

Abstract
The enzymatic transfer of the sugar mannose from activated sugar donors is central to the synthesis of a wide range of biologically significant polysaccharides and glycoconjugates. In addition to their importance in cellular biology, mannosyltransferases also provide model systems with which to study catalytic mechanisms of glycosyl transfer. Mannosylglycerate synthase (MGS) catalyzes the synthesis of α-mannosyl-d-glycerate using GDP-mannose as the preferred donor species, a reaction that occurs with a net retention of anomeric configuration. Past work has shown that the Rhodothermus marinus MGS, classified as a GT78 glycosyltransferase, displays a GT-A fold and performs catalysis in a metal ion-dependent manner. MGS shows very unusual metal ion dependences with Mg2+ and Ca2+ and, to a lesser extent, Mn2+, Ni2+, and Co2+, thus facilitating catalysis. Here, we probe these dependences through kinetic and calorimetric analyses of wild-type and site-directed variants of the enzyme. Mutation of residues that interact with the guanine base of GDP are correlated with a higher kcat value, whereas substitution of His-217, a key component of the metal coordination site, results in a change in metal specificity to Mn2+. Structural analyses of MGS complexes not only provide insight into metal coordination but also how lactate can function as an alternative acceptor to glycerate. These studies highlight the role of flexible loops in the active center and the subsequent coordination of the divalent metal ion as key factors in MGS catalysis and metal ion dependence. Furthermore, Tyr-220, located on a flexible loop whose conformation is likely influenced by metal binding, also plays a critical role in substrate binding.
Keywords: Carbohydrate Biosynthesis, Enzyme Catalysis, Enzyme Mechanisms, Enzyme Structure, Glycosylation, X-ray Crystallography, Carbohydrate-active Enzymes, Glycosyltransferase
Introduction
Mannose-containing oligosaccharides, polysaccharides, and glycoconjugates are ubiquitous across nature. The sugar is present in N-linked glycans and glycolipids, whereas mannose-containing polysaccharides represent key features of plant, fungal, and bacterial cell walls. In higher organisms, glycan decorations on glycoproteins have the potential to modulate many integral cellular processes, including protein trafficking, cell signaling, immune modulation, and host-pathogen recognition (1, 2). Reflecting their role in modulating many key cellular processes, mannose-processing enzymes, both glycoside hydrolases and glycosyltransferases (GTs),6 have considerable potential as cellular targets to probe both healthy and diseased cells.
To survive in the harsh conditions of their environments, extremophiles attach mannose to small negatively charged molecules such as glyceramide, glycerate, lactate, and phosphoglycerate, forming molecules which confer tolerance to osmotic stress and thermostability (3). These tolerance-conferring osmolytes function through balancing the ionic strength across membranes in saline-rich environments, as well as stabilizing proteins by reducing backbone motions, resulting in overall protein rigidification and melting temperature elevation (4, 5). Of these stabilizers, 2-O-α-d-mannosylglycerate appears to be both a potent and highly prevalent osmolyte in thermophilic and hyperthermophilic eubacteria and archaea (3). 2-O-α-d-Mannosylglycerate synthesis is catalyzed by a GDP-mannose-dependent GT, termed mannosyl-glycerate synthase (MGS), an enzyme that is central to the studies reported here.
Based on sequence similarities, GTs have been grouped into approximately 90 families in the carbohydrate-active enzyme database (6, 7), roughly 19 of which contain members that transfer mannosyl groups, although other donor substrate specificities are evident in many of these families. In the majority of these families, the donor substrate is GDPMan (2, 4, 15, 26, 32, 33, 55, 62, 69, 71, 78, 81, 91), although in GT families 22, 39, 50, 58, 76, and 87, the mannosyl residue is transferred from an activated phospholipid to its donor molecule. Surprisingly, given the sequence diversity represented across GT families, predominantly only two structural folds are adopted by nucleotide sugar-dependent GTs; termed GT-A and GT-B (6, 8). A third fold class, GT-C, has recently been proposed for the transmembrane-spanning GT family 66, which utilize lipid phosphate-activated sugars as the donor substrate (9, 10).
The majority of GT-A-type enzymes employ divalent cations to assist catalysis, whereas GT-B classified enzymes utilize positively charged side chains and/or hydroxyls and helix dipoles (reviewed in Ref. 11). The presence of the metal in the catalytic cycle is therefore pivotal to the major distinction between the GTs; yet the variation of this metal and the implications for its role have been explored little.
The overall architecture of both GT-A and GT-B fold types is similar in that each consists of a pair of β/α/β Rossmann-like domains. In the GT-A fold, the Rossmann-like domains are tightly associated, forming a centrally extended β-sheet architecture whose loops change conformation upon ligand binding, whereas in GT-Bs, the two domains are more loosely associated “facing” each other and often moving as whole domains in response to ligand binding (11). Mechanistically, GTs may also be divided into whether the reaction leads to net retention or inversion of the stereochemistry of the anomeric carbon of the donor. Representatives of both inverting and retaining GTs exist in both fold families. Analogous to the mechanism of inverting glycoside hydrolases, inverting GTs most likely follow a single SN2-like displacement mechanism where the acceptor molecule, which is activated generally be a catalytic base, acts as a nucleophile, attacking the sugar donor at the anomeric carbon (reviewed in Ref. 11). In contrast, the mechanistic details of retaining GTs are far less clear, although it has been proposed that these enzymes catalyze glycosidic bond formation through an SNi-like reaction of asynchronous “front face” departure and attack, which is mechanistically analogous to the well known reaction of alkyl chlorosulfites (Ref. 12 and recently discussed in a GT context in Refs. 11, 13). The continuing catalytic ambiguity of retaining GT highlights that, despite the importance of glycosyl transfer in nature and its potential for commercial application, there is a paucity of structural and mechanistic information on these enzymes.
The crystal structures have been reported for three mannosyltransferases that produce the protective osmolytes 2-O-α-d-mannosylglycerate (family 78) or mannosyl-3-phosphoglycerate (family 55 and 81) (14–16). The GT family 78 mannosyltransferase, MGS, is expressed by the hyperthermophilic bacterium Rhodothermus marinus. MGS adopts a GT-A fold and catalyzes glycosidic bond formation with retention of anomeric configuration of the transferred α-mannosyl moiety (Fig. 1a) (14). Employing a high throughput substrate screening strategy, MGS was shown to utilize GDP-α-mannose (and other GDP-sugars) as a donor substrate in a reaction in which the mannose is transferred not only to the C2-OH group of d-glycerate but also d-lactate or glycolate. Kinetically, MGS was shown to employ various divalent cations, with Mg2+ and Ca2+ and, to a lesser extent, Mn2+, Ni2+, and Co2+, facilitating catalysis.
FIGURE 1.

