding to CaV1.2e1b and CaV1.2e1c subunit protein were detected in lysates from human cerebral arteries.
Using the same custom antibodies, expression of Cav1.2 subunit N-terminal variants was examined in human small (<250-μm diameter) cerebral arteries. The anonymous donor was a 53-year-old Caucasian male with no history of hypertension. Both the CaV1.2e1b and CaV1.2e1c antibody detected CaV1.2 protein in human cerebral artery lysate (Fig. 1D). These data suggest that CaV1.2 splice variants containing amino acid sequences similar to those encoded by exon 1b and 1c are present in human cerebral arteries.
shRNA Targeting Exon 1b or 1c Induces Selective Knockdown of CaV1.2 α1 Subunit N-terminal Variants
To study physiological functions of CaV1.2e1b and CaV1.2e1c in arterial smooth muscle cells, we used shRNA to suppress expression of each splice variant. Silencing vectors encoding shRNA targeting either exon 1b (e1bshV) or exon 1c (e1cshV) nucleotide sequences or a vector encoding a control scrambled shRNA (scrmV) were inserted intracellularly into cerebral arteries using reverse permeabilization (23).
e1bshV and e1cshV reduced total arterial CaV1.2 subunit protein by ∼20 and 42%, respectively, when compared with scrmV (Fig. 2, A and B). Combining e1bshV and e1cshV further reduced total arterial CaV1.2 protein by ∼73%, when compared with scrmV (Fig. 2, A and B). Reprobing blots with N-terminal variant-specific antibodies revealed that e1bshV reduced CaV1.2e1b protein by ∼35% but increased CaV1.2e1c protein by ∼30%, when compared with scrmV. e1cshV reduced CaV1.2e1c protein by ∼65% but increased CaV1.2e1b protein by ∼18% when compared with scrmV (Fig. 2, A and B). These data indicate that shRNA against exon 1b or 1c reduces expression of the targeted CaV1.2 variant, induces compensatory up-regulation of the other variant, and reduces total CaV1.2 in cerebral arteries.
FIGURE 2.

shRNA targeting exon 1b or 1c selectively reduces arterial CaV1.2e1b and CaV1.2e1c subunit protein and inhibits smooth muscle cell CaV1.2 currents. A, representative Western blot showing results of reprobing lysates from scrmV (control), e1bshV-treated, and e1cshV-treated cerebral arteries for total CaV1.2, CaV1.2e1b, and CaV1.2e1c subunits and actin proteins. B, bar graph illustrating mean data for total CaV1.2, CaV1.2e1b, and CaV1.2e1c subunit protein in cerebral arteries that were treated with e1bshV (black bars), e1cshV (white bars), or a combination of e1bshV and e1cshV (gray bar) relative to scrmV (n = 4–5). C, left panel, representative whole cell voltage-dependent Ba2+ currents measured in cerebral artery smooth muscle cells isolated from arteries treated with scrmV (upper panel) or a combination of e1bshV and e1cshV (lower panel). Right panel, current-voltage relationships of voltage-dependent Ba2+ currents from scrmV (filled symbols) and e1bshV- and e1cshV-treated (open symbols) cerebral artery smooth muscle cells (n = 4 for each). *, significance compared with scrmV (p < 0.05); **, significance compared with e1bshV; #, significance compared with e1cshV (p < 0.05).
CaV1.2 Exon 1b and 1c Knockdown Reduces CaV1.2 Current Density in Arterial Myocytes
To validate the effects of exon 1 knockdown, voltage-dependent Ca2+ currents (Ba2+ as charge carrier) were measured from myocytes isolated from arteries treated with either scrmV or a combination of e1bshV and e1cshV. Combining e1bshV and e1cshV reduced mean CaV1.2 current density from 14.2 ± 0.2 pA/pF (scrmV) to 4.1 ± 1.2 pA/pF, or by ∼72% (Fig. 2C). This attenuation is similar to the ∼73% reduction in total CaV1.2 channel protein that occurred in arteries treated with both e1bshV and e1cshV (Fig. 2B).
