

Abstract
In the visual signal terminating transition state, the cyclic GMP phosphodiesterase (PDE6) inhibitory γ-subunit (PDEγ) stimulates GTPase activity of the α-subunit of transducin (αt) by enhancing the interaction between αt and its regulator of G protein signaling (RGS9), which is constitutively bound to the type 5 G protein β-subunit (β5). Although it is known from a crystal structure of partial molecules that the PDEγ C terminus contacts with both αt and RGS9, contributions from the intrinsically disordered PDEγ N-terminal half remain unclear. In this study, we were able to investigate this issue using a photolabel transfer strategy that allows for mapping the interface of full-length proteins. We observed label transfer from PDEγ N-terminal positions 50, 30, and 16 to RGS9·β5 in the GTPase-accelerating protein (GAP) complex composed of PDEγ·αt·RGS9·β5. In support of a direct PDEγ N-terminal interaction with RGS9·β5, the PDEγ N-terminal peptide PDEγ(1–61) abolished label transfer to RGS9·β5, and another N-terminal peptide, PDEγ(10–30), disassembled the GAP complex in label transfer and pulldown experiments. Furthermore, we determined that the PDEγ C-terminal interaction with αt was enhanced whereas the N-terminal interaction was weakened upon changing the αt conformation from the signaling state to the transition state. This “rearrangement” of PDEγ domain interactions with αt appears to facilitate the interaction of the PDEγ N-terminal half with RGS9·β5 and hence its contribution to optimal stabilization of the GAP complex.
Keywords: G Proteins, Phosphodiesterases, Phototransduction, Protein Conformation, Protein-Protein interactions, G Protein β5-Subunit, GTPase-accelerating Proteins, PDE6γ, Regulator of G Protein Signaling, Transition State Complex
Introduction
In vertebrate photoreceptor cells, interactions of the cyclic GMP (cGMP) phosphodiesterase (PDE6)2 inhibitory γ-subunit (PDEγ) with its targets switch on and off visual signal transduction (for review, see Refs. 1–4). The signaling is turned on when the GTP-bound α-subunit of transducin (αt), which is converted from the GDP-bound conformation by light-excited rhodopsin, activates PDE6 by interacting with PDEγ and displacing its C terminus from the PDE6 catalytic pocket (signaling state). Lowered cGMP levels then cause hyperpolarization of the plasma membrane by closing cGMP-gated channels, thus leading to signal transmission to the brain. Concomitantly, the GTPase activity of αt is accelerated to hydrolyze the bound GTP into GDP, via simultaneous αt interactions with PDEγ and the regulator of G protein signaling (RGS9) constitutively bound with the type 5 G protein β-subunit (β5) (transition state). Once the conformation of αt reverts back to the GDP-bound inactive form, PDEγ dissociates from αt and reinhibits PDE6. Visual transduction is thus turned off, and the signaling proteins are primed for the next round of the photoresponse.
Throughout this report, the two conformers of αt in the signaling state and transition state are represented respectively by αt-GTPγS (5) and αt-GDP-AlF4− (6). The four-component (αt·PDEγ·RGS9·β5) transition state complex is also referred to as the GAP (GTPase-accelerating protein) complex (2).
Our knowledge regarding the protein functions and interactions within the GAP complex has been learned largely from molecular and biochemical studies, in particular, the crystal structure of a partial GAP complex (7) that includes the GDP-AlF4−-bound αt/i1 chimera, the C-terminal half of PDEγ (residues 46–87), and the RGS9 catalytic core (RGS9d). RGS9d interacts intimately with the αt switch regions thus stabilizing its GTP hydrolysis conformation. The other domains of RGS9 together with β5 discriminate between the activated αt and the αt·PDEγ complex (8, 9). PDEγ enhances the affinity of RGS9·β5 with αt by interacting with both simultaneously (10). PDEγ is a small protein of 87 amino acids containing two major functional domains, the polycationic N-terminal domain (Gly19–Gly49) and the hydrophobic C-terminal domain (Thr62–Ile87) (3, 4). Both domains interact with αt as well as with PDEαβ (11–14) and likely in a complementary manner in the signaling state with the C-terminal domain favoring αt whereas the central domain binding more tightly with PDEαβ (13).
Although the partial GAP structure (7) has revealed critical atomic details for the transition state complex, our information regarding the interactions of PDEγ with its partners is still substantially missing. This is because the current structure contains only the C-terminal half of PDEγ and less than one-third of the RGS9 sequence with no β5 bound. Even though the recent crystal structure of the full-length RGS9·β5 (15) has greatly complemented our knowledge, important questions remain unanswered. One prominent question is in regard to the role of the PDEγ N-terminal half (hereafter defined as residues 1–50, as opposed to the C-terminal half residues 50–87 that are resolved in the partial GAP structure (7)). Although the PDEγ C-terminal peptide PDEγ(63–87) is reported to be fully potent in stimulating the GAP activity (10), the full-length PDEγ is needed for the high affinity GAP complex formation (8, 10). This implicates a necessary role for the PDEγ N-terminal portion. Additionally, previous studies indicate that practically all of the RGS9 domains as well as β5 participate in regulation of the PDEγ-stimulated GAP function (8, 9, 16). In particular, the noncatalytic constituent RGS9 domains and the N terminus of β5 play a decisive role in the RGS substrate selectivity toward the PDEγ complex with activated αt (16). In the partial GAP structure (7), however, only one direct contact between PDEγ and RGS9d has been discovered. It is therefore important to explore whether the PDEγ N-terminal half, which is absent in the existing crystal structures (7, 15, 17), also plays a role in the GAP complex by interacting with RGS9·β5.
In addition, with regard to the PDEγ interactions with αt and RGS9·β5 in the GAP complex, it is not known if PDEγ interacts with the transition state conformation in the same manner as with the signaling state conformation. Thus far there have been no atomic structures available for the full-length PDEγ in complex with either αt-GDP-AlF4− or αt-GTPγS. Although the crystal structure of the partial transition state complex (7) is often used to infer the signaling state PDEγ·αt interaction, it is not clear whether the PDEγ interactions with two different αt conformers are the same.
Answers to these questions are essential for a clear understanding of the PDEγ regulation of the photoresponse through dynamic interactions with its partners. However, the intrinsically disordered PDEγ, especially its N-terminal half (18), has been problematic in solving a crystal structure of αt and RGS9·β5 bound with the full-length PDEγ. As indicated in our previous reports (13, 19, 20), the label transfer approach has allowed for systematic mapping of interactions between full-length molecules, thus circumventing the problems of intrinsic disorder that can impede efforts in solving atomic structures. We have generated PDEγ photoprobes by derivatization at single-cysteine positions throughout the entire PDEγ molecule and used them in this study to investigate the interactions of PDEγ with full-length RGS9·β5 and αt in the GAP complex. We have observed an interaction between the N-terminal half of PDEγ and RGS9·β5, and differential PDEγ interactions with two αt conformers that rationalize the observed PDEγ N-terminal interaction with RGS9·β5. Our findings afford new insights into the structure and regulation of the GAP complex.
EXPERIMENTAL PROCEDURES
The chemicals and reagents used in this study were from the sources described previously (13, 19, 20) unless otherwise stated. The PDEγ peptides PDEγ(10–30) and PDEγ(15–26) were a generous gift from Dr. Rick Cote at the University of New Hampshire (12). The mouse monoclonal anti-His6 antibody and the rabbit polyclonal anti-αt N terminus antibody were purchased from Santa Cruz Biotechnology and Affinity Bioreagents, respectively. The Immobilon Western Chemiluminescent HRP substrate is a product of Millipore.
