

Abstract
Fluorinated nucleosides and nucleotides are of considerable interest to medicinal chemists due to their antiviral, anticancer, and other biological activities. However, their direct interactions at target binding sites are not well understood. A new class of 2′-deoxy-2′-fluoro-C6-substituted uridine and UMP derivatives were synthesized and evaluated as inhibitors of orotidine-5′-monophosphate decarboxylase (ODCase). These compounds were synthesized from the key intermediate, fully-protected 2′-deoxy-2′-fluorouridine. Among the synthesized compounds, 2′-deoxy-2′-fluoro-6-iodo-UMP covalently inhibited human ODCase with a second-order rate constant of 0.62 ± 0.02 M−1sec−1. Interestingly, the 6-cyano-2′-fluoro derivative covalently interacted with ODCase defying the conventional thinking, where its ribosyl derivative undergoes transformation into BMP by ODCase. This confirms that the 2′-fluoro moiety influences the chemistry at the C6 position of the nucleotides, thus interactions in the active site of ODCase. Molecular interactions of the 2′-fluorinated nucleotides are compared to those with the 3′-fluorinated nucleotides bound to the corresponding target enzyme, and the carbohydrate moieties were shown to bind in different conformations.
Introduction
Orotidine-5′-monophosphate decarboxylase (ODCase, 4.1.1.23) catalyzes the transformation of orotidine-5′-O-monophosphate (OMP, 1) to uridine-5′-O-monophosphate (UMP, 2) during the de novo synthesis of pyrimidine nucleotides (Figure 1).1,2 UMP is a precursor for the synthesis of other pyrimidine nucleoside triphosphates, which in turn are precursors for the synthesis of ribo and deoxyribo nucleic acids. In humans, ODCase is part of the bifunctional enzyme UMP synthase, and, in lower level organisms, it is a monofunctional enzyme.3,4,5
Figure 1.

Catalytic decarboxylation of OMP (1) to UMP (2) by ODCase for the synthesis of pyrimidine nucleotides in the de novo pathway.
In most species, including humans, pyrimidine nucleotides are obtained via de novo and salvage pathways. However, certain parasitic organisms, such as Plasmodia, are dependent on de novo synthesis of pyrimidine nucleotides because they do not have the necessary cellular machinery for the salvage of pyrimidine nucleotides.6 Therefore, de novo pyrimidine biosynthesis is a potential target for developing novel therapeutics. De novo synthesis of pyrimidine nucleotides is upregulated when the demand for pyrimidine nucleotides is high, for example, during the replication of cells and during abnormal cell growth.7,8 Thus, ODCase inhibitors could potentially exhibit a variety of biological activities including antiviral, antiplasmodial, and anticancer activities.9 Several 5′-monophosphate analogs of classic nucleoside derivatives, such as 6-hydroxyuridine (3), 6-thiocarboxamidouridine (4), 6-azauridine (5), pyrazofurin (6), and xanthosine (7), are potent inhibitors of ODCase (Figure 2).10,11,12 Interestingly, two non-nucleoside inhibitors, nifedipine and nimodipine, inhibit ODCase competitively with modest potency (Figure 2).13,14
Figure 2.

Structures of various nucleosides and nucleotides with ODCase inhibitory properties.
Since the discovery of novel C6-substituted UMP derivatives as potent antimalarial agents, there has been interest in exploring modified nucleosides as potential ODCase inhibitors.15,16,17,18,19,20 We reported that the monophosphate analog of 6-iodouridine (10) is a potent irreversible inhibitor of ODCase.21 We showed that the mononucleotide of 6-cyanouridine (11) was transformed into BMP by ODCase, and the latter is a tight-binding inhibitor of ODCase. This is a novel biotransformation that does not occur chemically and was catalyzed by ODCase exclusively.15,22,23
Introduction of fluorine atoms onto the carbohydrate ring gives rise to interesting antiviral and anticancer activities. A number of fluorinated nucleosides are currently used in clinics including gemcitabine (15), L-FMAU (16), and F-ara-C (17) (Figure 2).24,25,26,27 Gemcitabine and F-ara-C are successful anticancer agents, while L-FMAU is a potent anti-HBV drug.28,29,30 A fluoro substitution at the C5 position of the pyrimidine moiety led to novel anticancer agents; the mononucleotide derivative of 6-azido-5-fluorouridine (12) inhibited human ODCase irreversibly, and its 6-amino analog (13) competitively inhibited ODCase at submicromolar concentrations.17 Further investigation into 5-fluoro-6-substituted pyrimidine derivatives confirmed that 5-fluoro-6-azido-uridine and its 6-amino analog are potent compounds against various leukemia, multiple myeloma, and breast cancer cell lines.17 The effect of the fluoro substitution on the ribosyl moiety of UMP, its conformation, and the binding interactions with ODCase have not been investigated. In this report, we reveal the synthesis, enzyme inhibition studies, and effects of 2′-fluoro-C6-substituted uridine and UMP derivatives and provide insight into fluorinated nucleoside complexes and their binding interactions.