Anatomy of mannosylglycerate synthase. a, reaction catalyzed by mannosylglycerate synthase is the transfer of mannose, from GDP-mannose to d-glycerate to generate α-d-mannosyl 2-O-d-glycerate in a metal ion-dependent manner. b, composite diagram of Mg2+ + GDP-mannose + d-glycerate donor and acceptor substrates shown in ball-and-stick format. The flexibility of residues 215–222 in the MGS, the complex structures presented in Ref. 14 of Mn2+ + d-glycerate, citrate, Co2+ + GDP, and Mn2+ + GDP-mannose (PDB codes 2BO4, 2BO6, 2BO7, and 2BO8, respectively) are highlighted in red, blue, yellow, and magenta, respectively, overlapped with Mg2+ + GDP-mannose presented herein. This stretch of residues includes His-217 shown to be important for cation coordination in the active center. Positions of Gln-201 and Gln-202 variants, used to lower surface entropy of MGS, are shown in blue. c, composite diagram (divergent “wall-eyed” stereo) of the MGS active center with Mg2+ + GDP-mannose and d-glycerate shown in ball-and-stick format are colored gray. Some of the active center residues mutated and subsequently analyzed by kinetic methods, and in some cases biophysical methods (His-217 and Tyr-220), are shown in green. d, composite schematic diagram of the MGS active center based upon known GDP-mannose and acceptor complexes. Tyr-220 is present in a highly mobile loop with past crystal structures representing just one possible interpretation. The key structural feature is that Tyr-220 becomes well ordered in the GDPMan complex (blue) and then lies in a position where it would likely interact with the glycerate acceptor (colored red), which is consistent with kinetic data of variants presented here.
Here, we have used structural and biochemical approaches to probe the unusual variation in metal ion specificity displayed by MGS, as well as its donor recognition and acceptor substrate plasticity. The data revealed the structural features that determine the relative preferences for acceptor substrates, provided evidence for GDP interaction while also shedding light on the functional significance of His-217 in the selection of catalytic metal ions by the enzyme.
EXPERIMENTAL PROCEDURES
Materials
2-O-α-d-Mannosylglycerate for isothermal titration calorimetry (ITC) and product inhibition studies was synthesized and purified as described in the supplemental methods of Ref. 14.
Cloning, Site-directed Mutagenesis, and Expression of RmMGS
pJF1 and pJF2 (encoding full-length MGS and MGS with a 15-residue C-terminal truncation (MGS-Tr), respectively) were constructed by amplification of MGS from R. marinus genomic DNA as described previously (14). Mutants for kinetic studies were constructed by using the QuikChange site-directed mutagenesis kit (Stratagene) with pJF1 as template and primers listed in supplemental Table S1. MGS with a 15 residue, C-terminal, truncation was used for crystallization complexes with lactate. A variant protein was used for crystallization of other complexes (see under “Crystallization Data Collection and Structural Determination”). MGS wild-type and mutants were expressed and purified as described previously (14). Briefly, Escherichia coli (strain Tuner) transformed with appropriate plasmid was grown to an optical density, at 550 nm, of ∼0.6 in 1 liter of LB media with 50 μg/ml ampicillin and then incubated at 16 °C before induction of MGS expression with 0.2 mm isopropyl β-d-thiogalactopyranoside. Cell-free extracts were heat-treated at 65 °C for 20 min, and MGS was purified by immobilized metal ion affinity chromatography. Proteins for kinetic studies were sufficiently pure after immobilized metal ion affinity chromatography purification. When analyzing the influence of metals on enzyme function, MGS was treated with Chelex-100 (Bio-Rad) to unwanted metals. For crystallography, MGS was further purified by anion exchange.
MGS Surface Entropy Minimization Engineering
Large three-dimensional crystals of MGS-Tr readily develop in an array of crystallization conditions. However, the maximum resolution obtained during diffraction experiments of complex structures was of moderate quality (2.45–2.95 Å) (14). In an attempt to ameliorate MGS-Tr surface properties for crystallization, MGS-Tr was analyzed with SERp (17) to suggest segments of residues that make substantial surface entropy contributions. SERp highlighted three putative clusters at positions Glu-89/Glu-90, Glu-115/Glu-116, and Gln-253/Glu-254. An additional site was selected at Gln-201/Gln-202 through manual structural inspection (Fig. 1b). Each cluster was evaluated for solvent exposure and contribution to intra- and intermolecular contacts using known MGS-Tr structures. Based on this analysis, E89A/E90A and Q201A/Q202A MGS-Tr mutants were generated by site-directed mutagenesis of which only the Q201A/Q202A MGS surface variant expressed to an appreciable amount.
Crystallization Data Collection and Structural Determination
Recombinant MGS-Tr-Q201A/Q202A surface variant (MGS-Sv) was expressed and purified as described previously for MGS-Tr (14) with the exception that MGS-Sv was buffer exchanged into 25 mm Tris-HCl, 500 mm NaCl, pH 8.0, prior to crystallization. MGS-Sv crystals developed after ∼3 days in hanging drop setup at 19 °C in equal volumes of protein (15 mg/ml) and reservoir solution consisting of 4% (v/v) ethylene glycol, Bistris propane, pH 5.5, supplemented with either magnesium formate, manganese formate, or sodium malonate between 0.2 and 0.5 m, where appropriate. MGS-lactate crystals were grown in 0.3 m sodium d-lactate, 0.1 m sodium acetate trihydrate, pH 4.6, 40% (v/v) 2-methyl-1,3-propanediol, which acted as a cryoprotectant. GDP and GDPMan complexed crystals were obtained by soaking apo-crystals in mother liquor supplemented with 10 mm ligand for ∼10 min and subsequently cryoprotected in mother liquor supplemented with 35% (v/v) ethylene glycol.
MGS-Tr diffraction data were collected at beamlines ID-29 of the European Synchrotron Radiation Facility (lactate and Mg2+ + GDP), IO3 of Diamond Light Source (Mn2+ + GDP), and the York Structural Biology Laboratory home source (Mg2+ + GDPMan). In each case, MGS crystals were evaluated to belong to the space group P3221 and to contain two MGS molecules in the asymmetric unit. Data were processed with either the HKL2000 suite (18) or iMOSFLM/Scala (19, 20), and structural solutions were obtained by molecular replacement using PHASER (21). Ligands were subsequently incorporated followed by cycles of maximum likelihood refinement using REFMAC (22) interspersed with manual corrections of protein models using COOT (23). Overall, the surface entropy minimization of the MGS-Tr Q201A/Q202A mutant together with the identification of novel crystallization conditions resulted in a slight improvement to diffraction. Other computing used the CCP4 suite (24) unless otherwise stated. Data processing and refinement statistics are presented in Table 1. Structural figures were drawn with PyMol (DeLano Scientific LLC).
TABLE 1.