Preparation of Holotransducin, αt-GDP, and αt-GTPγS
Using frozen dark-adapted bovine retinas (J. A. & W. L. Lawson Co.), rod outer segment membranes were isolated, from which holotransducin was prepared as described previously (21). αt-GDP and βγt were then purified from holotransducin using a Blue-Sepharose CL-6B column. To prepare αt-GTPγS, GTPγS was added to rod outer segment membranes, αt-GTPγS was thus released and purified on a Blue-Sepharose CL-6B column. The purity of αt was determined to be >95% by SDS-PAGE and Coomassie staining. Correct conformations of αt-GTPγS and αt-GDP-AlF4− (prepared using αt-GDP) were confirmed by their resistance to trypsin digestion (22–24) (see supplemental Fig. S1).
Preparation of RGS9·β5
The RGS9·β5 protein samples that were prepared as described previously (25) were provided by Dr. Kirill Martemyanov and Dr. Vadim Arshavsky. Briefly, RGS9 and β5 were co-expressed in an Sf9/baculovirus expression system and then purified. Purified RGS9·β5 were gel-filtered on an NAP-10 column (Amersham Biosciences) equilibrated with a buffer containing 20 mm Tris-HCl (pH 8.0), 300 mm NaCl, 10 mm MgCl2, 1 mm dithiothreitol, and 10% glycerol. The purity of recombinant RGS9·β5 was no less than 80%.
Preparation of the PDEγ Photoprobes
The constructs for expressing the full-length wild-type (WT) PDEγ (single cysteine at position 68) and the PDEγ N-terminal peptide (PDEγ(1–61)) were reported previously (21). Single-cysteine PDEγ mutants were generated using the Strategene QuikChange method (19). These PDEγ variants were expressed in Escherichia coli and purified by chitin beads followed by reverse-phase HPLC using POROS 20 R2 resin (24). Greater than 95% pure PDEγ was used for the preparation of PDEγ photoprobes.
The biotin-tagged sulfhydryl-reactive and cleavable photoreactive compound, BBM (supplemental Fig. S2A) was obtained from Toronto Research Chemicals. The BBM-PDEγ photoprobes were prepared as described previously (20). Typically, a derivatization reaction contained 20 mm NaH2PO4 (pH 6.7), 100 mm NaCl, 50% acetonitrile, 150 μg of PDEγ, and BBM in a 10-fold molar excess over PDEγ. The reaction was incubated under argon for 3 h (22 °C, dark) and then loaded onto a POROS 20 R2 column for reversed-phase HPLC. An acetonitrile gradient of 0.125%/min (0.1% TFA, 1 ml/min) was applied to separate the PDEγ derivatives, which eluted at ∼44% acetonitrile. The radioactive [125I]ACTP-PDEγ photoprobes used in this study were from the same batch as reported previously (19).
Functional Assay of the PDEγ Photoprobes
Transducin GTPase activity was determined by using a single-turnover technique as described previously (25). The assays were conducted at room temperature (22–24 °C) in a buffer containing 25 mm Tris-HCl (pH 8.0), 140 mm NaCl, and 8 mm MgCl2. The urea-treated rod outer segment membranes, lacking endogenous activity of RGS9, were used as a source for the photoexcited rhodopsin required for transducin activation. The reactions were initiated by the addition of 10 μl of 0.6 μm [32P]GTP (∼105 dpm/sample) to 20 μl of urea-treated rod outer segment membranes (20 μm final rhodopsin concentration) reconstituted with transducin heterotrimer (1 μm) and recombinant RGS9·Gβ5 complex (0.5 μm). The reactions were performed in either the absence or presence of PDEγ derivatives (1 μm) and terminated by the addition of 100 μl of 6% perchloric acid followed by measurement of the 32P formation. The assays were conducted in the absence of reducing agents due to the presence of the disulfide linkage between the photoreactive group and PDEγ.
The assay of αt GTPase stimulation by BBM-PDEγ photoprobes indicated that the functional activities of the tested photoprobes were similar to WT-PDEγ (supplemental Fig. S2B). Furthermore, a native gel assay showed that interactions of the PDEγ photoprobes with αt-GDP-AlF4− were not markedly affected (supplemental Fig. S2C).
Photocross-linking/Label Transfer Using PDEγ Photoprobes
For the strategy of label transfer, please refer to the legend of supplemental Fig. S2A and our previous reports (13, 26). Photocross-linking reactions containing PDEγ photoprobes and target proteins at desired concentrations (for detailed conditions refer to corresponding figure legends) were dark-incubated on ice and then subjected to UV light. The reactions with BBM photoprobes were photolyzed at 5–10 °C for 2 × 5 min (with a 5-min dark interval on ice), in an RPR-100 Rayonet Photochemical Reactor equipped with 18 light bulbs of 350 nm (Southern New England Ultraviolet Company). The reactions using [125I]ACTP-PDEγ photoprobes were exposed to the UV light generated by an AH-6 water-jacketed 1000-watt high pressure mercury lamp for 5 s at a distance of 10 cm (19). Following photolysis, sample buffer was added immediately to the final concentrations of 1% SDS and 50 mm DTT. Proteins were then separated by SDS-PAGE. The gels were either used for Coomassie staining and autoradiography or for electrotransfer to PVDF membranes and subsequent far-Western detection of biotin label transfer. Autoradiography and far-Western blotting were performed as described previously (19, 20).
Pulldown Protein Binding Assay
Pulldown by PDEγ was performed using Ultra-Link Plus immobilized streptavidin gel (Pierce Biotechnology; hereafter termed streptavidin beads), following the previously reported method (13) with minor modifications. Biotinylated full-length PDEγ (Btn-PDEγ) or the PDEγ C-terminal half (PDEγ(46–87)-Btn) were prepared as described previously (13). For detailed experimental conditions please refer to the figure legends. Generally, prior to the experiments, streptavidin beads were equilibrated for 15 min at room temperature with the pulldown buffer indicated in a given experiment, in which a high concentration (0.5–1 μg/μl) of BSA was included to block possible nonspecific protein-bead interactions. The beads were then washed twice with 500 μl of buffer and were ready for use. In the pulldown experiments using Ni-NTA beads, the beads were also preincubated with BSA to eliminate possible protein-bead interactions. Incubation of the beads with the pulldown reactions was performed at 4 °C by rotating the microcentrifuge tubes. At the end of the pulldown experiments, proteins on the beads were eluted with the SDS/DTT-containing sample buffer at 80–90 °C for 5 min, resolved on a low cross-link 15% acrylamide gel (27), and then immunodetected by Western blotting using enhanced chemiluminescence.
RESULTS
Label Transfer from PDEγ to RGS9·β5 in the Transition State GAP Complex
To map the entire PDEγ·RGS9·β5 interaction interface in the GAP complex that is composed of the full-length molecules, photocross-linking experiments were performed using 13 PDEγ photoprobes prepared with BBM at positions throughout the PDEγ molecule. The BBM photolabel transfer to RGS9·β5 was then detected by far-Western blotting after DTT reversal. As shown in Fig. 1, A and B, prominent biotin label transfer onto RGS9 can be observed from PDEγ position 68, and interestingly, also from the PDEγ positions N-terminal to 68, such as 60, 50, 30, and 16. In contrast, there was no obvious label transfer observed from the positions C-terminal to 68. It is known that Val66, which is close to Cys68, makes direct contact with Trp362 in the RGS domain, as exhibited by the partial GAP structure that includes only the C-terminal half of PDEγ (7). Remarkably, from PDEγ N-terminal positions 30 and 16, label transfer was found on β5 as well (Fig. 1, A and B). Although the protein amount in each lane is approximately equal (Fig. 1A, lower panel), the labeling intensity of RGS9·β5 is PDEγ position-dependent, manifesting the specificity of the observed label transfer.