Experimental Section
Synthesis
General
All anhydrous reactions were performed under a nitrogen atmosphere. All solvents and reagents were obtained from commercial sources; anhydrous solvents were prepared following standard procedures. Reaction progress was monitored by thin-layer chromotography (TLC; Silica gel-60 F254) plates. Chromatographic purifications were performed using silica gel (60 Å, 70–230 mesh). Nuclear magnetic resonance (NMR) spectra were recorded on a Varian spectrometer (300 and 400 MHz for 1H, 75 and 100 MHz for 13C, 282 or 376 MHz for 19F, and 121 MHz for 31P). Chemical shifts were eported in δ ppm using tetramethylsilane (TMS) as a reference for the 1H NMR spectra and phosphoric acid as an external reference for 31P spectra. Purities for compounds 29–32 were evaluated on a Waters Delta 660 high-performance liquid chromotography (HPLC) system attached to a photodiode array (PDA) detector equipped with a Waters Symmetry® C18 column (4.6 × 100 mm length, 5 μM). Purities for compounds 27, 28, 33, 34, and 38 were evaluated on a Waters™ liquid chromotography/mass spectrometry LC/MS) system equipped with a PDA detector using an XBridge C18 column (4.6 mm × 150 mm, 5 μm). Two HPLC methods were used for the purity assessment of 29–32: method A used 15% methanol (MeOH) in H2O (1 mL/min, isocratic), and method B used 5% MeOH in H2O (0.5 mL/min, isocratic). Two of the following four methods were used for other compounds: method C used 10% MeOH in H2O (1 mL/min, isocratic, for 27, 28, and 33); method D used 20% MeOH in H2O (1 mL/min, isocratic, for 28 and 38); method E used 3% acetic acid (AcOH) in H2O (1 mL/min, isocratic, for 34 and 38); method F used 20% acetonitrile (CH3CN) (with 0.05% trifluoroacetic acid (TFA)) in H2O (1 mL/min, isocratic, for 29, 33, and 34). All HPLC solvents were filtered through Waters™ membrane filters (47 mm GHP 0.45 μm, Pall Corporation) and degassed with helium. Injection samples were filtered using Waters Acrodisc® syringe filters (4 mm, polytetrafluoroethylene (PTFE, 0.2 μm). All nucleotides that were tested against ODCase enzyme activity, have shown purity >95% except compound 33 exhibited upto 93% purity. Purity for compounds 35–37 was evaluated by elemental analysis (C, H, N) at the ANALEST Laboratory, University of Toronto, and the results were within 0.4% of the calculated values. All nucleosides evaluated in cell-based assays were >95% pure.
1-(2-Hydroxy-3, 5-di-O-tetrahydropyranyl-β-D-arabinofuranosyl)uracil (18)
This compound was synthesized as reported earlier.31
2′-Deoxy-2′-fluoro-3′,5′-di-O-tetrahydropyranyl-β-D-uridine (19)
Pyridine (24 mL) was added to a stirred solution of 18 (10 g, 24.3 mmol) in anhyd dichloromethane (120 mL) under a nitrogen atmosphere. The reaction mixture was cooled to 0°C and dimethyl aminosulfur trifluoride (DAST; 3.2 mL, 24.3 mmol) was added drop wise. After 30 min, an additional 5 equiv of DAST were added drop wise at 0°C. The reaction mixture was then stirred at 40°C overnight. The reaction mixture was cooled to 0°C, quenched with a saturated NaHCO3 solution, and then extracted with dichloromethane. The combined organic layers were washed with brine, dried (Na2SO4), and concentrated. The crude product was purified on a Biotage SP1™ chromatography unit using a normal-phase column (EtOAc: Hexanes, 3:7–4:6) to obtain compound 19 as a yellow solid (6 g, 60% yield). 1H NMR (CDCl3) δ 1.45–1.94 (broad m, 12H, THP-CH2), 3.48–4.87 (broad m, 10H, THP-CH-O, H-5″, H-3′, H-4′, H-5′), 4.90–5.19 (m, 1H, H-2′, 2JF = 52 Hz), 5.60–5.72 (4d overlap, 1H, H-5), 5.95–6.17 (4dd, 1H, H-1′, 3JF = 28 Hz), 7.94–8.10 (4d), 8.23 (broad s).
2′-Deoxy-2′-fluoro-6-iodo-3′,5′-di-O-tetrahydropyranyl-β-D-uridine (20)
Compound 19 (800 g, 1.9 mmol) was dissolved in anhyd tetrahydrofuran (THF; 40 mL), and cooled to −78°C. Lithium diisopropylamide (LDA; 0.7 g, 6.7 mmol, 3.4 mL of a 2M solution in THF) was added drop wise, and the reaction mixture was stirred at −78°C for 2 h. Then, iodine (960 mg, 3.8 mmol) dissolved in anhydrous THF (15 mL) was added to the reaction mixture and stirred. After 6 h, the reaction mixture was quenched with water (25 mL), concentrated, and re-dissolved in EtOAc (50 mL). The organic phase was washed with brine, dried (Na2SO4), concentrated, and purified by column chromatography (hexanes:EtOAc, 1:1) to give 20 as a yellow solid (450 mg, 45% yield). 1H NMR (CDCl3) δ 1.29 (m, 12H), 3.40–4.80 (m, 10H), 5.47 (m, 1H, H-2′, JH–F=55 Hz), 6.05 (4dd, 1H, H-1′, JH–F =27 Hz), 6.45 (s, 1H, H-5).
2′-Deoxy-2′-fluoro-3′,5′-di-O-tetrahydropyranyl-β-D-orotidine ethyl ester (21)
Compound 19 (500 mg, 1.2 mmol) in anhydrous THF (10 mL) was treated with LDA (2.5 mL, 4.6 mmol) at −78°C under a nitrogen atmosphere. Then, ethyl chloroformate (0.5 mL, 5 mmol) was added, and the reaction mixture was stirred for 2 days. The reaction was quenched with water, concentrated, and the crude product was purified on a Biotage SP1™ chromatography system using 0–50% EtOAc:hexanes as an eluent to obtain compound 21 as a yellow powder (175 mg, 30% yield). 1H NMR (CDCl3) δ 1.39 (t, 3H), 1.46–1.90 (broad m, 12H), 3.45–4.83 (broad m, 12H), 5.33–5.61 (m, 1H, H-2′, JH–F=54.7 Hz), 5.81–6.02 (m, 1H, H-1′, JH–F = 24.9 Hz), 6.13 (4s,1H); HRMS (ESI) for C22H31N2O9FNa (MNa+), calculated: 509.1911; observed: 509.1905.