CaV1.2 Exon 1b or 1c Knockdown Attenuates Myogenic Tone
The functional significance of targeting CaV1.2 exon 1b or 1c was studied by measuring arterial contractility. Endothelium-denuded cerebral arteries were pressurized to 20 and 60 mm Hg. Pressure-induced vasoconstriction (myogenic tone), depolarization-induced vasoconstriction, induced by an elevation in extracellular K+ (60 mm), and responses to nimodipine, a CaV1.2 channel blocker, were measured. Control (scrmV) arteries developed ∼13% myogenic tone at 20 mm Hg and ∼24% tone at 60 mm Hg (Fig. 3). 60 mm K+ constricted control arteries to ∼40% of passive diameter (Fig. 3). Arteries treated with e1bshV developed ∼33 and ∼34% less tone at 20 and 60 mm Hg, respectively, and the response to 60 mm K+ was reduced by ∼34% (Fig. 3). e1cshV reduced myogenic tone at 20 and 60 mm Hg by ∼43 and ∼57%, respectively, and attenuated the response to 60 mm K+ by ∼58% (Fig. 3). Nimodipine (1 μm) fully dilated pressurized arteries that had been treated with scrmV, e1bshV, or e1cshV, indicating that myogenic tone occurred because of Cav1.2 channel activation (Fig. 3). These data indicate that knockdown of either CaV1.2e1b or CaV1.2e1c reduces intravascular pressure- and depolarization-induced vasoconstriction.
FIGURE 3.

shRNA targeting CaV1.2e1b or e1c reduces myogenic tone in pressurized cerebral arteries. A, traces illustrating representative steady state myogenic tone and vasodilation to nimodipine (1 μm) and removal of bath Ca2+ at 60 mm Hg in arteries treated with scrmV, e1bshV, or e1cshV as indicated. B, bar graph summarizing mean data for steady state myogenic tone obtained at 20 and 60 mm Hg and in the presence of 60 mm K+ in scrmV-treated (black bars), e1bshV-treated (white bars), or e1cshV-treated (gray bars) cerebral arteries. *, significance compared with scrmV; #, significance compared with e1bshV-treated arteries (p < 0.05, n = 5–9).
α2δ-1 Knockdown Attenuates Plasma Membrane Insertion of CaV1.2 Subunits Containing Exon 1b- or 1c-encoded N Termini in Cerebral Artery Smooth Muscle Cells
α2δ-1 inserts CaV1.2 channels into the cerebral artery smooth muscle cell plasma membrane (22). CaV1.2 exon 1 splicing modifies membrane insertion of recombinant channels by α2δ-1 when these proteins are expressed in HEK293 and COS-1 cells (15). Co-expression of β1b normalizes this splice variant-dependent difference (15). Given that arterial smooth muscle cells express multiple β subunit isoforms and that the ratio of α2δ-1 to β subunits is unclear, we sought to examine whether α2δ-1 differentially regulates trafficking of endogenous CaV1.2 splice variants (7, 27–29). Therefore, we examined the effects of α2δ-1 knockdown on plasma membrane insertion of CaV1.2 N-terminal splice variants in cerebral arteries.
Silencing vectors encoding shRNA specific to α2δ-1 (α2δ-1shV) were used to knock down α2δ-1 in cerebral arteries (22). Plasma membrane and intracellular localization of endogenous CaV1.2 subunit N-terminal splice variants and α2δ-1 protein was determined using arterial surface biotinylation (22). α2δ-1shV reduced total α2δ-1 protein by ∼40% and increased total CaV1.2 protein by ∼48% when compared with scrmV control (Fig. 4, A and B). α2δ-1shV also reduced surface localization of α2δ-1 and total CaV1.2 proteins by ∼47 and ∼37%, respectively, when compared with scrmV (Fig. 4C). CaV1.2 subunit N terminus-specific antibodies revealed that α2δ-1shV increased total CaV1.2e1b subunit protein by ∼20% but reduced surface localization of CaV1.2e1b subunit protein by ∼14% (Fig. 4, A–C). α2δ-1shV also increased total CaV1.2e1c subunit protein by ∼33% but reduced surface localization by ∼43% (Fig. 4, A–C). Consistent with these data, α2δ-1shV increased cytoplasmic CaV1.2, CaV1.2e1b, and CaV1.2e1c, when compared with scrmV control (Fig. 4D). These data indicate that α2δ-1 subunits insert both endogenous CaV1.2 N-terminal variants into the cerebral artery smooth muscle cell plasma membrane but preferentially insert the CaV1.2e1c variant.
FIGURE 4.