FIGURE 1.

Profiling of label transfer from PDEγ to RGS9·β5 in the GAP complex. Photocross-linking/label transfer experiments were performed using the BBM-PDEγ photoprobes. One μm BBM-PDEγ was incubated with RGS9·β5 and αt at a stoichiometry of 1:1:2, in Buffer A containing 20 mm Tris-HCl, pH 7.9, 100 mm NaCl, 2 mm MgCl2, 0.25% lauryl sucrose, 20 mm imidazole, and 50 μg/ml BSA (49). 10 mm NaF and 30 μm AlCl3 were added when needed. After incubation on ice for 30 min, the reaction was subjected to UV light (350 nm), as described under “Experimental Procedures.” Ni-NTA beads of 3 μl were then added to the reaction, incubated on ice for 30 min with occasional shaking, and washed with 2 × 200 μl of reaction buffer. The proteins on the beads were eluted with the SDS/DTT-containing sample buffer, and the biotin label transfer to RGS9·β5 was detected by far-Western blotting using streptavidin-conjugated HRP. Each blot shown in the figure is a representative of 2–4 similar experiments. In the experiments of A, B, and C, to focus on profiling label transfer from PDEγ to RGS9·β5, Ni-NTA beads were used to “fish out” RGS9·β5 from the photocross-linked reaction mixture. This strategy allowed us to retain His-tagged RGS9·β5 on the beads and detect the biotin label transfer by far-Western blotting with a relatively clean background. During vigorous washing of the Ni-NTA beads, some αt may have been lost, making quantification of label transfer to αt and “within experiment” comparison with the label transfer to RGS9·β5 inappropriate. For this reason, the αt band is not shown in A, B, and C. The experiments in D were performed without using Ni-NTA beads, and biotin label was shown to be transferred to both RGS9 and αt from PDEγ position 68 (lane 2, left panel). Please note, however, that the αt band does not necessarily represent only the label transfer to the αt population that was in the GAP complex. There is a possibility that the label transfer to the αt population that was not in the GAP complex also mixed into the αt band on the blot, considering that the interaction of PDEγ with αt-GDP-AlF4− alone (without RGS9·β5) is strong (33). A, far-Western blot showing the biotin label transfer from BBM-PDEγ photoprobes to RGS9·β5 (upper panel) in the presence of αt-GDP-AlF4−. The same blot was stained with Amido Black to reveal equal amounts of RGS9·β5 proteins used in each reaction (lower panel). Alignment of the upper and lower panels indicates that the far-Western signal below the RGS9 band at position 30 was from β5, rather than from the asterisk-marked band, which is a commonly observed C-terminally truncated RGS9 species due to proteolysis (49). B, full spectrum label transfer to RGS9·β5 from PDEγ positions throughout the entire molecule in the presence of αt-GDP-AlF4−. The BBM derivatization positions on PDEγ are presented on top of the corresponding lanes. C, GDP-AlF4− dependence of label transfer to RGS9·β5. Experiments were performed as in A and B, but in the presence as well as absence of GDP-AlF4−. D, biotin label transfer to RGS9 from the PDEγ position 68 in the presence of αt-GDP (lane 1), αt-GDP-AlF4− (lane 2), or αt-GTPγS (lane 3). Experiments were performed essentially the same as described for A, B, and C, except that Ni-NTA beads were not used after UV photolysis. Following photocross-linking, the reactions were immediately quenched in the SDS/DTT-containing sample buffer and then subjected to SDS-PAGE and far-Western blotting (left panel). The Amido Black-stained PVDF membrane of the same blot is shown in the right panel.
Supporting the GAP-specific nature of the observed label transfer from PDEγ to RGS9·β5 (Fig. 1, A and B), prominent label transfer from positions 68, 60, 50, and 30 could be detected in the presence of αt-GDP-AlF4−, but not in the GDP-bound inactive conformation (Fig. 1C) or the GTPγS-bound form (Fig. 1D).
To answer the question of whether the label transfer to RGS9·β5 resulted from protein-protein interaction or was just due to close proximity of the PDEγ probe with RGS9·β5, label transfer experiments were performed using PDEγ peptides as competitors of the PDEγ probes (Fig. 2). The results indicate that the N-terminal peptide PDEγ(1–61) could efficiently eliminate RGS9 labeling from both positions 68 and 50 (see lanes 3 and 6). Furthermore, PDEγ(10–30), a shorter N-terminal peptide, also greatly reduced label transfer to RGS9 from position 50 (lane 8), but PDEγ(15–26) did not (lane 9). The C-terminal peptide PDEγ(62–87), although partially reducing label transfer from position 68 (lane 2), did not affect label transfer from position 50 (lane 5).
FIGURE 2.
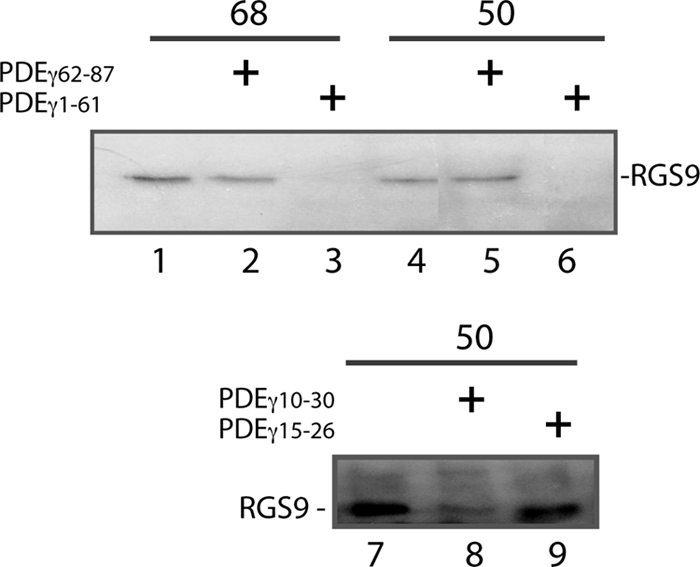
PDEγ N-terminal peptides PDEγ(1–61)and PDEγ(10–30) abrogate label transfer from PDEγ to RGS9·β5. Experiments were performed as described in Fig. 1. Shown in each panel is one of two similar experiments. Prior to UV photolysis, BBM-PDEγ was incubated with RGS9·β5 and αt-GDP-AlF4− in the absence (lanes 1, 4, and 7) or presence of PDEγ peptides in a 500 × molar excess over BBM-PDEγ. The following peptides were preincubated in the reactions prior to adding BBM-PDEγ: PDEγ(62–87), lanes 2 and 5; PDEγ(1–61), lanes 3 and 6; PDEγ(10–30), lane 8; PDEγ(15–26), lane 9.