5-Bromo-2′-deoxy-2′-fluoro-3′,5′-di-O-tetrahydropyranyl-β-D-uridine (22)
NaN3 (5 g, 77 mmol) in water (20 mL) was added to a stirred solution of 19 (8.0 g, 19.4 mmol) in 1,2-dimethoxyethane (90 mL) at 22°C, followed by N-bromosuccinimide (NBS; 5.1 g, 29 mmol). The reaction mixture was stirred for 18 h. Then, the solvent was removed in vacuo, and the crude product was dissolved in water and extracted with EtOAc (300 mL × 4). The combined organic layers were dried (Na2SO4) and concentrated under vacuum to obtain the crude product 22 (10.8 g) as a yellow solid, which was used for the next reaction without further purification.
6-Cyano-2′-deoxy-2′-fluoro-3′, 5′-di-O-tetrahydropyranyl-β-D-uridine (23)
NaCN (2.0 g, 41.5 mmol) was added at 22°C to a stirred solution of the crude compound 22 (6.8 g, 13.8 mmol) in anhydrous dimethylformamide (DMF; 80 mL), and the reaction mixture was stirred for 18 h. The reaction mixture was then neutralized with glacial AcOH and was concentrated under vacuum. The crude reaction mixture was dissolved in water and extracted with EtOAc (200 mL × 4). The combined organic layers were dried (Na2SO4) and concentrated in vacuo to obtain the crude product 23 (5.1 g) as a yellow solid, which was used in the next reaction without further purification. 1H NMR (CDCl3) δ 1.40–1.94 (broad m, 12H), 3.47–4.84 (broad m, 10H), 5.34–5.82 (m, 1H, H-2′, JH–F = 55 Hz), 5.84–6.06 (m, 1H, H-1′, JH–F = 26 Hz), 6.30 (d, 1H); HRMS (ESI) for C20H26N3O7FNa (MNa+), calculated: 462.1633; observed: 462.1646.
6-Amido-2′-deoxy-2′-fluoro-3′,5′-di-O-tetrahydropyranyl-β-D-uridine (24)
Compound 23 (1.1 g, 2.5 mmol) was dissolved in 1N NaOH (10 mL), and the reaction mixture was stirred at 22°C for 1.5 h. The reaction mixture was brought to pH 6 using a 5% HCl solution and was concentrated. The concentrated reaction mixture was dissolved in water, extracted with EtOAc (100 mL × 4), the combined organic layers were dried (Na2SO4), concentrated, and the crude product was purified by silica gel column chromatography (5% MeOH:CH2Cl2) to yield compound 24 as a yellow powder (970 mg, 84% yield). 1H NMR (CDCl3) δ 1.40–1.94 (m, 12H), 3.45–4.88 (m, 10H), 5.34–5.82 (m, 1H, H-2′, JH–F = 55 Hz), 5.70–5.95 (m, 2H); HRMS (ESI) for C20H28N3O8FNa (MNa+), calculated: 480.1737; observed: 480.1752.
6-Azido-2′-deoxy-2′-fluoro-3′,5′-di-O-tetrahydropyranyl-β-D-uridine (25)
Compound 20 (406 mg, 0.8 mmol) in anhydrous DMF (3.4 mL) was treated with sodium azide (60 mg, 0.9 mmol). The reaction mixture was stirred for 6 h at room temperature, concentrated, and dissolved in EtOAc (30 mL). The combined organic phases were washed with water (20 mL), brine (20 mL), dried (Na2SO4), concentrated, and purified in the dark by silica gel column chromatography (hexanes:EtOAc, 6:4→3:7) to obtain compound 25 as a yellow solid (292 mg, 85% yield). 1H NMR (CDCl3) δ 1.34 (m, 12H), 3.40–4.77 (m, 10H); 5.31–5.49 (m, 2H, H-5, H-2′, JH–F=55 Hz), 6.04 (m, 1H), 8.56 (broad s, 1H).
5-Cyano-2′-deoxy-2′-fluoro-3′,5′-di-O-tetrahydropyranyl-β-D-uridine (26)
Sodium cyanide (1.1 g, 22.2 mmol) was added to a stirred solution of compound 22 (3.7 g, 7.4 mmol) in anhydrous DMF (50 mL), and the reaction was carried out in a microwave for 5 min at 150°C. The reaction mixture was diluted with water, neutralized (AcOH), concentrated, and extracted with EtOAc (100 mL × 4). The combined organic layers were dried (Na2SO4) and concentrated to obtain the crude product 26 (1.2 g), which was used without further purification. HRMS (ESI) for C20H26N3O7FNa (MNa+), calculated: 462.1644; observed: 462.1646.