Knockdown of α2δ-1 attenuates plasma membrane insertion of CaV1.2e1b and CaV1.2e1c channels. A, representative Western blots illustrating total, intracellular, and surface CaV1.2, CaV1.2e1b, CaV1.2e1c, and α2δ-1 proteins and effects of α2δ-1 knockdown. B–D, bar graphs show changes in α2δ-1 (black bars), CaV1.2, (white bars), CaV1.2e1b (light gray bars), and CaV1.2e1c (dark gray bars) subunit total protein (B), surface protein (C), and cytosolic protein (D) levels in α2δ-1shV-treated cerebral arteries relative to scrmV-treated arteries (n = 4 for each). *, significance compared with scrmV (p < 0.05); #, significance compared with CaV1.2e1b (p < 0.05).
CaV1.2 Exon 1 Overexpression Reduces Surface Trafficking of CaV1.2 Channels
The data indicate that exon 1 splice variation alters CaV1.2 channel plasma membrane insertion. Therefore, we investigated the functional effects of inducing an elevation in CaV1.2 subunit N termini. Vectors encoding either exon 1b or 1c tagged to EGFP (e1b-EGFP and e1c-EGFP) were constructed and inserted into cerebral arteries using reverse permeabilization. To verify that exon 1b and 1c were expressed, transfected arteries were imaged using confocal microscopy. EGFP fluorescence was clearly visible in smooth muscle cells of arteries transfected with e1b-EGFP or e1c-EGFP but was absent in mock transfected control arteries (Fig. 5A). To determine expression levels of e1b-GFP and e1c-GFP in cerebral arteries, the lysates were probed with a GFP antibody. e1b-EGFP expression and e1c-EGFP expression were similar (e1c-GFP protein was 103 ± 3% of e1b-EGFP, n = 4, p > 0.05; Fig. 5B). Furthermore, e1b-EGFP or e1c-EGFP expression did not alter CaV1.2 channel protein (e1c-EGFP and e1b-EGFP were 113 ± 15 and 114 ± 8% of control (pEGFP-N3 vector), respectively; n = 6 for each, p > 0.05; Fig. 5B).
FIGURE 5.

Overexpression of exon 1b or 1c in cerebral arteries. A, confocal images of mock treated arteries (control) or arteries in which vectors expressing e1b-EGFP or e1c-EGFP were inserted illustrating EGFP fluorescence in smooth muscle cells. EGFP, Differential Interference Contrast (DIC) and merged images are shown. The scale bar represents 50 μm. B, representative Western blots showing detection of EGFP in e1b-EGFP- and e1c-EGFP-treated but not in mock treated (control) cerebral artery lysates. The Western blots also indicate that e1b-EGFP and e1c-EGFP expression does not alter CaV1.2 channel expression. The blots are representative of four experiments.
Next, using arterial biotinylation, we tested the hypothesis that exon 1b or 1c overexpression alters plasma membrane localization of CaV1.2 subunits (22). e1b-EGFP overexpression reduced CaV1.2 subunit surface expression by ∼24%, when compared with control (pEGFP-N3 vector; Fig. 6A). e1c-EGFP overexpression attenuated CaV1.2 surface expression by ∼46% (Fig. 6A). In contrast, exon 1 overexpression did not alter surface expression of α2δ-1 or total or surface expression of large conductance Ca2+-activated K+ (BKCa) channels (Fig. 6, A and B). These data indicate that CaV1.2 N termini are involved in forward trafficking of CaV1.2 subunits to the plasma membrane and that exon 1 overexpression interferes with this process. These data also suggest that exon 1c overexpression more effectively reduces membrane localization of CaV1.2 than does exon 1b overexpression.
FIGURE 6.

Exon 1 overexpression inhibits surface trafficking and whole cell CaV1.2 currents in cerebral artery smooth muscle cells. A, upper panel, representative Western blot illustrating surface CaV1.2 and α2δ-1 proteins in lysates from control arteries (pEGFP-N3 vector) or arteries expressing e1b-EGFP or e1c-EGFP. Lower panel, bar graph illustrating the percentage of CaV1.2 (black bars) and α2δ-1 (white bars) surface protein remaining relative to control (pEGFP-N3 vector) in arteries expressing e1b-EGFP or e1c-EGFP. *, significance compared with control (pEGFP-N3 vector); #, significance compared with arteries overexpressing e1b-EGFP (p < 0.05, n = 4 for each). B, representative Western blot showing that e1c-EGFP overexpression does not alter total or surface expression of BKCa channel α subunits when compared with control (pEGFP-N3 vector) in cerebral arteries. C, exon 1c overexpression inhibits CaV1.2 currents in cerebral artery smooth muscle cells. Upper panel, representative whole cell currents from smooth muscle cells isolated from arteries treated with either control vector (pEGFP-N3) or vectors that express e1c-EGFP. Lower panel, current-voltage relationships of voltage-dependent Ba2+ currents from control (pEGFP-N3 vector, filled symbols) and e1c-EGFP treated (open symbols) cerebral artery myocytes (n = 4–5 for each). *, significance compared with scrmV (p < 0.05).