PDEγ N-terminal Peptide PDEγ(10–30) Disrupts the RGS9 Pulldown from the Transition State GAP Complex
To confirm further a direct PDEγ N-terminal interaction with RGS9·β5 in the GAP complex that was determined from the label transfer approach, we performed pulldown experiments using Btn-PDEγ and streptavidin beads (Fig. 3). The GAP complex was first reconstituted by incubating Btn-PDEγ with RGS9·β5 and αt-GDP-AlF4− and then captured on the streptavidin beads followed by Western blotting analyses of the co-precipitated proteins. As can be seen in Fig. 3A, RGS9(·β5) was efficiently pulled down by Btn-PDEγ in the presence of αt-GDP-AlF4− (lane 3, compared with the input in lane 5), which was not a result of nonspecific interaction with the beads (no pulldown in lane 1). However, in the absence of either AlF4− (lane 2) or αt (lane 4), RGS9 pulldown was significantly reduced, indicating a GAP complex-dependence of the observed RGS9 pulldown by PDEγ (lane 3). Consistently, the pulldown of αt was also AlF4−-dependent (lane 3 compared with lane 2 in B).
FIGURE 3.
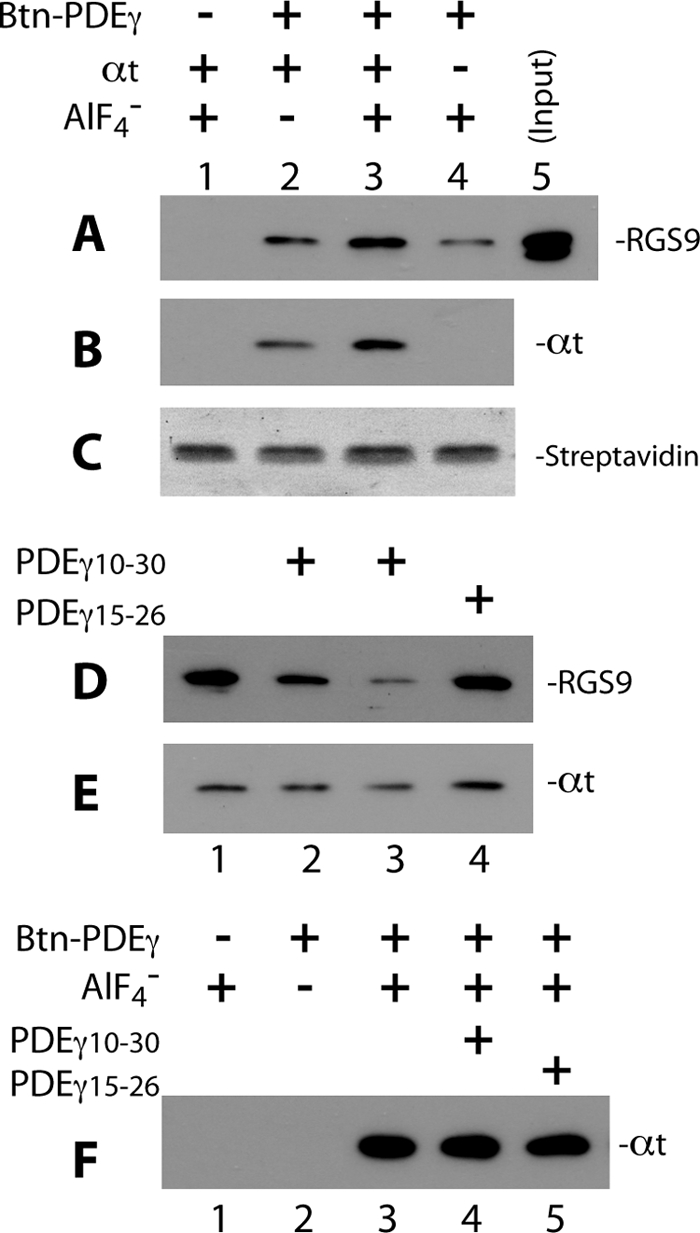
PDEγ N-terminal peptide PDEγ(10–30) disrupts Btn-PDEγ-mediated pulldown of RGS9·β5. Pulldown of RGS9·β5 and αt in the GAP complex was performed using Btn-PDEγ and streptavidin beads. To pulldown RGS9·β5 efficiently, Btn-PDEγ and αt were used in molar excess over the RGS9·β5 heterodimer (molar ratio 25:2.5:1). In each reaction, 2.5 μm Btn-PDEγ was first incubated with RGS9·β5 and αt-GDP on ice for 20 min in Buffer A, the reaction was then mixed with 0.5 μl of streptavidin beads and rotated at 4° C for 20–30 min. 10 mm NaF and 30 μm AlCl3 were added for the plus AlF4− conditions. At the end of incubation, the beads were washed twice with 100 μl of Buffer A. Proteins pulled down on the beads were analyzed by Western blotting. Each blot represents 2–4 similar experiments. A and B, specificity of the pulldown of RGS9 (A) and αt (B), as detected on the same blot, is indicated by lack of signal in the absence of Btn-PDEγ (lane 1) or low signal in the absence of AlF4− (lane 2). Western blotting was first performed with the mouse monoclonal anti-His6 antibody to visualize RGS9, and a secondary probing of αt was done with the rabbit polyclonal anti-αt antibody. C, Amido Black-stained streptavidin bands on the PVDF membrane indicate equal volume of beads used in each reaction. D and E, PDEγ(10–30) but not PDEγ(15–26) reduced RGS9 (D) and αt (E) pulldown. Btn-PDEγ, RGS9·β5, and αt-GDP-AlF4− were present in all conditions. Lane 1 represents exactly the same conditions as those of lane 3 in A and B. PDEγ(10–30) in a molar excess of 150 × (lane 2) or 1,500 × (lane 3) over PDEγ, or PDEγ(15–26)of 150 × (lane 4) was preincubated with RGS9·β5 and αt for 20 min on ice prior to addition of Btn-PDEγ. F, in the absence of RGS9·β5, PDEγ(10–30) (lane 4) or PDEγ(15–26) (lane 5) did not affect αt pulldown by Btn-PDEγ (compared with lane 3). Experiments were conducted under the same conditions as in A–E, except that RGS9·β5 was not added. The −Btn-PDEγ and −AlF4− controls are shown in lanes 1 and 2, respectively.
Importantly, pulldown of RGS9 decreased in the presence of PDEγ(10–30) at a 150-fold molar excess over Btn-PDEγ (lane 2 compared with lane 1 in D) and was almost completely abolished when PDEγ(10–30) was in a 1500-fold molar excess (lane 3). This indicates that PDEγ(10–30), which is part of the PDEγ N-terminal sequence, competed with the N-terminal interaction of the full-length Btn-PDEγ with RGS9·β5, further supporting direct interactions between RGS9·β5 and the PDEγ N-terminal positions that were observed from the label transfer (Fig. 1). In contrast to PDEγ(10–30), the peptide PDEγ(15–26), which has similar positive charge properties, however, did not reduce the level of RGS9 pulldown at all (lane 4). This result argues against the possibility that PDEγ(10–30) eliminated RGS9 pulldown (lane 3) nonspecifically by a charge effect, and it also supports the sequence specificity of the PDEγ(10–30) result. A consistent result was also obtained in the label transfer experiments (Fig. 2, lanes 7–9). Because PDEγ(10–30) contains two of the positions (16 and 30) on PDEγ that were shown to interact with RGS9 as well as β5 (Fig. 1, A and B), it is likely to be much more potent than PDEγ(15–26) in competing with the full-length PDEγ interaction.