Data collection and refinement statistics
| MGS + lactate | MGS Mg2+ + GDP | MGS Mn2+ + GDP | MGS Mg2+ + GDP-mannose | |
|---|---|---|---|---|
| Data collection | ||||
| Space group | P3221 | P3221 | P3221 | P3221 |
| Cell dimensions | ||||
| a, b, c | 149.0, 149.0, 155.0 Å | 149.6, 149.6, 154.1 Å | 149.8, 149.8, 154.7 Å | 149.5, 149.5, 154.3 Å |
| α, β, γ | 90, 90, 120° | 90, 90, 120° | 90, 90, 120° | 90, 90, 120° |
| Resolution | 67 to 2.30 Å (2.42 to 2.30 Å) | 50 to 2.45 Å (2.54 to 2.45 Å) | 50 to 2.80 Å (2.90 to 2.80 Å) | 23.4 to 2.70 Å (2.85 to 2.70 Å) |
| Rmerge | 0.10 (0.37) | 0.072 (0.52) | 0.075 (0.59) | 0.12 (0.55) |
| I/σI | 7.6 (2.4) | 17.5 (2.9) | 22.3 (3.4) | 9.5 (2.8) |
| Completion | 99.7% (96.8%) | 100% (100%) | 99.9% (100%) | 96.8% (98.8%) |
| Redundancy | 2.6 (2.6) | 6.1 (6.3) | 10.9 (10.7) | 5.4 (5.3) |
| Refinement | ||||
| Rwork/Rfree | 0.20/0.22 | 0.20/0.23 | 0.19/0.22 | 0.19/0.22 |
| Root mean square deviations | ||||
| Bond lengths | 0.017 Å | 0.015 Å | 0.016 Å | 0.016 Å |
| Bond angles | 1.58° | 1.52° | 1.67° | 1.62° |
| PDB codes | 2y4j | 2y4k | 2y4l | 2y4m |
Mass Spectrometric Activity Screening through Direct Detection of Products
Initial rates were determined using a quantitative mass spectrometry-based assay as described previously (14, 25). Briefly, a calibration curve for mannosylglycerate was constructed by measuring the ratios of the total ion count of mannosylglycerate at varying concentrations (1, 3, 5, 7, and 9 μm) to the total ion count of pseudo-internal standard UDP (100 μm) in 1 mm Tris-HCl buffer, pH 7.8. Enzyme-catalyzed reactions were performed in 1 mm Tris-HCl buffer, pH 7.8, at 37 °C, using final concentrations of 100 μm GDPMan, 100 μm d-glycerate, 50 μm Ca2+, and 1 μm enzyme. Initial rates were determined from mannosylglycerate concentrations calculated every 30 min over 480 min from total ion count measurements of a 10-μl aliquot mixed with 10 μl of UDP (100 μm). Initial rates determined by MS methods were within error of those determined by linked methods thus eliminating the possibility of artifacts from the latter.
Continuous Enzyme-linked Assay
Initial rates were also determined using indirect detection of GDP by coupling the release of GDP to NADH oxidation via pyruvate kinase and lactate dehydrogenase developed from Ref. 26. Assays were performed in final concentrations as follows: 10 mm HEPES, pH 7.0, 50 mm KCl, 0.1% (w/v) BSA, 0.15 mm NADH, 0.7 mm phosphoenolpyruvate potassium salt, 6 units/ml pyruvate kinase, and 30 units/ml lactate dehydrogenase (both Sigma, rabbit muscle-type II, ammonium sulfate suspension) with 75–500 nm enzyme at 23 °C. NADH oxidation was monitored by the decrease in absorbance at 340 nm. Either 2 mm MgCl2 or 2 mm MnCl2 was added to each assay to test the effect of the metal ion. For each variant of MGS assayed, the reaction was carried out in the absence of acceptor and showed no GDP release. These experiments confirmed that there was no enzyme-mediated hydrolysis of the donor substrate.
For pseudo-single substrate kinetics, the fixed substrate concentration used was >3 times the Km value. kcat and Km values were calculated by fitting the Michaelis-Menten equation to the initial rates (GraphPad Prism version 4.03, GraphPad Software, San Diego). Bi-substrate kinetics used concentrations of each substrate corresponding to ½, 1, 2, 4, and 10 times the Km value determined in the pseudo-single substrate kinetics. Data were analyzed for a general bi-substrate system using Equation 1,
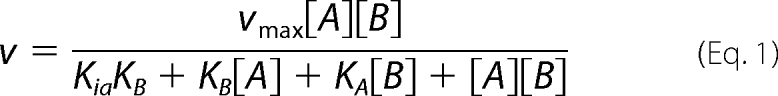 |
KA and KB are the limiting Michaelis constant for acceptor (A) and donor (B), respectively, when the other substrate is saturating. Ki,a and Ki,b are the dissociation constants between enzyme and acceptor or donor respectively. KA, KB, Ki,a, and Ki,b values were calculated as described previously (27).
Metal Ion Dependence and Product Inhibition Analysis
The metal ion dependences of MGS WT and the H217A variant were determined by adding 0.001–0.5 μm enzyme at 37 °C to (final concentration) 100 μm d-glycerate and 500 μm GDP-mannose in 13 mm HEPES, pH 7.0, 1 mm EDTA, 0.1% (w/v) BSA with 3 mm of either CaCl2, CoCl2, MnCl2, or NiCl2. Aliquots were mixed with EDTA (final concentration 10 mm) to stop the reaction. Product inhibition reactions were carried out in 10 mm HEPES, pH 7.0, 50 mm KCl, 2 mm CaCl2, 0.1% (w/v) BSA, and 0.1 μm enzyme at 23 °C. The concentration of d-glycerate was fixed at 500 μm, whereas the concentration of GDPMan covered a range from 50 to 4000 μm. GDP was added to give a final concentration of 10–100 μm. Aliquots were mixed with the same volume of 10 mm EDTA as a metal chelator to inactivate MGS.
Direct Measurement of GDP Release
The release of GDP was determined by HPLC (Gemini C18, 5 μm, 250 × 4.60 mm column (Phenomenex), Dionex ICS-3000 HPLC, 100-μl aliquots) equilibrated with buffer A (50 mm KH2PO4, 5 mm tetrabutylammonium hydrogen sulfate, 2% (v/v) acetonitrile). GDP and GDPMan were eluted using a linear gradient (0–20% of buffer B (100% (v/v) acetonitrile)) and monitored by UV detection (260 nm) at 25 °C (Dionex AD20 absorbance detector).
Isothermal Titration Calorimetry
ITC measurements were performed on a VP-ITC microcalorimeter from MicroCal Inc. (Northampton, MA). All titrations were carried out at 25 °C in 20 mm Tris-HCl, pH 8.0. The concentrations of ligand (GDPMan, d-glycerate (calcium salt), GDP, and mannosylglycerate) used were 0.5–1 mm and MGS wild-type and H217A mutant in the reaction cell were at 40–55 μm. All protein was dialyzed in Chelex-treated buffer, and stock solutions of GDP, CaCl2, MgCl2, and MnCl2 were prepared in the same dialysis buffer. Background titrations with GDP or GDPMan into buffer were subtracted from the raw data of titrations of MGS with GDP or GDPMan, respectively. The determined KA and ΔH were used to derive ΔS from standard thermodynamic equations.
RESULTS
Three-dimensional Structure of MGS and Ligand Complexes
The general structure of MGS is composed of a mixed α/β, GT-A fold catalytic domain (residues 2–262) and a six-helix C-terminal domain (residues 263–381) thought to be important for oligomerization (MGS is a tetramer) (Fig. 1b) (14). In previous work, we provided a preliminary indication that perhaps the active center coordination of the nucleotide portion of the sugar donor species was as important as the nature of the donor sugar itself (14). Here, we have probed the donor and acceptor subsites, primarily through kinetic analysis of enzyme variants.