2′-Deoxy-2′-fluoro-6-iodo-β-D-uridine (27)
Compound 20 (380 mg, 1.7 mmol) was dissolved in aqueous MeOH and treated with Amberlite (H+) (150 mg). The reaction mixture was stirred in the dark overnight at room temperature, filtered, and concentrated. The crude product was purified by silica gel column chromatography (15% CH3OH:CHCl3) in the dark to obtain compound 27 as a light brown solid (200 mg, 78%). UV λmax (MeOH) =267 nm (ε=10,856 mol−1cm−1); 1H NMR (CD3OD) δ 3.67 (dd, 1H), 3.87 (m, 2H), 4.53 (ddd,1H, H-3′, JH–F=21 Hz), 5.43 (dd,1H, H-2′, JH–F=57 Hz), 6.08 (dd, 1H, H-1′, JHF=27 Hz), 6.43 (s, 1H). 19F NMR (CD3OD) δ −193.27 (ddd, JH–F =57, 27, 21 Hz). HRMS (ESI) calculated for C9H10N2O5FNaI (M+Na): 394.9510; observed: 394.9516.
Deprotection of 5- and 6-substituted 2′-fluoro nucleosides
Compounds 21, 23–26 were deprotected using the procedure described for 27 to yield compounds 28–32, respectively.
6-Azido-2′-deoxy-2′-fluoro-β-D-uridine (28)
Compound 25 was deprotected to obtain compound 28 as a pale yellow solid (100 mg, 54% yield). UV λmax (MeOH) = 283 nm (ε=10,691 mol−1cm−1). 1H NMR (CD3OD) δ 3.61 (dd, 1H), 3.71 (m, 2H), 4.43 (ddd, 1H, H-3′, JH–F=21 Hz), 5.24 (dd,1H, H-2′, JH–F=56.1 Hz), 5.46 (s, 1H), 5.94 (dd, 1H, H-1′, JH–F=27.5 Hz). 19F NMR (CD3OD) δ −193.32 (ddd, J =56.1, 20.1, 27.5 Hz). HRMS (ESI) calculated for C9H11FN5O5 (MH+): 288.0745, observed: 288.0748; calculated for C9H10FN5NaO5 (MNa+): 310.0564, observed: 310.0564.
6-Cyano-2′-deoxy-2′-fluoro-β-D-uridine (29)
Compound 23 was deprotected to yield product 29 as a pale yellow solid (1.86 g, 82% yield). UV λmax (MeOH)=278 nm ε=3209 mol−1 cm−1); 1H-NMR (CD3OD) δ 3.69 (dd, 1H), 3.84–3.94 (m, 2H), 4.55 (ddd, 1H, H3′, JH–F = 20 Hz), 5.45 (dd, 1H, H2′, JH–F = 55 Hz), 5.88 (d, 1H, H1′, JH–F = 25 Hz), 6.46 (s, 1H); 13C NMR (CD3OD) δ 61.4, 68.8, 83.7, 91.9, 92.6, 93.7, 111.1, 113.0,128.1, 161.7; 19F NMR (CD3OD) δ − 195.5 (ddd, J = 54.9, 25.0, 19.8 Hz); HRMS (ESI) for C10H10N3O5FNa (MNa+), calculated: 294.0506; observed: 294.0496.
6-Amido-2′-deoxy-2′-fluoro-β-D-uridine (30)
Compound 24 was deprotected to yield product 30 as a white solid (212 mg, 61% yield). UV λmax (MeOH) = 264 nm (ε 2,655 mol−1cm−1). 1H NMR (CD3OD) δ 3.68 (dd, 1H), 3.79–3.88 (m, 2H), 4.49 (ddd, 1H, H-3′, JH–F =19 Hz), 5.38 (ddd, 1H, H-2′, JH–F =56 Hz), 5.73 (dd, 1H, H-1′, JH–F =26 Hz), 5.81(s, 1H); 13C NMR (CD3OD) δ 61.9, 69.0, 83.7, 92.0, 94.5, 101.7, 150.3, 150.5, 164.0, 164.7; 19F NMR (CD3OD) δ − 195.2 (ddd, J = 55.5, 25.8, 19.0 Hz); HRMS (ESI) for C10H12N3O6FNa (MNa+), calculated: 312.0589; observed: 312.0602.
5-Cyano-2′-deoxy-2′-fluoro-β-D-uridine (31)
Product 31 was obtained as a yellow solid (532 mg, 87% yield) from compound 26. UV λmax (MeOH) = 274 nm (ε 4066 mol−1cm−1); 1H NMR (CD3OD) δ 3.79 (dd, 1H), 3.99–4.09 (m, 2H), 4.23 (ddd, 1H, H-3′, JH–F=24 Hz), 5.0 (dd, 1H, H-2′, JH–F=53 Hz), 5.95 (d, 1H, H-1′, JH–F=16 Hz), 9.0 (s, 1H, H-6); 13C NMR δ 58.7, 67.1, 83.9, 89.0, 92.7, 95.2, 113.4, 149.39, 149.6, 160.9; 19F NMR (CD3OD) δ 145.3 (ddd, J =52.1, 23.2, 16.0); HRMS (ESI) for C10H11N3O5F (MH+), calculated: 272.0666; observed: 272.0677.
2′-Deoxy-2′-fluoro-β-D-orotidine ethyl ester (32)
Compound 21 was deprotected to yield compound 32 as a white solid (93 mg, 81% yield). UV λmax (MeOH) = 272 nm (ε 2887 mol−1cm−1). 1H NMR (CD3OD) δ 1.36 (t, 3H), 3.62–3.69 (m, 1H), 3.80–3.84 (m, 2H), 4.35–4.48 (m, 3H), 5.34 (ddd, 1H, H-2′, JH–F = 55 Hz), 5.85 (dd, 1H, H-1′, JH–F =25 Hz), 6.07 (s, 1H); 19F NMR (CD3OD) δ − 195.54 (ddd, J = 55.4, 25.5, 19.1 Hz); HRMS (ESI) for C12H15N2O7FNa (MNa+), calculated: 341.0756; observed: 341.0755.