An alternative explanation for the PDEγ(10–30) effect is that in the GAP complex PDEγ(10–30) may have competed with the interaction between PDEγ and αt and thus indirectly removed the RGS9 that was bound to αt. The data in Fig. 3E, however, shows only a slight decrease of αt-GDP-AlF4− in the presence of 1500-fold of PDEγ(10–30), whereas there was nearly complete loss of RGS9 pulldown (Fig. 3D). Considering that Btn-PDEγ was in a molar excess (25 mol of PDEγ versus 1 mol of RGS9·β5 dimer) and αt was also in excess (2.5-fold over the RGS9·β5 heterodimer), only a portion of αt was bound in the GAP complex whereas the majority of αt molecules were bound to Btn-PDEγ with no RGS9·β5 bound. Therefore, the observed reduction of αt pulldown in the presence of PDEγ(10–30) (Fig. 3E, lane 3) was most likely due to the depletion of RGS9·β5 to which αt was bound (Fig. 3D, lane 3). This further supports the conclusion that PDEγ(10–30) competed with the Btn-PDEγ N-terminal interaction with RGS9·β5 rather than that with αt-GDP-AlF4−. Indeed, in the absence of RGS9·β5, PDEγ(10–30) in 1500-fold molar excess did not reduce αt pulldown (Fig. 3F). This result is also consistent with a previous mutational study indicating that the three lysine residues (Lys41, Lys44, and Lys45) outside the Ile10–Phe30 region were mainly responsible for the PDEγ N-terminal interaction with activated αt (28).
Differential PDEγ Domain Interactions with the αt Conformers of the Transition State and Signaling State
Based on the new observation of a PDEγ N-terminal interaction with RGS9·β5 (Figs. 1–3), it is important to explore further whether the PDEγ·αt interaction is dynamically adjusted upon a change from the signaling state with PDE6 being a partner (13, 14), to the transition state when RGS9·β5 becomes a target for PDEγ (8, 10).
We asked the question whether PDEγ interacts with two conformers of αt, αt-GTPγS and αt-GDP-AlF4− differentially. We compared label transfer from the same PDEγ position to the two conformers of αt side by side on the same gel under exactly the same conditions using [125I]ACTP-PDEγ photoprobes (19) (Fig. 4). With this side-by-side comparison on the same gel we were able to eliminate variations caused by different gels, etc.
FIGURE 4.

Comparison of label transfer to αt-GTPγS and to αt-GDP-AlF4− from the same PDEγ position. Photocross-linking/label transfer experiments with [125I]ACTP-PDEγ and αt were conducted as described previously (13) (also see “Experimental Procedures”). The same batch of [125I]ACTP-PDEγ photoprobes reported previously (13, 19) was used in this study. The same [125I]ACTP-PDEγ photoprobe (at 0.8 μm) was incubated with 1 μm αt-GTPγS or 1 μm αt-GDP-AlF4− in Buffer B (20 mm HEPES, pH 7.5, 120 mm NaCl, 2 mm MgCl2) and then UV-photolyzed. The reactions with αt-GTPγS and αt-GDP-AlF4− were run side by side on the same gel, and intensities of the radiolabel transferred to the two conformers of αt were detected by autoradiography and compared after normalization with the protein amounts in the αt bands. αt-GDP-AlF4− was prepared by adding 10 mm NaF and 30 μm AlCl3 into holotransducin. βt thus served as an internal control. BSA was included in the reactions as another internal control. A, autoradiogram of label transfer from each PDEγ position to a pair of αt-GTPγS and αt-GDP-AlF4− (upper panel) and the corresponding αt bands on the Coomassie-stained gel (lower panel). Duplicates are shown for position 50. B, comparison of label transfer to the two αt conformers from the same PDEγ position. A labeling difference between the αt conformers from each PDEγ position is expressed as the percentage of the radiolabel on αt-GDP-AlF4− versus the label on αt-GTPγS minus 100%. Therefore, a positive value indicates more label on αt-GDP-AlF4− over αt-GTPγS, and a negative value is the reverse. Each bar is an average of 3–5 experiments (±S.D. (error bars)) except position 21 (one experiment). Student's t test indicates a very significant (**, p < 0.01) difference of percent labeling change (αt-GDP-AlF4− versus αt-GTPγS) between the PDEγ N-terminal region (positions 21, 30, 40, 50, and 60) and the C-terminal domain (positions 70, 73, 76 and 87). The photoprobe derivatization positions on PDEγ are listed under the figure.
As indicated in our previous study (13), these [125I]ACTP-PDEγ photoprobes did not have a major impact on the PDEγ·αt interaction and photolabeled αt in a specific manner. Consistently, in this study, although radiolabel was transferred from PDEγ to αt, both of the internal controls BSA and βt were not labeled (Fig. 4A), indicating the specificity of the label transfer.
Interestingly, as indicated by the quantitative results presented in Fig. 4B, compared with the photolabel transfer to αt-GTPγS, label transfer to αt-GDP-AlF4− from the PDEγ N-terminal positions 21, 30, 50, and 60 decreased, whereas label transfer from the C-terminal positions 70–87 increased. Although the difference in label transfer to the two αt conformers appeared less remarkable from position 40 or position 70, statistical analysis indicated a very significant overall difference between the PDEγ N-terminal 21–60 region and the C-terminal 70–87 region in terms of a change of label transfer to the two αt conformers. In other words, upon a change from the signaling state conformation to the transition state conformation of αt, the PDEγ C-terminal interaction with αt was enhanced whereas the N-terminal interaction with αt was weakened. The same pattern of change was also observed using the BBM photoprobes (data not shown).
This observation was further confirmed using pulldown as a secondary approach (Fig. 5). Because the N-terminal peptide PDEγ(1–61) could not readily pull down αt-GTPγS due to the weak interaction (13), we used the biotin-tagged PDEγ C-terminal half (PDEγ(46–87)-Btn), which was previously shown to pull down αt-GTPγS (13). As indicated by the data in Fig. 5B, PDEγ(46–87)-Btn pulled down αt-GDP-AlF4− ∼2-fold more efficiently than it did αt-GTPγS at the stoichiometry. Statistical analysis of pulldown of αt by PDEγ(46–87)-Btn indicated a significant difference between the two αt conformers (Fig. 5C). Considering that the PDEγ C-terminal domain (residues 62–87) contributes the major strength to binding with αt (13), these data are consistent with the label transfer results indicating an enhanced PDEγ C-terminal interaction with αt-GDP-AlF4− compared with that with αt-GTPγS (Fig. 4B).
FIGURE 5.