Exon 1 Overexpression Reduces CaV1.2 Current Density in Arterial Myocytes
Exon 1c overexpression reduced mean CaV1.2 current density from 15.8 ± 2.2 pA/pF (pEGFP-N3 vector) to 7.5 ± 1.2 pA/pF (e1c-EGFP) or by ∼53% (Fig. 6C). This reduction is similar to the decrease in surface CaV1.2 protein (∼50%) induced by exon 1c overexpression (Fig. 6A). Therefore, these data indicate that exon 1c overexpression impairs surface expression of CaV1.2 channels (Fig. 6C).
Exon 1b or 1c Overexpression Dilates Cerebral Arteries
Endothelium-denuded control (pEGFP-N3 vector), e1b-EGFP-transfected, and e1c-EGFP-transfected arteries were pressurized to 60 mm Hg, and myogenic tone was compared. Exon 1b overexpression reduced myogenic tone by ∼26%, whereas exon 1c overexpression gave a significantly greater (∼45%) reduction in tone when compared with control (Fig. 7). These data indicate that exon 1b or 1c overexpression dilates cerebral arteries. In summary, the data indicate that when compared with exon 1b overexpression, exon 1c overexpression more effectively inhibits plasma membrane localization of CaV1.2 channels and therefore is a more effective vasodilator.
FIGURE 7.
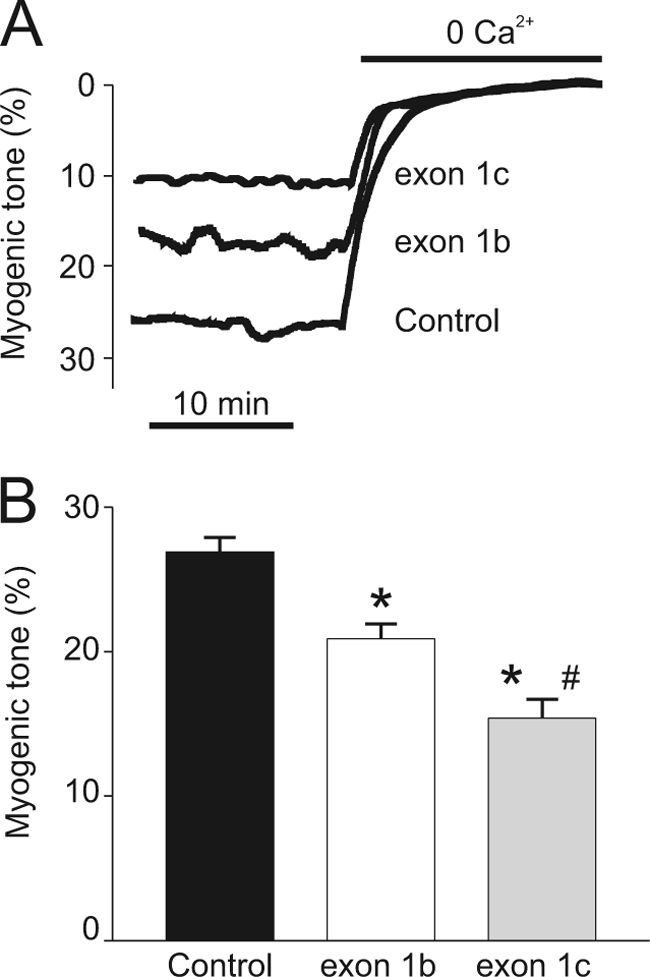
Overexpression of CaV1.2 exon 1b or 1c dilates pressurized cerebral arteries. A, representative traces illustrating myogenic tone at 60 mm Hg in control (pEGFP-N3 vector) arteries and in arteries overexpressing e1b-EGFP or e1c-EGFP. B, bar graph indicating effects of e1b-EGFP, e1c-EGFP, and control (pEGFP-N3 vector) overexpression on steady state myogenic tone at 60 mm Hg (n = 18–23). *, p < 0.05 compared with control; #, p < 0.05 compared with arteries overexpressing e1b-EGFP (p < 0.05).