2′-Deoxy-2′-fluoro-6-iodo-β-D-uridine-5′-O-monophosphate (33)
Compound 27 (100 mg, 0.27 mmol) was added to a solution of water (14 μL, 0.78 mmol), CH3CN (2.5 mL), pyridine (0.1 mL, 1.28 mmol), and POCl3 (164 mg, 0.1 mL, 1.1 mmol) at 0°C. The reaction mixture was stirred for 5 h at 0°C. The reaction was quenched with cold water (2 mL), concentrated, and the crude product was purified by Dowex (H+) resin using 100% water followed by 5% formic acid as an eluent. The product was then dissolved in 3 mL of cold water, neutralized to pH 7 with a saturated NH4OH solution, and lyophilized to obtain compound 33 as a white solid (54 mg, 41% yield). UV λmax (MeOH) =267 (ε 948 mol−1cm−1); 1HNMR (D2O) δ 3.89 (dd, 1H), 4.02 (m, 2H), 4.65 (m, 1H), 5.51 (dd, 1H, H-2′, JH-F=57 Hz), 6.08 (s, 1H), 6.26 (d, 1H, H-1′, JH–F=30 Hz). 31P NMR (D2O) δ 1.04 (s); HRMS (ESI) calculated for C9H10N2O8P (M−): 305.0180; observed: 305.0191.
6-Azido-2′-deoxy-2′-fluoro-β-D-uridine-5′-O-monophosphate (34)
A procedure similar to that for compound 33 was used. The reaction was carried out in the dark, and product 34 was obtained as a white solid (23 mg, 37% yield) from compound 28. UV λmax(MeOH) =283 nm (ε 9,179 mol−1cm−1); 1H NMR (D2O) δ 4.00 (m, 2 H), 4.14 (m, 1 H), 4.62 (ddd, 1H, H-3′, JH–F=20.6 Hz), 5.44 (dd, 1H, H-2′, JH–F=55 Hz), 5.69 (s, 1H), 6.18 (dd, 1H, H-1′, JH–F=27.5 Hz),. 19F NMR (D2O) δ − 192.2 (ddd, JH–F=55, 20.7, 25.8 Hz). 31P NMR (D2O) δ 1.13 (s); HRMS (ESI) for C9H11FN5NaO8P+ (M-2NH4+Na+2H+), calculated: 390.0227; observed: 390.0226.
6-Cyano-2′-deoxy-2′-fluoro-β-D-uridine-5′-O-monophosphate (35)
Compound 29 (154 mg, 0.6 mmol) was added to a stirred solution of POCl3 (0.2 mL, 2.3 mmol), H2O (0.03 mL, 1.6 mmol), CH3CN (3 mL), and pyridine (0.2 mL, 2.7 mmol) at 0°C and stirred for 7 h. The reaction mixture was quenched with ice, concentrated, and the crude product was purified using a Biotage SP1™ column chromatography system (C18 column, 100% H2O) and lyophilized to give 35 as a white foam (83 mg, 38% yield). UV λmax (H2O) =278 nm (ε 2,793 mol−1cm−1); 1H NMR (D2O) δ 3.80–3.88 (m, 1H), 3.94–4.03 (m, 2H), 4.51 (ddd, 1H, H-3′, JH–F =21 Hz), 5.40 (ddd, 1H, H-2′, JH–F =55 Hz), 5.88 (dd, 1H, H-1′, JH–F=26 Hz), 6.46 (s, 1H); 19F NMR (D2O) δ − 194.0 (ddd, J= 54.4, 25.3, 20.5 Hz); 31P NMR (D2O) δ 3.80 (s); HRMS (ESI) for C10H10N3O8FP (M−), calculated: 350.0207; observed: 350.0195.
6-Amido-2′-deoxy-2′-fluoro-β-D-uridine-5′-O-monophosphate (36)
A procedure similar to that for compound 35 was used. The product was obtained as a white solid (70 mg, 31% yield) from compound 30. UV λmax (MeOH) = 264 nm (ε 2,414 mol−1cm−1); 1H NMR (D2O) δ 3.77–3.88 (m, 1H), 3.88–4.0 (m, 2H), 4.43 (ddd, 1H, H-3′, JH–F =20 Hz), 5.35 (dd, 1H, H-2′, JH–F =55 Hz), 5.65 (d, 1H, H1′, JH–F =26 Hz), 5.94 (s, 1H), 8.31 (s, 1H); 19F NMR (D2O) δ −194.1 (ddd, J =55.1, 25.5, 20.2 Hz); 31P NMR (D2O) δ 3.94 (s); HRMS (ESI) for C10H12N3O9FP (M−), calculated: 368.0305; observed: 368.0300.
5-Cyano-2′-deoxy-2′-fluoro-β-D-uridine-5′-O-monophosphate (37)
A stirred solution of POCl3 (0.2 mL, 2.3 mmol) and H2O (0.03 mL, 1.6 mmol) in CH3CN (3 mL) was treated with pyridine (0.02mL, 2.7 mmol) at 0°C, 31 (150 mg, 0.57 mmol) was added, and the reaction mixture was stirred at 0°C for 7 h. The reaction mixture was quenched with cold water and continuously stirred overnight at 22°C. Following evaporation of the solvent, the reaction mixture was brought to pH 6 with NH4OH, purified on a Biotage SP1™ chromatography system (C18, mobile phase: 100% H2O), concentrated, and lyophilized to obtain compound 37 as a solid (79 mg, 36% yield). UV λmax (MeOH)=274 nm (ε 3412 mol−1cm−1); 1H NMR (D2O) δ 3.79 (dd, 1H), 3.97–4.16 (m, 2H), 4.23 (ddd, 1H, H-3′, JH–F=23 Hz), 5.0 (dd, 1H, H-2′, JH–F=53 Hz), 5.86 (d, 1H, H-1′, JH–F=19 Hz), 8.3 (s, 1H, H6); 19F NMR (D2O) δ 148.6 (ddd, J=52.8, 23.0, 18.8 Hz); 31P (D2O) δ 4.97 (s); HRMS (ESI) for C10H10N3O8FP (M−), calculated: 350.0190; observed: 350.0195.