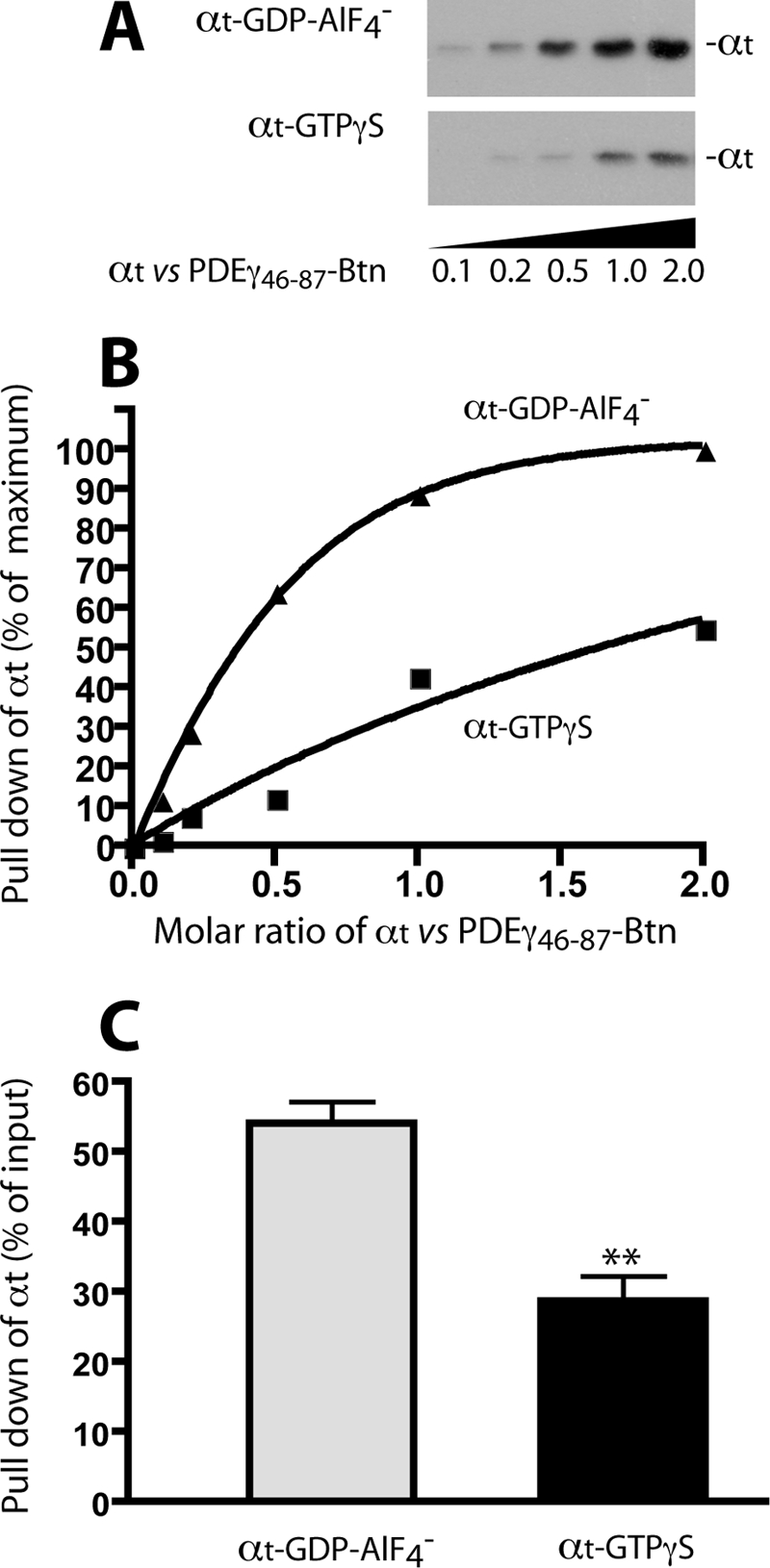
PDEγ(46–87)-Btn pulls down αt-GDP-AlF4− more efficiently than αt-GTPγS. A, pulldown experiments were performed essentially as described under “Experimental Procedures.” To each 0.4 μl of streptavidin beads 0.5 μg of PDEγ(46–87)-Btn was immobilized. PDEγ(46–87)-Btn was bound completely to the beads (data not shown) and then incubated with various concentrations of αt-GDP-AlF4− or αt-GTPγS for 1.5 h at 4° C in Buffer B. After washing the beads twice with 100 μl of Buffer B, αt remaining on the beads was eluted with the SDS/DTT-containing sample buffer and detected by Western blotting using the polyclonal rabbit antibody against the N terminus of αt. The molar ratios of αt versus PDEγ(46–87)-Btn are shown under the blot. B, quantification of αt pulldown in the Western blot in A. Background in the blot was subtracted, and the intensity of each band is expressed as a percentage of the highest signal (the last lane of the upper panel). C, statistical comparison of pulldown of the two αt conformers by PDEγ(46–87)-Btn (at the molar ratio of 2 αt versus 1 PDEγ). Each bar represents a mean (±S.D. (error bars), n = 3) of percent αt pull down relative to the input. **, t test, p < 0.01.
DISCUSSION
PDEγ N-terminal Interaction with RGS9·β5 Contributes to PDEγ Function in Stabilizing the GAP Complex
A PDEγ N-terminal interaction with RGS9β5 has not been reported previously. In fact, the PDEγ N-terminal half has been missing in the relevant atomic structures (7, 15, 17). Using a label transfer method, we detected interactions of PDEγ N-terminal positions with RGS9·β5 in the GAP complex (Figs. 1 and 2). This result was then confirmed by the effective competition of PDEγ(10–30) with the full-length Btn-PDEγ when pulling down RGS9·β5 from the GAP complex (Fig. 3). Importantly, our data indicate that the PDEγ N-terminal interaction with RGS9·β5 contributes to the role of PDEγ as an enhancer of the RGS9·β5·αt interaction in the transition state complex.
The label transfer approach applied here is validated by the existing crystal structure of the partial GAP complex that includes the C-terminal half of PDEγ (7). In this structure, Val66 on PDEγ, which is near position 68, makes contact with Trp362 of RGS9d. This hydrophobic interaction is complemented by an electrostatic RGS9 Arg360 interaction with PDEγ Glu52, which is close to Phe50. Thus, the photoprobe placed at position 68 or 50 on PDEγ was able to reach (as measured by PyMOL) and cross-link with RGS9, resulting in the label transfer to RGS9 that we have observed (Fig. 1, A and B).
Moreover, previous studies using an evolutionary trace method combined with mutational analysis predicted that residues 314, 353, 357–360, and 362–364 in the catalytic core of RGS9 form part of the effector-GAP interface (29, 30). Yet only one direct contact, that is between Trp362 and Val66 (on PDEγ), was resolved in the partial GAP structure (7). This raises the real possibility that the remainder of the predicted interface between RGS9 and PDEγ, i.e. the PDEγ N-terminal half that is missing in this crystal structure, was not revealed due to the PDEγ truncation.
Essentially, all the domains within the RGS9·β5 complex contribute to the PDEγ-regulated GAP function. In particular, the noncatalytic RGS9 domains together with β5 that are missing in the partial GAP structure (7), play an important role in discriminating between the activated αt alone and the PDEγ·αt complex (8, 9). This evidence also supports the likelihood that PDEγ N-terminal interactions with RGS9 and β5 occur but are missing in the partial GAP structure.
Indeed, our data are consistent with a direct interaction of the PDEγ N-terminal half with RGS9·β5, because the PDEγ N-terminal peptide PDEγ(1–61) effectively reduced RGS9 labeling (Fig. 2). Another N-terminal peptide, PDEγ(10–30), also eliminated pulldown of RGS9·β5 by directly competing with the PDEγ·RGS9·β5 interactions (see Fig. 3 and corresponding comments under “Results”).
More importantly, the fact that the N-terminal PDEγ peptides could effectively disassemble the GAP complex (Figs. 2 and 3) indicates that the N-terminal half of PDEγ contributed considerably to enhancing the affinity between RGS9·β5 and αt. The greater potency of PDEγ(1–61) than PDEγ(62–87) in preventing label transfer from the PDEγ photoprobes to RGS9 (Fig. 2) suggests that the N-terminal half of PDEγ plays an important role for the function of PDEγ as an affinity enhancer to tighten the binding of αt with RGS9·β5 in the GAP complex. As shown in previous studies, whereas the PDEγ C-terminal peptide PDEγ(63–87) was equally potent in stimulating the GAP activity, the full-length PDEγ was needed to maintain a high affinity GAP complex (10). We propose that whereas the PDEγ C-terminal interaction with RGS9 (in particular, PDEγ Val66 with Trp362 in the RGS domain) is essential to secure a correct conformation of αt for efficient GTP hydrolysis (7), the PDEγ N-terminal interactions with RGS9β5 complement the C-terminal interaction to ensure sufficient affinity between αt and RGS9·β5.