DISCUSSION
Here, we studied physiological functions of CaV1.2 exon 1 splice variants in cerebral artery smooth muscle cells and tested the hypothesis that targeting CaV1.2 exon 1 is a viable strategy to induce vasodilation. We show for the first time that CaV1.2 channel proteins containing N termini encoded by exon 1b or 1c are both present in rat and human resistance size cerebral arteries. shRNA specific to exon 1b or 1c reduced expression of each variant, up-regulated expression of the other variant, reduced total CaV1.2 subunit protein, reduced whole cell CaV1.2 currents, and induced vasodilation. Exon 1c knockdown caused a greater reduction in total CaV1.2 protein and induced a larger vasodilation than did knockdown of exon 1b. Exon 1b or 1c overexpression did not alter total CaV1.2 protein but reduced CaV1.2 channel plasma membrane localization and inhibited CaV1.2 currents. Exon 1b or 1c expression also reduced myogenic tone. Although exon 1c and 1b expression was similar, exon 1c overexpression better suppressed plasma membrane expression of CaV1.2, leading to a larger vasodilation. α2δ-1 knockdown reduced surface expression of both CaV1.2 variants, with the reduction in surface expression of CaV1.2e1c greater than for CaV1.2e1b. Taken together, the data indicate that both N termini regulate plasma membrane localization of CaV1.2 channels, thereby modulating resistance size cerebral artery contractility. The data also suggest that the exon 1c splice variant plays a more prominent signaling and functional role in the regulation of arterial contractility than the exon 1b variant. These data are the first to determine the expression and physiological functions of CaV1.2 N termini in arterial smooth muscle cells.
Quantitative real time PCR previously demonstrated that exon 1b and 1c are the only CaV1.2 N-terminal splice variant transcripts present in smooth muscle cells of rat resistance size cerebral arteries (15). Exon 1c exhibited far higher mRNA expression (∼96% of total mRNA) than exon 1b, with exon 1a absent (15). In contrast, in cardiac myocytes exon 1a was predominant (∼82% of total mRNA) (15). The tissue selective expression of exon 1c in arterial smooth muscle cells could provide a means to selectively target this CaV1.2 channel variant and thus modulate vascular contractility. However, it was unclear whether CaV1.2 channel proteins containing the two different N termini encoded by exon 1b and 1c were expressed in arterial smooth muscle cells. Here, using selective antibodies, we demonstrate that the CaV1.2e1b and CaV1.2e1c channel proteins are present in rat cerebral arteries. Although not quantitative, Western blots suggested that the ratio of CaV1.2e1b to CaV1.2e1c was higher than expected based on previous evidence that CaV1.2e1b message is small (∼4%) and that CaV1.2e1c message is the majority (15). Explanations for the higher level of CaV1.2e1b protein than would be expected from relative message include that CaV1.2e1b mRNA or protein may be more stable than CaV1.2e1c.
Splice variant-specific antibodies also detected bands corresponding to CaV1.2e1b and CaV1.2e1c in human cerebral artery lysate. Exon 1b has previously been reported in humans (30), but to date no study has reported a sequence similar to rodent exon 1c in humans. Our data suggest that an amino acid sequence similar to that encoded by rodent exon 1c is present in human cerebral artery CaV1.2 channels. A nucleotide sequence identical to the rat exon 1b sequence is present in the human genome, but a sequence identical to 1c is not found. However, a sequence that shares high homology with the 5′-untranslated region upstream of exon 1c is present in the human genome (p13.33 of chromosome 12). Multiple codons can encode the same amino acid. Therefore, a dissimilar exon 1c nucleotide sequence in rats and humans could encode an identical amino acid sequence. It was not a goal of this study to clone human CaV1.2 exon 1c. Given the difficulty in obtaining live human resistance size arteries for study, such a determination will be the focus of a future investigation.