6-Amino-2′-deoxy-2′-fluoro-β-D-uridine (38)
Compound 28 (84 mg, 0.29 mmol) was dissolved in 50% aqueous MeOH (6 mL) in the dark and was treated with 10% Pd/C (9 mg). The reaction mixture was stirred for 3 h under a hydrogen atmosphere at 1 atm at room temperature and filtered through a pad of Celite™. The solvent was evaporated under vacuum to yield compound 38 (59 mg, 78% yield) as a pale yellow solid. UV λmax (MeOH)=270 nm (ε 17,147 mol−1cm−1); 1H NMR (D2O) δ 3.74 (dd, 1H), 3.90 (dd, 1H), 3.97 (m, 1H), 4.51 (ddd, 1H, H-3′, JH–F=18.3 Hz), 4.93 (s, 1H), 5.46 (dd, 1H, H-2′, JH–F=54.4 Hz), 5.94 (dd, 1H, H-1′, JH–F=25.8 Hz),. 19F NMR (D2O) δ −193.68 (ddd, J=54.4, 18.3, 25.8 Hz). HRMS (ESI) for C9H13FN3O5 (MH+), calculated: 262.0840, observed: 262.0841; for C9H12FN3NaO5 (MNa+), calculated: 282.0659, observed: 284.0659.
Enzymology
Assays were performed on a VP-ITC microcalorimeter (MicroCal/GE Healthcare) using established protocols.15,20 Mt ODCase was used as a model enzyme, and assays with human ODCase were conducted when possible.
Reversible, competitive inhibition of ODCases
Concentrated enzyme stock solutions were prepared in 50 mM Tris, pH 7.5, 20 mM DTT, and 40 mM NaCl. Concentrated stocks of the substrate, OMP, and inhibitors were prepared in 50 mM Tris. The stock concentration of Mt ODCase was 20 μM and that of human ODCase was 60 μM. The assay temperatures for the human and Mt enzymes were 37 and 55°C, respectively. The final concentration of Mt ODCase in the control reaction was 20 nM, and the final substrate concentration was 40 μM. The final concentration of human ODCase was 60 nM in the assay mixture, and the final concentration of the substrate was 20 μM.
Time-dependent inactivation of human and Mt ODCases
Enzyme stocks (70 and 25 μM for human and Mt, respectively) were prepared in 50 mM Tris, pH 7.5, 20 mM DTT, and 40 mM NaCl and were incubated overnight at room temperature prior to evaluation using the assay. The substrate and the inhibitor were prepared in 50 mM Tris buffer. Assay samples were prepared in a degassed buffer (50 mM Tris, 1 mM DTT, pH 7.5).
Human enzyme (60 μM) or Mt ODCase was incubated at room temperature in the presence of inhibitor 33 at various concentrations. Aliquots of 2.5 μL of the incubation mixture were diluted to a final volume of 2.5 mL in 50 mM Tris and 1 mM DTT buffer. The remaining enzyme activity was then measured by introducing 5 mM substrate (5.7 and 11.4 μL for human and Mt ODCases, respectively) via a single injection into the sample cell of the calorimeter. Samples containing 20 μM enzyme and compound 35 at various concentrations were incubated at room temperature for up to 48 h. The control reaction contained no inhibitor. For the time-dependent assay using the 6-azido derivative 34 against Mt ODCase, the enzyme (20 μM) was incubated at room temperature in the presence of various concentrations of 34. The remaining enzyme activity was measured in the control and inhibitor-treated samples at various time points.
Mass spectral analyses
Mass spectra for the enzymes and the complexes were obtained at the Mass Spectrometry-AIMS Laboratory, Department of Chemistry, University of Toronto on a AB/Sciex QStar mass spectrometer with an electrospray ionization (ESI) source and on an Agilent 1100 capillary LC (MDS Sciex). All samples were prepared in 50 mM Tris (pH 7.5), 20 mM DTT, and 40 mM NaCl. Human ODCase was exposed to 29 mM 34, and Mt ODCase was incubated in the presence of 25 mM 34 overnight at room temperature in the dark. The reaction mixture of human ODCase and 33 was measured after 2 h of incubation of the enzyme (60 μM) with the inhibitor (3 mM).
For compound 35, the inhibitor was incubated overnight with either human or Mt ODCase. The concentrations of the human ODCase and compound 35 were 100 μM and 437.5 mM, and those for Mt ODCase and 36 were 200 μM and 388 mM, respectively.