Differential PDEγ Interactions with the Two αt Conformers Suggest a “Rearrangement” That Primes the PDEγ N-terminal Half for Interaction with RGS9·β5
In this study, we also identified differential PDEγ domain interactions with αt in two different signal transduction states, the signaling state conformation preserved in αt-GTPγS (5) and the transition state conformation mimicked by αt-GDP-AlF4− (6). The interaction of the PDEγ N-terminal domain with αt-GDP-AlF4− was determined to be weaker compared with that with αt-GTPγS, whereas the PDEγ C-terminal domain bound to αt-GDP-AlF4− stronger than with αt-GTPγS (Figs. 4 and 5). Differential PDEγ domain interactions with the two αt conformers may not be readily detectable using methods other than the label transfer approach applied here. Given the high similarities between the two αt conformers in their crystal structures (5, 6) and their affinities with the full-length PDEγ (31–34), this issue appears to have been previously overlooked.
Although the overall differences between the crystal structures of αt-GTPγS and αt-GDP-AlF4− are not remarkable (5, 6), a considerable difference occurs in the αt Switch II region, which is flexible and contains primary binding sites for both RGS9d and the PDEγ C-terminal domain (7, 33, 35). Interestingly, a single mutation in the αt Switch II region (E203A) turned αt constitutively active for PDE6 activation even in the GDP-bound form (36). Another Switch II mutant, W207F, underwent the same type of conformational change of Switch II as did the wild-type αt upon activation, yet had ∼100-fold lower affinity with PDEγ (37). These examples together with a systematic mutational analysis of the Switch II region (35) strongly argue for the notion that even a minor structural difference in αt Switch II is sufficient to mediate significant changes in the αt·PDEγ interactions. Moreover, the RGS domain binds to the two similar αt conformers with drastically different affinities with a preference for the transition state conformation (35, 38–40). Further, PDEγ together with RGS9 allosterically changes the αt Switch II region into a stable conformation optimal for GTP hydrolysis (7). Allosteric interplay between the RGS and effector binding sites via the Switch II region appears possible for all Gα subunits that bind RGS proteins. Thus, subtle changes in the conformation of Gα Switch II region could have profound effects on the affinity of effectors and GAPs (41).
Accordingly, the observed changes in the binding strength of the PDEγ N-terminal and C-terminal domains with αt-GDP-AlF4− relative to that with αt-GTPγS (Figs. 4 and 5) may be rationalized by the function of PDEγ in the transition state complex. Because PDEγ acts by physically enhancing the interaction between αt and RGS9·β5 (8, 10), the interaction of the PDEγ C terminus with αt is an important binding force for stabilizing the GAP complex. An enhanced binding of the PDEγ C terminus with αt in the transition state complex is therefore induced (Figs. 4 and 5) after its removal from PDEαβ in the signaling state (7, 17). On the other hand, the PDEγ N-terminal interaction with αt in the transition state is reduced compared with that in the signaling state (Fig. 4). Thus, in the transition state the most reasonable role for the PDEγ N-terminal regions is to bind RGS9·β5, providing additional strength to stabilize the GAP complex.
Therefore, we propose that upon a change of the αt conformation from the signaling state to the transition state, a “rearrangement” of the PDEγ domain interactions with αt occurs, even though the overall PDEγ affinity with αt is not significantly altered (31–34). In the signaling state, a “transducisome” is formed by the complementary PDEγ C-terminal interaction with αt-GTP and the N-terminal interaction with PDEαβ (13). Evidenced by a high sensitivity to trypsinization (42, 43) and an extended structure in the PDEαβ-bound state (20), the PDEγ N-terminal half is likely exposed on the surface of PDEαβ GAF domains (44), providing an interface to interact with αt simultaneously in the transducisome. In the ensuing transition state, RGS9·β5 recognizes and binds to the PDEγ C-terminal complex with αt. This may allosterically change αt into a conformation that tightens its binding with the PDEγ C-terminal domain while loosening its interaction with the PDEγ N-terminal half (Fig. 4), which now becomes available to bind to RGS9·β5 (Figs. 1–3). With the C-terminal domain tightly bound to αt and the N-terminal regions (and Val66) interacting with RGS9·β5, PDEγ likely serves as a molecular “clasp” to stabilize the αt interaction with RGS9. Indirect evidence suggests that PDEγ stays associated with PDEαβ in the transition state (45), perhaps binding to both PDEαβ and RGS9·β5 via alternate N-terminal residues in different orientations. Although the PDEγ C-terminal domain together with the RGS domain induce an optimal αt Switch II structure for GTP hydrolysis (7, 15), the role of the PDEγ N-terminal domain is most reasonably to be an interaction with RGS9·β5 to secure a stable GAP complex, which is critical for efficient visual signal termination.
As an intrinsically disordered small protein (18), PDEγ plays important physiological roles in the photoreceptor neurons (46–48) via interactions with PDE6, transducin, as well as RGS9·β5 (3, 4). In the context of activation and ensuing termination of phototransduction, it remains enigmatic as to how PDEγ alternates/adapts to different targets to regulate the amplitude and duration of the photoresponse. Future studies on comparison of PDEγ interactions in the signaling state and transition state are warranted.
Supplementary Material
Acknowledgments
We thank Drs. A. R. Hajipour and M. Arbabian for assistance in radiosynthesis and preparation of transducin samples.
This work was supported, in whole or in part, by National Institutes of Health Grant GM-33138 (to A. E. R. and L.-W. G. and a Retina Research Foundation Edwin and Dorothy Gamewell Professorship (to A. E. R.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- PDE6
- rod photoreceptor cGMP phosphodiesterase
- ACTP
- N-[3-iodo-4-azidophenylpropioamido-S-(2-thiopyridyl)]cysteine
- αt
- transducin α-subunit
- β5
- type 5 G protein β-subunit
- BBM
- 2-[Nα-benzoylbenzoicamido-N6-(6-biotinamidocaproyl)-l-lysinyamido] ethyl methanethiosulfonate
- βt
- transducin β-subunit
- Btn
- biotinylated
- GAP
- GTPase-activating protein
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- NTA
- nitrilotriacetic acid
- PDEαβ
- PDE6 catalytic heterodimer
- PDEγ
- PDE6 inhibitory subunit
- RGS9
- ninth member of regulators of G protein signaling in photoreceptors
- RGS9d
- the RGS9 catalytic domain.