CaV1.2e1b or CaV1.2e1c knockdown resulted in a compensatory increase in the other splice variant. α2δ-1 knockdown inhibited CaV1.2 channel surface expression and also up-regulated CaV1.2 expression in cerebral arteries, consistent with a previous study (22). Transcriptional compensation has been described for a variety of proteins. In rat cardiac myocytes, knockdown of the SR Ca2+ ATPase was associated with up-regulation of TRPC4 and TRPC5 channel protein and Na+/Ca2+ exchanger expression (31). TRPC3 channel protein was up-regulated in vascular smooth muscle cells of TRPC6-deficient mice (32). TRPC1 knockdown in A7r5 vascular smooth muscle cells caused a compensatory increase in TRPC6 expression (33). Medulla chromaffin cells from N-type calcium channel α1B subunit-deficient mice express elevated N-type α1A and β4 calcium channel subunits (34). The mechanism by which CaV1.2 targeting elevates CaV1.2 transcription in cerebral arteries is unclear, but several possibilities exist. CaV1.2 expression may be up-regulated in response to a reduction in plasma membrane CaV1.2 protein and the ensuing fall in intracellular Ca2+ concentration. In addition, CaV1.2 knockdown may lead to a reduction in the truncated CaV1.2 subunit C terminus, which has been demonstrated to suppress CaV1.2 expression in cardiac smooth muscle cells (35). Thus, our data suggest that CaV1.2 channels may self-regulate expression in arterial smooth muscle cells. Identification of the CaV1.2 channel self-transcriptional feedback mechanism could provide a novel approach by which to alter CaV1.2 channel expression and modulate arterial contractility.
CaV1.2 channel activation is essential for intravascular pressure- and depolarization-induced vasoconstriction in small arteries and arterioles (6, 10). Although knockdown of one CaV1.2 splice variant led to compensatory up-regulation of the other variant, total CaV1.2 protein was reduced, leading to a decrease in both myogenic tone and depolarization-induced vasoconstriction. Targeting of both exon 1b and 1c in knockdown experiments similarly reduced CaV1.2 protein and current density, in agreement with previous findings that >95% of CaV1.2 channel protein is expressed at the cell surface (22). e1cshV reduced CaV1.2 subunit protein and myogenic tone more so than did e1bshV. This result is consistent with the concept that transcriptional inhibition of CaV1.2e1c, the predominant arterial smooth muscle cell CaV1.2 mRNA variant, produces the largest reduction in protein (15). It is also possible that the exon 1c shRNA sequence used may be more effective than the shRNA used to target exon 1b. Given that CaV1.2 exon 1c is encoded by a 30-nucleotide sequence and CaV1.2 exon 1b is encoded by a 60-nucleotide sequence, there was limited latitude for shRNA sequence variability to study this hypothesis in detail. These data raise the possibility that targeting of exon 1c is more effective than targeting of exon 1b at reducing CaV1.2 expression and arterial contractility.
As an alternative approach to study physiological functions of CaV1.2 exon 1, EGFP-tagged exon 1b and 1c were overexpressed in smooth muscle cells of resistance size cerebral arteries. Exon 1b or 1c overexpression did not alter total CaV1.2 expression but reduced CaV1.2 plasma membrane localization. Whole cell CaV1.2 currents were also reduced in arterial myocytes overexpressing exon 1c. Myogenic tone was reduced in arteries overexpressing exon 1b or 1c by amounts comparable with the loss of surface CaV1.2 channel protein. Because exon 1 overexpression did not alter surface α2δ-1 or BKCa channel total protein or surface expression, these data suggest that exon 1-encoded peptides act as competitive antagonists of CaV1.2 membrane insertion. Thus, we have identified CaV1.2 exon 1 peptides as novel vasodilators that act by inhibiting CaV1.2 surface expression.
Experiments performed using heterogeneous expression of recombinant subunits in HEK293 and COS-1 cells indicated that α2δ-1 promoted surface expression of CaV1.2e1b more so than CaV1.2e1c (15). Here, experiments studying endogenous CaV1.2 subunits in cerebral arteries indicated that α2δ-1 knockdown reduced surface expression of CaV1.2e1c more so than CaV1.2e1b, a result that is in contrast to observations obtained using recombinant protein expression. These data highlight mechanistic differences that can occur in simplified heterologous overexpression systems and illustrate the importance of studying functions of endogenous proteins in native cells. It is also important to note that β1b, β2a, or β3 subunit overexpression equalized differential surface expression of recombinant CaV1.2e1b and CaV1.2e1c channels induced by α2δ-1 (15). A major difference between data obtained in an earlier study and here is that arterial smooth muscle cells express multiple β subunit isoforms. The ratio of endogenous α2δ-1 to β subunits is unclear. Here, α2δ-1 knockdown alone was sufficient to differentially alter CaV1.2e1b and CaV1.2e1c surface expression. We have previously demonstrated that α2δ-1 knockdown does not alter β subunit expression, suggesting that changes in β subunits cannot explain this result in arterial smooth muscle cells (22). Therefore, our data support the concept that exon 1-encoded N termini promote CaV1.2 surface expression. Furthermore, we show that endogenous β subunits do not normalize α2δ-1-mediated differential surface expression of N-terminal variants in arterial smooth muscle cells.