Data analyses
The raw data were analyzed using Origin 7.0 software. First, the data were adjusted for the time delay (16 sec) for the start of the reaction in the isothermal calorimeter (ITC), and the baseline was adjusted using the “baseline” function. These data were then converted into rate versus [S] plots. All data sets were fitted to equation 1:
| Equation 1 |
The data sets were further analyzed using Grafit 5.0 to derive the inhibition parameters for competitive inhibitors. The inhibition constant, Ki, for reversible (competitive) inhibition was determined by fitting the rate versus [S] data to equation 2:
| Equation 2 |
To determine the inactivation parameters for the inhibition of human and Mt ODCases by 6-iodo derivative 34, kobs was first computed at each inhibitor concentration from the slope of ln(% remaining enzyme activity) versus time. The calculated kobs and the inhibitor concentrations [I] were used to calculate the second-order rate constant of inactivation (kobs/[I]). The inactivation of Mt ODCase due to inhibitors 35 and 36 followed first-order reaction kinetics. The KI and kianct were derived for each inhibitor from the nonlinear least squares fit of the kobs versus the inhibitor concentration to the following equation:
| Equation 3 |
X-Ray Crystallography
Crystallization and collection of diffraction data
All human ODCase concentrations were determined using a BioRAD protein assay kit and bovine serum albumin (BSA) as a standard. Crystals were grown at room temperature using the hanging-drop method; 2 μL of protein solution containing 10 mg/mL enzyme, 10 mM 6-iodo derivative 33 in 25 mM HEPES pH 7.5, 500 mM NaCl, 5% glycerol, and 25 mM EDTA, were mixed with 2 μL of the respective reservoir solution. Preliminary hits were optimized. For data collection, the crystals were cryo-protected by bringing the mother liquor to 20% glycerol before flash-freezing in a stream of boiling nitrogen. Diffraction data were collected at 100 K and λ = 0.97934 Å on beamline 08ID-1 at the Canadian Macromolecular Crystallography Facility at the Canadian Light Source, Inc. Data were reduced and scaled using HKL2000.32
The structure of the enzyme complex was determined using molecular replacement techniques with the program package MOLREP; subsequent refinement was performed with Refmac-5.2, and model building used COOT.33,34,35 Statistics for data collection and refinement are given in Table 3. Atomic coordinates and structure factors have been deposited into the Protein Data Bank (PDB IDs: ****).
Table 3.
X-ray diffraction data for the co-crystals of 2′-fluoro-6-iodo-UMP (33) bound to human ODCase.
| Diffraction data | ||
| Resolution (Å) | 1.35(1.40–1.35) | |
| Measured reflections (n) | 58,884 | |
| Unique reflections (n) | 56,107 | |
| Completeness (%) | 94.1(97.7) | |
| Rsym (%) | 7.3 (47.6) | |
| Space group | C2221 | |
| Cell dimensions (Å); | a | 78.499 |
| b | 116.0 | |
| c | 62.1 | |
| Molecules in asymmetric unit (n) | 1 | |
| Refinement statistics | ||
| Resolution (Å) | 50–1.35 (1.35–s1.38) | |
| Protein atoms (n) | 2,300 | |
| Water molecules (n) | 280 | |
| Reflections used for Rfree (n) | 2,994 (220) | |
| Rwork (%) | 15.5 (23.8) | |
| Rfree (%) | 17.5 (25.6) | |
| Root mean square deviation bond length (Å) | 0.010 | |
| Root mean square deviation bond angle (°) | 1.44 | |
| Average B-factor (Å2) | 14.0 | |
Results and Discussion
Nucleosides with modified carbohydrates carrying fluoro and difluoro substitutions have shown potent antiviral and anticancer activities.36,37 This is due to the structural and conformational influences introduced by the fluoro moiety onto the carbohydrate moiety of the nucleosides as well as the improved metabolic stabilities of such modified nucleosides.24,38,39,40 Incorporation of the fluorine moiety at the 2′-position of the ribofuranosyl uridine derivatives is based on the idea that a fluorine atom (radius of 1.47 Å) could mimic the features of an oxygen or a hydrogen atom (radii of 1.52 and 1.20 Å, respectively). Despite a long history of incorporation of fluorines onto carbohydrate moieties and nucleosides, there are no general principles linking the substitution of fluorines onto nucleosides and the potential improvements in the corresponding biological activities, specifically in relation to their binding to the target site. To investigate the effect of the 2′-fluoro moiety on the binding of ligands to ODCase, we synthesized a series of nucleoside derivatives in which the 2′-hydroxyl moiety was substituted with a fluorine atom in addition to the substitutions at the C-5 or C-6 positions of the uridine moiety (Figure 2).
The target compounds were synthesized from the key intermediate 18 (Scheme 1).31 First, the arabinosyl moiety in compound 18 was transformed into the 2′-fluoro ribosyl configuration by treatment with diethylamino sulfur trifluoride to yield the fluoro derivative 19. This derivative was then treated with LDA followed by iodine or ethyl chloroformate to obtain compounds 20 and 21, respectively.41 Compound 22, carrying a C5-bromo substitution, was obtained by the treatment of fully protected uridine 19 with N-bromosuccinimide. Compound 22 was then transformed into the 6-cyano derivative 23 exclusively by treatment with sodium cyanide at 22°C.42
Scheme 1.

Synthesis of 2′-fluoro-6-substituted nucleosides. Reagents: (a) DAST, Pyridine, CH2Cl2, 40°C, overnight; (b) LDA, THF, −70°C followed by I2 or ClCO2Et; (c) NBS, NaN3, 1,2-dimethoxyethane, rt, overnight; (d) NaCN, DMF, overnight, rt; (e) 1N NaOH, rt, 1 h; (f) NaN3, DMF; (g) NaCN, DMF, μW @ 150°C, 5 min.