REFERENCES
- 1. Arshavsky V. Y., Lamb T. D., Pugh E. N., Jr. (2002) Annu. Rev. Physiol. 64, 153–187 [DOI] [PubMed] [Google Scholar]
- 2. Burns M. E., Arshavsky V. Y. (2005) Neuron 48, 387–401 [DOI] [PubMed] [Google Scholar]
- 3. Cote R. H. (2008) in Visual Transduction and Non-Visual Light Perception (Tombran-Tink J., Barnstable C. J. eds) pp. 141–169, Humana Press, Totawa, NJ [Google Scholar]
- 4. Guo L. W., Ruoho A. E. (2008) Curr. Protein Pept. Sci. 9, 611–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Noel J. P., Hamm H. E., Sigler P. B. (1993) Nature 366, 654–663 [DOI] [PubMed] [Google Scholar]
- 6. Sondek J., Lambright D. G., Noel J. P., Hamm H. E., Sigler P. B. (1994) Nature 372, 276–279 [DOI] [PubMed] [Google Scholar]
- 7. Slep K. C., Kercher M. A., He W., Cowan C. W., Wensel T. G., Sigler P. B. (2001) Nature 409, 1071–1077 [DOI] [PubMed] [Google Scholar]
- 8. Skiba N. P., Martemyanov K. A., Elfenbein A., Hopp J. A., Bohm A., Simonds W. F., Arshavsky V. Y. (2001) J. Biol. Chem. 276, 37365–37372 [DOI] [PubMed] [Google Scholar]
- 9. Martemyanov K. A., Arshavsky V. Y. (2002) J. Biol. Chem. 277, 32843–32848 [DOI] [PubMed] [Google Scholar]
- 10. Skiba N. P., Hopp J. A., Arshavsky V. Y. (2000) J. Biol. Chem. 275, 32716–32720 [DOI] [PubMed] [Google Scholar]
- 11. Artemyev N. O., Rarick H. M., Mills J. S., Skiba N. P., Hamm H. E. (1992) J. Biol. Chem. 267, 25067–25072 [PubMed] [Google Scholar]
- 12. Mou H., Cote R. H. (2001) J. Biol. Chem. 276, 27527–27534 [DOI] [PubMed] [Google Scholar]
- 13. Guo L. W., Hajipour A. R., Ruoho A. E. (2010) J. Biol. Chem. 285, 15209–15219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang X. J., Skiba N. P., Cote R. H. (2010) J. Biol. Chem. 285, 4455–4463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheever M. L., Snyder J. T., Gershburg S., Siderovski D. P., Harden T. K., Sondek J. (2008) Nat. Struct. Mol. Biol. 15, 155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. He W., Lu L., Zhang X., El-Hodiri H. M., Chen C. K., Slep K. C., Simon M. I., Jamrich M., Wensel T. G. (2000) J. Biol. Chem. 275, 37093–37100 [DOI] [PubMed] [Google Scholar]
- 17. Barren B., Gakhar L., Muradov H., Boyd K. K., Ramaswamy S., Artemyev N. O. (2009) EMBO J. 28, 3613–3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Song J., Guo L. W., Muradov H., Artemyev N. O., Ruoho A. E., Markley J. L. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 1505–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guo L. W., Grant J. E., Hajipour A. R., Muradov H., Arbabian M., Artemyev N. O., Ruoho A. E. (2005) J. Biol. Chem. 280, 12585–12592 [DOI] [PubMed] [Google Scholar]
- 20. Guo L. W., Muradov H., Hajipour A. R., Sievert M. K., Artemyev N. O., Ruoho A. E. (2006) J. Biol. Chem. 281, 15412–15422 [DOI] [PubMed] [Google Scholar]
- 21. Grant J. E., Guo L. W., Vestling M. M., Martemyanov K. A., Arshavsky V. Y., Ruoho A. E. (2006) J. Biol. Chem. 281, 6194–6202 [DOI] [PubMed] [Google Scholar]
- 22. Fung B. K., Nash C. R. (1983) J. Biol. Chem. 258, 10503–10510 [PubMed] [Google Scholar]
- 23. Mazzoni M. R., Malinski J. A., Hamm H. E. (1991) J. Biol. Chem. 266, 14072–14081 [PubMed] [Google Scholar]
- 24. Guo L. W., Assadi-Porter F. M., Grant J. E., Wu H., Markley J. L., Ruoho A. E. (2007) Protein Expr. Purif. 51, 187–197 [DOI] [PubMed] [Google Scholar]
- 25. Martemyanov K. A., Arshavsky V. Y. (2004) Methods Enzymol. 390, 196–209 [DOI] [PubMed] [Google Scholar]
- 26. Guo L. W., Hajipour A. R., Gavala M. L., Arbabian M., Martemyanov K. A., Arshavsky V. Y., Ruoho A. E. (2005) Bioconjug. Chem. 16, 685–693 [DOI] [PubMed] [Google Scholar]
- 27. Baehr W., Devlin M. J., Applebury M. L. (1979) J. Biol. Chem. 254, 11669–11677 [PubMed] [Google Scholar]
- 28. Brown R. L. (1992) Biochemistry 31, 5918–5925 [DOI] [PubMed] [Google Scholar]
- 29. Sowa M. E., He W., Wensel T. G., Lichtarge O. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 1483–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sowa M. E., He W., Slep K. C., Kercher M. A., Lichtarge O., Wensel T. G. (2001) Nat. Struct. Biol. 8, 234–237 [DOI] [PubMed] [Google Scholar]
- 31. Slepak V. Z., Artemyev N. O., Zhu Y., Dumke C. L., Sabacan L., Sondek J., Hamm H. E., Bownds M. D., Arshavsky V. Y. (1995) J. Biol. Chem. 270, 14319–14324 [DOI] [PubMed] [Google Scholar]
- 32. Skiba N. P., Artemyev N. O., Hamm H. E. (1995) J. Biol. Chem. 270, 13210–13215 [DOI] [PubMed] [Google Scholar]
- 33. Skiba N. P., Bae H., Hamm H. E. (1996) J. Biol. Chem. 271, 413–424 [DOI] [PubMed] [Google Scholar]
- 34. Artemyev N. O. (1997) Biochemistry 36, 4188–4193 [DOI] [PubMed] [Google Scholar]
- 35. Natochin M., Granovsky A. E., Artemyev N. O. (1998) J. Biol. Chem. 273, 21808–21815 [DOI] [PubMed] [Google Scholar]
- 36. Mittal R., Erickson J. W., Cerione R. A. (1996) Science 271, 1413–1416 [DOI] [PubMed] [Google Scholar]
- 37. Faurobert E., Otto-Bruc A., Chardin P., Chabre M. (1993) EMBO J. 12, 4191–4198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Natochin M., Granovsky A. E., Artemyev N. O. (1997) J. Biol. Chem. 272, 17444–17449 [DOI] [PubMed] [Google Scholar]
- 39. McEntaffer R. L., Natochin M., Artemyev N. O. (1999) Biochemistry 38, 4931–4937 [DOI] [PubMed] [Google Scholar]
- 40. Skiba N. P., Yang C. S., Huang T., Bae H., Hamm H. E. (1999) J. Biol. Chem. 274, 8770–8778 [DOI] [PubMed] [Google Scholar]
- 41. Shankaranarayanan A., Thal D. M., Tesmer V. M., Roman D. L., Neubig R. R., Kozasa T., Tesmer J. J. (2008) J. Biol. Chem. 283, 34923–34934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hurley J. B., Stryer L. (1982) J. Biol. Chem. 257, 11094–11099 [PubMed] [Google Scholar]
- 43. Artemyev N. O., Hamm H. E. (1992) Biochem. J. 283, 273–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Muradov H., Boyd K. K., Artemyev N. O. (2004) Vision Res. 44, 2437–2444 [DOI] [PubMed] [Google Scholar]
- 45. Wensel T. G. (2008) Vision Res. 48, 2052–2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tsang S. H., Burns M. E., Calvert P. D., Gouras P., Baylor D. A., Goff S. P., Arshavsky V. Y. (1998) Science 282, 117–121 [DOI] [PubMed] [Google Scholar]
- 47. Tsang S. H., Gouras P., Yamashita C. K., Kjeldbye H., Fisher J., Farber D. B., Goff S. P. (1996) Science 272, 1026–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tsang S. H., Woodruff M. L., Janisch K. M., Cilluffo M. C., Farber D. B., Fain G. L. (2007) J. Physiol. 579, 303–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Baker S. A., Martemyanov K. A., Shavkunov A. S., Arshavsky V. Y. (2006) Biochemistry 45, 10690–10697 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.