Previous studies performed in other cell types have not focused specifically on the significance of CaV1.2 exon 1 but have identified functions of the N terminus. The proximal CaV1.2 N terminus regulates CaV1.2 sensitivity to intracellular anions (36). The more distal CaV1.2 N terminus can interact with regulatory proteins, including KChip, calmodulin, auxiliary β subunits, and calcium-binding protein-1 (37–40). Our study is the first to report that the CaV1.2 N terminus is a critical element required for endogenous channel surface trafficking in cerebral artery smooth muscle cells and that exon 1 knockdown and CaV1.2 exon 1 overexpression is a novel approach that can be utilized to induce vasodilation.
In summary, we demonstrate that cerebral artery smooth muscle cells contain CaV1.2e1b and CaV1.2e1c subunits, and both of these CaV1.2 splice variants are located in the plasma membrane. shRNA targeting exon 1b or 1c selectively reduces the expression of each variant, leading to vasodilation. α2δ-1 inserts both CaV1.2 N-terminal variants into the arterial smooth muscle cell plasma membrane but preferentially inserts CaV1.2e1c subunits. Exon 1b or 1c overexpression inhibited CaV1.2 surface trafficking, leading to vasodilation. These data indicate that exon 1-encoded N termini promote plasma membrane insertion of CaV1.2 subunits in arterial smooth muscle cells and indicate that exon 1 targeting is a novel approach that can be used to induce vasodilation.
This work was supported, in whole or in part, by National Institutes of Health Grants HL67061 and HL094378 (to J. H. J.) and KO1 HL096411 (to A. A.).
- CaV1.2
- voltage-dependent L-type Ca2+
- EGFP
- enhanced GFP.
REFERENCES
- 1. Arikkath J., Campbell K. P. (2003) Curr. Opin. Neurobiol. 13, 298–307 [DOI] [PubMed] [Google Scholar]
- 2. Catterall W. A., Perez-Reyes E., Snutch T. P., Striessnig J. (2005) Pharmacol. Rev. 57, 411–425 [DOI] [PubMed] [Google Scholar]
- 3. Dolphin A. C. (2009) Curr. Opin. Neurobiol. 19, 237–244 [DOI] [PubMed] [Google Scholar]
- 4. Gollasch M., Nelson M. T. (1997) Kidney Blood Press. Res. 20, 355–371 [DOI] [PubMed] [Google Scholar]
- 5. Navedo M. F., Amberg G. C., Westenbroek R. E., Sinnegger-Brauns M. J., Catterall W. A., Striessnig J., Santana L. F. (2007) Am. J. Physiol. Heart Circ. Physiol 293, H1359–H1370 [DOI] [PubMed] [Google Scholar]
- 6. Nelson M. T., Patlak J. B., Worley J. F., Standen N. B. (1990) Am. J. Physiol. 259, C3–C18 [DOI] [PubMed] [Google Scholar]
- 7. Sonkusare S., Palade P. T., Marsh J. D., Telemaque S., Pesic A., Rusch N. J. (2006) Vascul. Pharmacol. 44, 131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wamhoff B. R., Bowles D. K., Owens G. K. (2006) Circ. Res. 98, 868–878 [DOI] [PubMed] [Google Scholar]
- 9. Berridge M. J., Lipp P., Bootman M. D. (2000) Nat. Rev. Mol. Cell Biol. 1, 11–21 [DOI] [PubMed] [Google Scholar]
- 10. Moosmang S., Schulla V., Welling A., Feil R., Feil S., Wegener J. W., Hofmann F., Klugbauer N. (2003) EMBO J. 22, 6027–6034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pratt P. F., Bonnet S., Ludwig L. M., Bonnet P., Rusch N. J. (2002) Hypertension 40, 214–219 [DOI] [PubMed] [Google Scholar]
- 12. Pesic A., Madden J. A., Pesic M., Rusch N. J. (2004) Circ. Res. 94, e97–e104 [DOI] [PubMed] [Google Scholar]
- 13. Triggle D. J. (2007) Biochem. Pharmacol. 74, 1–9 [DOI] [PubMed] [Google Scholar]
- 14. Abernethy D. R., Soldatov N. M. (2002) J. Pharmacol. Exp. Ther. 300, 724–728 [DOI] [PubMed] [Google Scholar]