In the previous report on the crystal structure of MGS, there were several crystal forms, some of which had many molecules in the asymmetric unit. In this study, we have used a combination of site-directed mutagenesis and modifications to the original crystallization conditions to generate crystals of MGS in the space group P3221 with two molecules in the asymmetric unit (Table 1), which was associated with a high solvent content (∼75%). The description of the crystal structures of MGS in complex with various ligands is based partly on data described previously by Flint et al. (14) (GDPMan-Mn2+, GDP-Mn2+, and Mn2+-glycerate) and also new complexes (GDPMan-Mg2+, GDP-Mg2+, GDP-Mn2+/malonate, and lactate) that are reported here. Together, these survey structures in which both hard and soft metals are bound.
The active center is located almost in the protein core, with residues shown to make important donor/acceptor interactions contributed from various interlacing loops of the catalytic domain. Because of the high crystal solvent content (previous crystal structures and new structures reported here), the MGS structures have high molecular mobility. Although this characteristic has the effect of diminishing crystal order and therefore diffraction resolution limits, it does provide an opportunity to characterize some of the highly dynamic regions of MGS. B-factor analysis suggests residues in regions 1–66, 100–140, 215–222, and 250–270 have high mean atomic displacements. Many of these residues are located in the active center. Interestingly, these regions remain flexible even when MGS is in complex with donor or acceptor substrates and products, suggesting that protein flexibility is important for all stages along the reaction coordinate.
Donor Substrate Recognition
The MGS structure(s) revealed that the guanine base of GDP is housed in a hydrophobic pocket between the side chains of Lys-9 and Tyr-37 on one face and the aliphatic portion of the side chain of Lys-76 on the opposing face, with Gln-66 accepting hydrogen bonds from the endocyclic N1 and exocyclic N2 (Fig. 1, c and d). To explore the functional significance of guanine recognition, the properties of the alanine mutants K76A, Y37A, K9A, and Q66A were evaluated (Table 2). Determination of kinetic parameters in the presence of excess co-substrate revealed that mutation of Lys-76 to alanine resulted in ∼3–4-fold increase in kcat coupled with an elevation in Km for d-glycerate and GDPMan of 15- and 3-fold, respectively. Similarly, the Y37A and K9A mutations also caused a significant increase in kcat (4–10-fold) in the presence of saturating co-substrate. Notably, the two mutations had little effect on the Km value for GDPMan. The Km value toward the acceptor substrate glycerate for the K9A mutant was also little affected, whereas the Y37A mutant showed an 8-fold increase in the Km value compared with wild-type MGS (Table 2). As a consequence, K9A has a catalytic efficiency (kcat/Km) that is almost an order of magnitude higher than wild-type enzyme for both substrates. The Q66A mutation resulted in substantial increases in kcat (up to 72-fold) and Km (∼30-fold) for both the acceptor and donor substrates (Table 2).
TABLE 2.
Kinetic parameters of MGS variants containing mutations in the donor substrate binding region
When glycerate was the varied substrate, GDP-mannose was at 500 μm except for the Q66A mutation where GDPMan was at 2 mm. When GDP-mannose was the varied substrate, glycerate was at 100 μm except for Q66A, K9A, and H217A, where glycerate was at 1 mm, 10 μm, and 10 mm, respectively.
| Enzyme | Varied substrate, glycerate |
Varied substrate, GDPMan |
||
|---|---|---|---|---|
| kcat | Km | kcat | Km | |
| min−1 | μm | min−1 | μm | |
| Wild type | 1.9 (±0.2) | 2.2 (±0.3) | 1.1 (±0.1) | 3.1 (±0.9) |
| W189A | 4.6 (±0.1) | 15 (±1.9) | 4.0 (±1.5) | 2.8 (±0.4) |
| M229A | 8.1 (±1.8) × 10−1 | 7.8 (±1.4) | 7.6 (±0.4) | 2.8 (±0.2) |
| D192A | Inactivea | Inactive | Inactive | Inactive |
| K76A | 4.5 (±0.05) | 30 (±3.4) | 4.1 (±0.5) | 11 (±0.9) |
| E166A | 1.4 (±0.1) | 32 (±2.1) | 1.7 (±0.02) | 9.6 (±2.2) |
| Q66A | 1.4 (±0.09) × 102 | 70 (±5.6) | 58 (±0.4) | 1.0 (±0.03) × 102 |
| Y37A | 11 (±0.06) | 17 (±3.9) | 9.8 (±0.7) | 2.7 (±0.1) |
| K9A | 6.9 (±0.4)b | 1.4 (±1.0) | 4.9 (±0.08) | 1.7 (±0.3) |
| R73A | 3.3 (±0.5) | 3.8 (±0.3) | 2.8 (±0.07) | 1.0 (±0.4) |
| R73G | 3.6 (±0.02) | 4.1 (±0.3) | 3.6 (±0.04) | 4.1 (±0.2) |
| R218A | 15 (±0.6) | 5.8 (±1.2) | 11 (±0.8) | 33 (±3.1) |
| H217A | 16 (±0.6) | 1.9 (±0.3) × 103 | 29 (±1.6) | 12 (±0.8) |
a Activity was less than 2.8 × 10−4 min−1.
b High degree of substrate inhibition was observed.
The mutation data described above suggest that altered binding to the guanine base leads to an increase in the turnover rate of the enzyme. The elevation in kcat through mutation of regions remote from the catalytic residues (and hence less likely to directly influence the transition state of transfer) suggests that the rate of nucleotide product release limits the turnover rate of the reaction. Mutations that weaken the interaction of GDP with the enzyme facilitate product departure and, in so doing, increase the catalytic rate. It should be emphasized that the actual rate-limiting step is unclear. It may comprise the direct release of the nucleotide product, although it is also possible that the turnover rate is limited by conformational changes required as a prelude to ejecting GDP from the active site.
Ribose recognition in the donor subsite is provided through interaction with the side chain of Glu-11, the main chain amides of Lys-9 and Ala-101, and main chain carbonyl of Pro-7. Arg-218 and Tyr-220 interact with the α- and β-phosphates of GDPMan, respectively, and have highly mobile side chains across MGS structures (Fig. 1). Previous mutational analysis (14) showed that the E11A mutation caused a modest increase in kcat and Km values (for GDPMan). By contrast, the R218A substitution resulted in a significant increase in kcat and Km values for GDPMan but had only a modest influence on the Km value for the acceptor substrate (Table 2). The functional significance of mutants of Tyr-220 is discussed with respect to acceptor recognition below.
The mannosyl moiety of GDPMan is observed in 4C1 conformation and is coordinated in a pocket with Met-229 and Trp-189 providing a hydrophobic platform below the pyranoside ring and side chains of Lys-76, Asp-100, and Asp-192, together with the main chain carbonyl of Leu-163, interacting with O3 (Lys-76 and Asp-100), O4 (Lys-76 and Asp-192), O6 (Asp-192), and O2 (Leu-163) (Fig. 1). A hydrogen-bonding network from Asp-192 via Lys-76 to Asp-100 is established in a system analogous to the UDP-galactosyltransferase LgtC (28). Previous mutational analysis showed that Asp-100 plays a critical role in substrate binding as the D100A mutant was completely inactive (14). The importance of polar interactions with mannose is reinforced by the observation that D192A is also completely inactive (Table 2). It is somewhat surprising that the K76A mutation did not have a detrimental effect on activity as the Nζ of Lys-76 appears to make hydrogen bonds with O3 and O4, while also making electrostatic interactions with Asp-192. It would appear that these polar contacts are less important than those mediated by the carboxylate side chains. Indeed, we have previously speculated that a basic residue in a position equivalent to Lys-76 was a conserved feature of GT-A GTs; however, the functional significance of this amino acid is rather opaque. It is possible that its contribution to the guanine binding pocket may be more important than its polar contact with the donor sugar. The minor influence of the M229A mutation on catalytic performance suggests that the methionine also does not play an important role in substrate recognition despite its suggested role in the hydrophobic “base” of the donor site. The W189A mutant displayed, respectively, a 3- and 7-fold increase in kcat and Km for d-glycerate, compared with the wild-type enzyme, but it did not influence utilization of GDPMan. Thus, the hydrophobic platform-mediated mannose binding under these conditions does not appear critical, which is in contrast to many carbohydrate-binding proteins (both catalytic and noncatalytic) where apolar interactions with sugar rings represent an important binding mechanism.
MGS Acceptor Site
Previously (14), two MGS complexes with molecules in the acceptor-binding site were obtained, MGS with the natural substrate d-glycerate and the substrate analog citrate. In both cases, ligand coordination featured main chain interactions complemented with the side chains of only Arg-131 and Thr-139. To investigate further interactions at the mannose acceptor site, we obtained complexes of MGS with an additional substrate d-lactate (Fig. 2). Comparison of this structure with the d-glycerate complex highlights that coordination of the acceptor carboxylate effectively anchors each molecule at an equivalent position in the acceptor site. Arg-131 sits “below” the acceptor position, with the side chain guanidino NH2 groups forming two polar interactions with one of the carboxylate oxygens. This oxygen makes an additional interaction with the hydroxyl side chain of Thr-139. The partnering carboxylate oxygen forms hydrogen bonds with backbone amide hydrogens of Met-137, Ile-138, and Thr-139 in an overall pyramidal arrangement. Additionally, the C3-OH of d-glycerate forms a hydrogen bond with the backbone amide of Ala-136, stabilizing the substrate and helping to correctly orient the C2-OH for mannose addition. Overall, with the exception of the orientation of the Arg-131 side chain, the MGS acceptor site undergoes very little change across all the obtained structures. To investigate the importance of the contribution of Thr-139 to the acceptor site, the activity of T139A was analyzed (previously the R131A variant was shown to be >1,000 less active than wild-type MGS (14)). The data showed that the T139A mutation resulted in a modest decrease in kcat but a 1500-fold increase in the Km values for d-glycerate (Table 3). The mutant, similar to the wild-type enzyme, was ∼500-fold less active when d-lactate, rather than d-glycerate, was the acceptor substrate.
FIGURE 2.

Electron density for diverse MGS complexes. a, MGS active center complex with the mannose acceptor d-lactate shown (as a composite with Mg2+-GDP-mannose). 2Fo − Fc electron density is presented for d-lactate shown at 1.0σ. Helix 222–240 is omitted for clarity. The coordinates of glycerate from PDB 2BO6 are shown in cyan for reference. b, as a but with electron density shown for malonate (glycerate shown in blue). c, GDP-divergent metal complexes (divergent wall-eyed stereo) with both Mg2+ (green/gray) and Mn2+ (purple). Cation coordinating active center residues Asp-102 and His-217 are shown. With Mg2+ as the divalent metal, His-217 is disordered in the GDP complexes and does not interact with the metal.
TABLE 3.
Kinetic parameters (Mn2+ as metal) of MGS variants containing mutations in the acceptor substrate binding region
| Enzyme | Varied substrate, d-glyceratea,b or d-lactateb,c |
[d-glycerate] | Varied substrate, GDPMan |
||
|---|---|---|---|---|---|
| kcat | Km | kcat | Km | ||
| min−1 | μm | mm | min−1 | μm | |
| Wild-typea | 1.9 (±0.2) | 2.2 (±0.3) | 0.1 | 1.1 (±0.1) | 3.1 (±0.9) × 10−1 |
| Wild-typec | 1.0 | 7.2 × 102 | |||
| T139Aa | 1.0 (±0.1) | 3.3 (±0.5) × 103 | 6 | 5.1 (±0.1) × 10−1 | 1.1 (±0.8) × 10−1 |
| T139Ac | >100 mm | ||||
| Y220Aa | 4.0 (±0.1) | 1.1 (±0.06) × 103 | 100 | 2.5 (±0.04) | 2.9 (±0.8) |
| Y220Fa | 4.2 (±0.1) | 3.8 (±0.2) × 103 | 100 | 2.2 (±0.05) | 6.5 (±0.2) |
| D135Aa | 3.9 (±0.02) | 1.3 (±0.05) × 104 | 35 | 2.7 (±0.2) | 6.3 (±2.1) |
a The varied acceptor substrate was glycerate.
b When the acceptor substrates were varied, the concentration of the donor substrate (GDPMan) was 500 μm.
c The varied acceptor substrate was lactate. See supplemental material for further kinetic parameters.
Asp-135, although positioned in the vicinity of the C3-OH of glycerate, does not appear to make any direct interactions with either d-glycerate or d-lactate. However, Asp-135 does form an ion pair with one of the guanidino NH2 groups of Arg-266 in the structures examined, potentially contributing to the stability of the structure and helping to preserve the topology of the acceptor site. D135A had an extremely high Km value for d-glycerate (5600-fold higher than wild-type) and displayed a slight increase in kcat (2-fold) compared with wild-type enzyme (Table 3). Structural alignment of the MGS ligand structures of acceptor (d-glycerate, PDB 2BO6) and donor (GDPMan/Mn2+, PDB 2BO8) suggests that Tyr-220 could also contribute to the binding of C2-OH of the acceptor. Tyr-220 is part of the highly mobile flexible loop that forms part of the active center. Exact delineation of its position in crystal structures has proved extremely difficult with at best poor, and often absent, electron density. Yet the 500- and 1500-fold increase in Km for d-glycerate of Y220A and Y220F in the presence of saturating GDPMan, respectively, strongly suggests that Tyr-220 constitutes an essential part of the acceptor binding site. A role of Tyr-220 in acceptor binding perhaps comes from the GDPMan (donor) complex published previously; here, the loop and the whole of the Tyr-220 side chain become ordered with the Tyr-220 hydroxyl interaction with the β-phosphate. In this position, it is highly likely that Tyr-220 would interact with glycerate bound in the acceptor site providing a rationally compelling argument for both the importance of GDPMan binding in defining the acceptor site and thus also for the importance of Tyr-220 to glycerate binding reflected in the kinetic analyses.