- 15. Cheng X., Liu J., Asuncion-Chin M., Blaskova E., Bannister J. P., Dopico A. M., Jaggar J. H. (2007) J. Biol. Chem. 282, 29211–29221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cheng X., Pachuau J., Blaskova E., Asuncion-Chin M., Liu J., Dopico A. M., Jaggar J. H. (2009) Am. J. Physiol. Heart Circ. Physiol 297, H680–H688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jurkat-Rott K., Lehmann-Horn F. (2004) J. Physiol 554, 609–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tang Z. Z., Hong X., Wang J., Soong T. W. (2007) Cell Calcium 41, 417–428 [DOI] [PubMed] [Google Scholar]
- 19. Tang Z. Z., Liang M. C., Lu S., Yu D., Yu C. Y., Yue D. T., Soong T. W. (2004) J. Biol. Chem. 279, 44335–44343 [DOI] [PubMed] [Google Scholar]
- 20. Liao P., Yu D., Lu S., Tang Z., Liang M. C., Zeng S., Lin W., Soong T. W. (2004) J. Biol. Chem. 279, 50329–50335 [DOI] [PubMed] [Google Scholar]
- 21. Henkel A. W., Bieger S. C. (1994) Anal. Biochem. 223, 329–331 [DOI] [PubMed] [Google Scholar]
- 22. Bannister J. P., Adebiyi A., Zhao G., Narayanan D., Thomas C. M., Feng J. Y., Jaggar J. H. (2009) Circ. Res. 105, 948–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lesh R. E., Somlyo A. P., Owens G. K., Somlyo A. V. (1995) Circ. Res. 77, 220–230 [DOI] [PubMed] [Google Scholar]
- 24. Zhao G., Adebiyi A., Blaskova E., Xi Q., Jaggar J. H. (2008) Am. J. Physiol. Cell Physiol. 295, C1376–C1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jaggar J. H. (2001) Am. J. Physiol. Cell Physiol. 281, C439–C448 [DOI] [PubMed] [Google Scholar]
- 26. Adebiyi A., McNally E. M., Jaggar J. H. (2008) Mol. Pharmacol. 74, 736–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marsh J. D., Telemaque S., Rhee S. W., Stimers J. R., Rusch N. J. (2008) Trans. Am. Clin. Climatol. Assoc. 119, 171–183 [PMC free article] [PubMed] [Google Scholar]
- 28. Obermair G. J., Tuluc P., Flucher B. E. (2008) Curr. Opin. Pharmacol. 8, 311–318 [DOI] [PubMed] [Google Scholar]
- 29. Reimer D., Huber I. G., Garcia M. L., Haase H., Striessnig J. (2000) FEBS Lett. 467, 65–69 [DOI] [PubMed] [Google Scholar]
- 30. Powers P. A., Gregg R. G., Lalley P. A., Liao M., Hogan K. (1991) Genomics 10, 835–839 [DOI] [PubMed] [Google Scholar]
- 31. Seth M., Sumbilla C., Mullen S. P., Lewis D., Klein M. G., Hussain A., Soboloff J., Gill D. L., Inesi G. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 16683–16688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dietrich A., Mederos Y., Schnitzler M., Gollasch M., Gross V., Storch U., Dubrovska G., Obst M., Yildirim E., Salanova B., Kalwa H., Essin K., Pinkenburg O., Luft F. C., Gudermann T., Birnbaumer L. (2005) Mol. Cell. Biol. 25, 6980–6989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Selli C., Erac Y., Kosova B., Tosun M. (2009) Vascul. Pharmacol. 51, 96–100 [DOI] [PubMed] [Google Scholar]
- 34. Takahashi E., Nagasu T. (2006) Comp Med. 56, 168–175 [PubMed] [Google Scholar]
- 35. Schroder E., Byse M., Satin J. (2009) Circ. Res. 104, 1373–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Babai N., Kanevsky N., Dascal N., Rozanski G. J., Singh D. P., Fatma N., Thoreson W. B. (2010) PLoS One 5, e8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thomsen M. B., Wang C., Ozgen N., Wang H. G., Rosen M. R., Pitt G. S. (2009) Circ. Res. 104, 1382–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dick I. E., Tadross M. R., Liang H., Tay L. H., Yang W., Yue D. T. (2008) Nature 451, 830–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bichet D., Cornet V., Geib S., Carlier E., Volsen S., Hoshi T., Mori Y., De Waard M. (2000) Neuron 25, 177–190 [DOI] [PubMed] [Google Scholar]
- 40. Zhou H., Yu K., McCoy K. L., Lee A. (2005) J. Biol. Chem. 280, 29612–29619 [DOI] [PubMed] [Google Scholar]