Substrate Binding Order
Inhibitors that compete with substrates, typically products of the reaction, can be used to probe the binding order of reactants in reactions catalyzed by GTs. GDP inhibited MGS by competing with GDPMan with a Ki of ∼36 μm (supplemental Fig. 1). Pseudo-single substrate kinetics with Mn2+ showed significant substrate inhibition with d-glycerate, which excluded the possibility of a rapid equilibrium random ordered mechanism (29). d-Glycerate can only function as a substrate inhibitor if it is able to bind to MGS-GDP, forming a stable nonproductive complex. This is consistent with the observation that d-glycerate will bind to MGS in the presence of GDP and a metal ion (KD ∼20 μm measured by ITC) but not in the absence of the nucleotide diphosphate (Fig. 3). These results support a compulsory order binding mechanism in which GDPMan and d-glycerate bind successively to the enzyme. Mannosylglycerate was also evaluated as a product inhibitor; however, no inhibition was observed in the millimolar range, and ITC analysis revealed no significant binding of the molecule. By contrast, ITC showed that GDPMan and GDP bind to MGS, in the absence of d-glycerate, with KD values of 200 and 500 nm, respectively. These data are consistent with the view that the conjugated reaction product binds weakly to the enzyme and is likely to depart the active site prior to the other reaction product GDP. The relatively tight binding to GDP is consistent with departure of the nucleotide diphosphate limiting the turnover rate of the enzyme.
FIGURE 3.

Analysis of ligand binding by ITC. In the example titrations, the reaction cell contained wild-type MGS at 50 μm in 20 mm Tris-HCl, pH 8.0, containing MnCl2 at 1 mm. The title of the different panels identify the titrant used, which was at 2 mm in the syringe. In the panel entitled glycerate (GDP), the nucleotide was included in the reaction cell. The top half of each panel shows the raw ITC heats; the bottom half shows the integrated peak areas fitted using a one single binding site model by MicroCal Origin software.
Role of Metal Ions in Substrate Recognition
A key feature of GT-A glycosyltransferases, including MGS, is the role of metals in substrate recognition. Thus, notably, with GDP and the transition series metals Co2+ or Mn2+, the divalent cation is coordinated by one of the imidazole nitrogens of His-217 and the second aspartate of the DXD motif, Asp-102 (11). The DXD motif is highly conserved in GT-A GTs and is implicated in divalent metal ion binding, which coordinates with the donor substrate. These residues aid in the positioning of the metal between O2 and O1 of the α- and β-phosphates of GDP. Unusually, MGS displays a much higher kcat when group II metal ions (Ca2+ and Mg2+) function as co-factors, compared with when transition series metal ions fulfill this role (Table 4 and supplemental Fig. 2). The contribution of Asp-102 and His-217 to ion selection was explored through the characterization of the mutants D102A and H217A. The data (see Table 4) show that although D102A was completely inactive in the presence of all metals tested, H217A displays a significant change in metal preference. Thus, the H217A mutant was essentially inactive in the presence of Ca2+ (specific activity >1000-fold lower than wild-type MGS), whereas the kcat value was reduced 23-fold in the presence of Mg2+ (compared with wild-type enzyme). In the presence of Mn2+, the removal of the imidazole side chain had the opposite effect; the kcat value increased 16-fold. The Km value for d-glycerate (Ka) increased 34- and >1600-fold for Mg2+ and Mn2+, respectively, and a similar increase was observed for the dissociation constant of d-glycerate (Ki,a) in the presence of Mg2+. The kinetic parameters Ka and Kb (Km for GDPMan) for wild-type MGS in the presence of Mn2+ could only be estimated as the values are >1 μm. Thus, Ki,a and Ki,b values (dissociation constant for GDPMan) could not be calculated for wild-type MGS when Mn2+ was the co-factor. To summarize, the H217A mutation causes a substantial reduction in the affinity for the acceptor substrate d-glycerate in the presence of both Mg2+ and Mn2+. Although the mutation did not influence donor substrate binding in the presence of Mg2+, the extremely tight interaction between the enzyme and GDPMan in the presence of Mn2+ was greatly reduced in the H217A mutant. Direct comparison of the activity of H217A in the presence of both metals also highlights that its binding of GDPMan is increased with Mn2+. Together, these data suggest that His-217 may act as a functional “trigger” of hard versus soft metal usage.
TABLE 4.
Bi-substrate kinetics of wild type and variants of MGS
Subscript A indicates parameters determined with respect to d-glycerate (acceptor), and subscript B indicates those for GDP-mannose (donor).
| Enzyme | Metal ion | kcat | KA | KB | Ki,a | Ki,b |
|---|---|---|---|---|---|---|
| min−1 | μm | μm | μm | μm | ||
| Wild type | Mg2+ | 115 ± 17 | 3.9 ± 7 | 59 ± 16 | 5 ± 1 | 7 ± 2 |
| Wild type | Mn2+ | 2 ± 0.4 | <1a | <1a | –b | –b |
| Wild type | Ca2+c | 36 | 166 | – | 157 | – |
| H217A | Mg2+ | 5 ± 1 | 1322 ± 438 | 46 ± 19 | 230 ± 31 | 8 ± 2 |
| H217A | Mn2+ | 35 ± 2 | 1610 ± 286 | 14 ± 1 | 159 ± 87 | 2 ± 1 |
| H217A | Ca2+ | No activity detected | ||||
| Y220A | Mn2+ | 4 ± 0.1 | 1990 ± 340 | <1a | –b | –b |
a Value was below detection limit.
b Data cannot be calculated because Ka was <1 μm or Kb was <1 μm.
c The kinetics parameters in the presence of calcium were determined under pseudo-single substrate conditions where the reaction product was quantified by HPLC.
Crystal Structure of MGS Bound to Different Metals
To explore the structural basis for the metal preference of MGS, the crystal structure of the enzyme was determined in complex with GDP-Mg2+ and GDPMan-Mg2+ (Fig. 2). Diffraction data were collected for three different GDP/Mg2+ crystals, but in each case, side chain density corresponding to His-217 was absent, even though magnesium was present, consistent with a poor, indeed absent, interaction between nitrogen and these “harder” cations (Fig. 2). Whether cause or effect, the presence of the manganese ion positions GDP and GDP-mannose “deeper” in the transfer center with most of the active center side chains translated slightly to accommodate this association. This translation positions the O1 of the donor sugar 0.94 Å closer to the O2 of glycerate compared with the GDPMan-Mg2+ complex. The α-carbon positions of the flexible loop, comprising residues 215–222, vary in all four structures, making it difficult to evaluate if loop positions are directly affected by different cations. However, this inherent plasticity, even in substrate-loaded forms, emphasizes the flexibility of MGS, which is likely to be important for substrate recognition and product release. As demonstrated in multiple GDP/Mg2+ crystal structures, cation coordination does not require His-217, potentially mimicking the H217A mutant (Fig. 2). In the GDPMan/Mg2+ structure, cation coordination with Asp-102 results in the magnesium ion being pulled away from the transfer center relative to Mn2+ coordinated with His-217 (Fig. 2). This could result in the donor sugar being less tightly packed in the active center, potentially facilitating product release in a proposed rate-limiting step as discussed below.
Calorimetric Studies of Nucleotide Binding
To explore the influence of metal ions on nucleotide binding, the capacity of MGS to bind GDP in the presence of different metals was explored. Titration of GDP into MGS without addition of metal ion showed weak enthalpies and poor affinity (data not shown), suggesting that a cation was critical for GDP binding. GDP, however, bound to MGS in the presence of Ca2+, Mg2+, or Mn2+, with binding driven by both enthalpic and entropic processes (Fig. 3 and Table 5). The affinity (KD) of MGS for GDP was in the low micromolar to high nanomolar range (10–100-fold tighter binding than observed for α3GT (30)). MGS showed the strongest affinity toward GDP-Mn2+ of the three tested cations (Ca2+, Mg2+, or Mn2+) (Table 5), which could correlate to the more buried position for GDP in the transfer center in MGS within the GDP-Mn2+ complex structure, compared with those obtained with Mg2+ present. Metal ion coordination in the H217A variant displayed an ∼3-fold decrease in binding of GDP-Mn2+ and GDP-Mg2+, whereas the mutation had no significant effect on affinity for GDP-Ca2+.
TABLE 5.
The use of ITC to measure binding of GDP and GDPMan to wild type and variants of MGS in the presence of metals
| Enzyme | Ligand | Metal ion | KA |
|---|---|---|---|
| m−1 | |||
| Wild type | GDPMan | Ca2+ | 9.0 (±0.1) × 105 |
| Wild type | GDPMan | Mg2+ | 2.9 (±0.3) × 106 |
| Wild type | GDPMan | Mn2+ | 5.4 (±0.6) × 106 |
| Wild type | GDP | Ca2+ | 1.5 (±0.3) × 105 |
| Wild type | GDP | Mg2+ | 2.8 (±0.3) × 105 |
| Wild type | GDP | Mn2+ | 2.0 (±0.4) × 106 |
| H217A | GDPMan | Ca2+ | 1.4 (±0.4) × 105 |
| H217A | GDPMan | Mg2+ | 1.0 (±0.1) × 105 |
| H217A | GDPMan | Mn2+ | 6.3 (±0.8) × 105 |
| H217A | GDP | Ca2+ | 1.2 (±0.02) × 105 |
| H217A | GDP | Mg2+ | 8.9 (±0.1) × 104 |
| H217A | GDP | Mn2+ | 6.1 (±0.7) × 105 |
To explore the influence of the nucleotide-sugar donor on metal ion coordination, binding of GDPMan in the presence of the three metals was determined by ITC (Fig. 3). GDPMan bound 2–10-fold tighter to wild-type MGS than GDP (Table 5 and supplemental Table 2), suggesting that mannose makes a contribution to donor substrate binding. Addition of Mn2+ resulted in a higher KA value for GDPMan, compared with when the donor substrate was titrated with the enzyme in the presence of Mg2+ or Ca2+; however, the difference was smaller than with GDP. Similar to the data obtained for the GDP-metal complexes, the H217A mutation significantly reduced affinity for GDP/Mn2+ and GDP/Mg2+, but not for GDP/Ca2+ (Table 5).
The ITC data indicated that although His-217 contributes to GDP/Mn2+ and GDP/Mg2+ metal coordination, the relatively modest reduction in affinity for these ligands in the H217A mutant suggests that the two metals can adopt alternative coordinate bonds between the protein and nucleotide. This is consistent with the observation that in the GDP/Mg2+ crystal structure the metal makes a bidentate coordination with Asp-102, although His-217 is highly disordered. Indeed, the similar affinity of GDP/Ca2+ for wild-type MGS and the H217A mutant suggests that the histidine does not contribute to calcium coordination with the nucleotide product. Although it is possible that the metals are making nucleotide-independent contact with the enzyme, this is unlikely as titration with the metal in the absence of GDP or GDPMan revealed no binding (data not shown).
DISCUSSION
This study shows that the introduction of the H217A mutation into MGS induces a shift in metal preference from the hard group II metals, which are typically less commonly used by GTs, to softer transition metals. Structural data reveal subtle yet clear differences in the position adopted by the donor substrate in complex with different metals, which may influence catalytic efficiency. The metal coordination observed here, in which His-217, Asp-102 (the second aspartate in the DXD motif) and oxygens from both the α- and β-phosphates are ligand, is a common feature of GT-A enzymes, although examples of enzymes in which both acid residues of the DXD motif bind the metal co-factor have been observed in blood group enzymes (31). It is possible that the H217A mutation alters the coordination of the group II metals such that they are unable to induce the conformational changes required for catalysis. By contrast, the transition metals may be able to participate in different coordination chemistries, possibly involving both carboxylates in the DXD motif, which may facilitate the formation of a ternary complex. Such changes in metal preference in GTs are unusual, although the work reported here has considerable resonance with a study by Qasba and co-workers (32) on bovine β-1,4-galactosyltransferase. The normal metal in this GT-A-inverting GT is Mn2+, with no detectable activity with Mg2+. In this case, mutation of a methionine that made an unusual coordinate bond with Mn2+ to histidine generated a variant whose loops were able to close (allowing access to a “closed” three-dimensional structure) while reversing the metal ion specificity from Mn2+ to Mg2+ (32). This differential use of metals and their different coordination and activation also has strong resonance with chemical methods for the activation of glycosyl donors. In these chemical systems, the use of the appropriate Lewis acid is a common strategy for “tuning” reactivity of a given glycosyl donor to higher or lower levels. It may be that, in a similar manner, when nature uses diphosphate nucleotides as leaving groups she has evolved the appropriate combination of amino acids and metal to achieve activation. Thus, harder metals, such as Ca(II), which in its binding to GDPMan here was little affected by His-217, may be capable of direct activation of the phosphate in MGS with the need for only Asp-102 as a stabilizing/coordinating ligand (and hence lead to higher activity). In such a scenario the DXD motif may act as a “soft” motif, although the Asp-102 alone acts as “hard”; here, His-217 may then be playing the role of a metal mimic to engage the second Asp. However, increasingly softer metals Mg(II) and then Mn(II) may require additional activation, perhaps via coordination of His-217 that might tune the metal reactivity through both σ donation and π back-bonding. In this way, and through such hard-soft tuning methods, if harder (Ca(II)) metals were environmentally unavailable, softer metals could be used as surrogates. Over time, a greater abundance of softer metals may have led to GTs evolving to use Mn(II) without the need for such additional “Lewis acidity tuning” or, indeed, this may have reinforced the DXD motif. MGS, an archaeal enzyme, may therefore represent a historically early “Ca(II)-affluent ancestor” on this progression.
Donor substrate binding is associated with a significant change in the conformation of a highly flexible loop extending from Pro-212 to Gly-221. How this flexible loop confers acceptor binding and catalysis is central to understanding how retaining GT-A GTs mediate glycosyl transfer. Within this loop, the phenolic ring of Tyr-220 adopts a different orientation in complexes where MGS is bound to GDP and GDPMan, respectively. In one conformation, the phenol hydroxyl makes polar interactions with the α-phosphate of GDPMan, although in its other conformation (adopted in the MGS-GDP complex) it is pointing into the acceptor site and likely makes a hydrogen bond with O2 of glycerate. The observed substantial increase in Km for glycerate when the tyrosine is mutated, which is not mirrored by a change in affinity for GDPMan, is consistent with its role within the acceptor site and suggests that the latter interaction with O2 of glycerate is more functionally relevant than the former interaction with phosphate. Thus, we propose that the primary mechanism by which the donor substrate facilitates glycerate binding is through conformational changes in the highly mobile loop. The interaction of Tyr-220, in this loop, with the donor, results in the presentation of the hydroxyl of this residue into the acceptor binding site.
To conclude, this study reveals the importance of metal ion coordination not only in donor substrate binding but also in the assembly of the ternary complex, with leaving group departure defining the maximum catalytic rate. This study provides a foundation for manipulating the activity of GTs by engineering active site metal ion recognition and may reflect an intriguing mechanism by which GTs utilize available metals to tune their Lewis acid hardness to make them suitable activators for nucleotide diphosphate leaving groups in a manner akin to strategies used in synthetic chemical glycosylation chemistry (33).
Supplementary Material
Acknowledgment
Monica Palcic (Carlsberg Laboratory) is thanked for assistance and advice with kinetics.
This work was supported in part by Biotechnology and Biological Sciences Research Council of the United Kingdom.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2 and Tables 1 and 2.
The atomic coordinates and structure factors (codes 2y4j, 2y4k, 2y4l, and 2y4m) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- GT
- glycosyltransferase
- MGS
- mannosylglycerate synthase
- PDB
- Protein Data Bank
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- ITC
- isothermal titration calorimetry.
REFERENCES
- 1. Ohtsubo K., Marth J. D. (2006) Cell 126, 855–867 [DOI] [PubMed] [Google Scholar]
- 2. Parodi A. J. (2000) Annu. Rev. Biochem. 69, 69–93 [DOI] [PubMed] [Google Scholar]
- 3. Martins L. O., Empadinhas N., Marugg J. D., Miguel C., Ferreira C., da Costa M. S., Santos H. (1999) J. Biol. Chem. 274, 35407–35414 [DOI] [PubMed] [Google Scholar]
- 4. Borges N., Ramos A., Raven N. D., Sharp R. J., Santos H. (2002) Extremophiles 6, 209–216 [DOI] [PubMed] [Google Scholar]
- 5. Pais T. M., Lamosa P., Garcia-Moreno B., Turner D. L., Santos H. (2009) J. Mol. Biol. 394, 237–250 [DOI] [PubMed] [Google Scholar]
- 6. Coutinho P. M., Deleury E., Davies G. J., Henrissat B. (2003) J. Mol. Biol. 328, 307–317 [DOI] [PubMed] [Google Scholar]
- 7. Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., Henrissat B. (2009) Nucleic Acids Res. 37, D233–D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bourne Y., Henrissat B. (2001) Curr. Opin. Struct. Biol. 11, 593–600 [DOI] [PubMed] [Google Scholar]
- 9. Igura M., Maita N., Kamishikiryo J., Yamada M., Obita T., Maenaka K., Kohda D. (2008) EMBO J. 27, 234–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maita N., Nyirenda J., Igura M., Kamishikiryo J., Kohda D. (2010) J. Biol. Chem. 285, 4941–4950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lairson L. L., Henrissat B., Davies G. J., Withers S. G. (2008) Annu. Rev. Biochem. 77, 521–555 [DOI] [PubMed] [Google Scholar]
- 12. Lewis E. S., Boozer C. E. (1952) J. Am. Chem. Soc. 74, 308–311 [Google Scholar]
- 13. Errey J. C., Lee S. S., Gibson R. P., Martinez Fleites C., Barry C. S., Jung P. M., O'Sullivan A. C., Davis B. G., Davies G. J. (2010) Angew. Chem. Int. Ed. Engl. 49, 1234–1237 [DOI] [PubMed] [Google Scholar]
- 14. Flint J., Taylor E., Yang M., Bolam D. N., Tailford L. E., Martinez-Fleites C., Dodson E. J., Davis B. G., Gilbert H. J., Davies G. J. (2005) Nat. Struct. Mol. Biol. 12, 608–614 [DOI] [PubMed] [Google Scholar]
- 15. Gonçalves S., Borges N., Esteves A. M., Victor B. L., Soares C. M., Santos H., Matias P. M. (2010) J. Biol. Chem. 285, 17857–17868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fulton Z., McAlister A., Wilce M. C., Brammananth R., Zaker-Tabrizi L., Perugini M. A., Bottomley S. P., Coppel R. L., Crellin P. K., Rossjohn J., Beddoe T. (2008) J. Biol. Chem. 283, 27881–27890 [DOI] [PubMed] [Google Scholar]
- 17. Goldschmidt L., Cooper D. R., Derewenda Z. S., Eisenberg D. (2007) Protein Sci. 16, 1569–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 19. Leslie A. G. (2006) Acta Crystallogr. D Biol. Crystallogr. 62, 48–57 [DOI] [PubMed] [Google Scholar]
- 20. Evans P. (2006) Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 21. McCoy A. J. (2007) Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 23. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 24. Collaborative Computational Project Number 4 (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–76315299374 [Google Scholar]
- 25. Yang M., Brazier M., Edwards R., Davis B. G. (2005) ChemBioChem 6, 346–357 [DOI] [PubMed] [Google Scholar]
- 26. Gosselin S., Alhussaini M., Streiff M. B., Takabayashi K., Palcic M. M. (1994) Anal. Biochem. 220, 92–97 [DOI] [PubMed] [Google Scholar]
- 27. Seto N. O., Palcic M. M., Compston C. A., Li H., Bundle D. R., Narang S. A. (1997) J. Biol. Chem. 272, 14133–14138 [DOI] [PubMed] [Google Scholar]
- 28. Persson K., Ly H. D., Dieckelmann M., Wakarchuk W. W., Withers S. G., Strynadka N. C. (2001) Nat. Struct. Biol. 8, 166–175 [DOI] [PubMed] [Google Scholar]
- 29. Cornish-Bowden A. (2004) in Fundamentals of Enzyme Kinetics (Copeland R. A. ed) 3rd Ed., Portland Press, London [Google Scholar]
- 30. Boix E., Zhang Y., Swaminathan G. J., Brew K., Acharya K. R. (2002) J. Biol. Chem. 277, 28310–28318 [DOI] [PubMed] [Google Scholar]
- 31. Gastinel L. N., Bignon C., Misra A. K., Hindsgaul O., Shaper J. H., Joziasse D. H. (2001) EMBO J. 20, 638–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramakrishnan B., Boeggeman E., Qasba P. K. (2004) Biochemistry 43, 12513–12522 [DOI] [PubMed] [Google Scholar]
- 33. Davis B. G. (2000) J. Chem. Soc. Perkin Trans. 1, 2137–2160 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.