The 6-cyano derivative 23 was treated with sodium hydroxide to obtain 6-amido derivative 24 (Scheme 1A). Treatment of 6-iodo derivative 20 with sodium azide yielded the 6-azido derivative 25 (Scheme 1B). The 5-cyano derivative 26 was obtained by the treatment of 5-bromo derivative 22 at 150°C for 5 min under microwave conditions using the modified procedure of Torrence et al. (Scheme 1C).43 Compounds 20, 21, and 23–26 were deprotected using Amberlite® (H+), yielding the corresponding 6-substituted-2′-fluoro nucleosides 27–32 (Scheme 2A). These nucleosides were phosphorylated at the 5′-position to obtain the corresponding nucleotides 33–37. 6-Amino uridine derivative 38 was obtained by the reduction of the corresponding azido derivative 28 (Schemes 2B).
Scheme 2.

Synthesis of 2′-fluoro nucleosides and mononucleotides. Reagents: (a) Amberlite (H+), H2O:MeOH (1:1); (b) H2O/Pyridine/POCl3, CH3CN, 0°C; (c) H2, Pd/C.
First, 6-cyano derivative 35 was investigated as a reversible inhibitor of ODCase (35 is also a time-dependent inactivator of ODCase, and its characteristics are presented after this discussion). Compound 35 exhibited moderate inhibition of Mt ODCase as a competitive inhibitor with an inhibition constant (Ki) of 37.0 ± 2.5 μM, but it did not inhibit the catalytic activity of human ODCase up to a concentration of 2 mM (Table 1). In comparison, 6-cyano ribosyl derivative 11 is a moderate inhibitor of Mt ODCase with an inhibition constant of Ki = 29 μM.16 The 6-amido-2′-deoxy-2′-fluoro analog 36 was a poor inhibitor of both human and Mt ODCases (Ki >3 mM) (Table 1). Compound 37, carrying a cyano group at C5 position, exhibited weaker inhibition of the catalytic activity of ODCase (Ki = 360 ± 23 and 759 ± 88 μM, against Mt and human ODCases, respectively; Table 1). 6-Amido-UMP, a close analog of the substrate OMP, and its 2′-deoxy-2′-fluoro analog 36 were poor inhibitors of Mt ODCase, with Kis of 1.32 ± 0.04 and 3.32 ± 0.18 mM, respectively. In general, competitive inhibition of ODCases by 2′-deoxy-2′-fluoro uridine derivatives was either similar to or weaker than the corresponding ribosyl derivatives.16,17,21 Compounds with 2′-fluoro substitution did not necessarily improve the potency of the synthesized nucleotides against ODCase as competitive inhibitors. At the outset, the loss of potency by one or more folds in the 2′-fluorinated nucleotides may have been due to the loss of hydrogen-bonding interactions when the 2′-hydroxyl moiety was replaced with the fluorine atom. However, there were other significant reactivities that were introduced onto the 2′-fluorinated nucleotides (vide infra), despite the loss of hydrogen-bonding strength. Among the 2′-fluorinated derivatives, the inhibitory constants (Kis) against the human and Mt ODCases varied up to 3 orders of magnitude (Table 1). Such variations were due to differences in the species that existed between the Mt and human ODCases and are commonly seen with other classes of inhibitors.20
Table 1.
Inhibition kinetics of Mt and human ODCases using various 2′-fluorinated uridine nucleotides.
Three of the derivatives, 33, 34, and 35, carrying the iodo, azido, and cyano moieties, respectively, at the C6 position of the nucleotide exhibited time-dependent inactivation of ODCases, which was discovered during the enzyme kinetics experiments. While compounds 33 and 34 were expected to behave as covalent and time-dependent inhibitors, compound 35, carrying a 6-cyano moiety, displayed surprising behavior; it inactivated ODCase irreversibly, forming a covalent bond. 6-Iodo derivative 33 inactivated human ODCase irreversibly with a second-order rate constant (Kobs/[I]) of 2.0 ± 0.01 M−1sec−1 and with a three-fold increase in the rate of inactivation against Mt ODCase (0.62 ± 0.02 M−1sec−1) (Table 2). 6-Azido derivative 34 and 6-cyano derivative 35 were weaker inactivators, with equilibrium inhibition constants (KI) of 1.2 ± 0.1 and 30 ± 6 mM, respectively, against Mt ODCase (Table 2). The first-order rates of inactivation (kinact) were 4.0 ± 0.2 h−1 for 34 and 2.0 ± 0.2 h−1 for compound 35. Interestingly, 6-azido-5-fluoro-UMP (12) exhibited higher affinity (KI) than the corresponding 2′-deoxy-2′-fluoro derivative 34. In comparison, 6-azido-UMP (14) was a potent compound, with an equilibrium inhibition constant of KI = 6.3 ± 1.1 × 10−4 mM, which was about 30-fold higher than that due to the 5-fluoro derivative 12 and about 2000-fold more potent than the corresponding 2′-deoxy-2′-fluoro derivative 34 (Table 2). The ribofuranosyl analog, 6-iodo-UMP (10), was the most potent of all these molecules, with a second-order rate constant ~5 orders of magnitude higher than that of the corresponding 2′-deoxy-2′-fluoro analog 33 (kinact =2.6 × 105 h−1).
Table 2.
Irreversible inactivation kinetics of Mt and human ODCases by 2′-fluoro-6-substituted nucleotides.
| Inhibitor | kobs/[I] (M−1 sec−1) | ||
|---|---|---|---|
| M. thermoautotrophicum ODCase | Human ODCase | ||
| 6-I-UMP (10)16,22 | 2.6 × 105 | ND | |
| 2′-F-6-I-UMP (33) | 2.0 ± 0.01 | 0.62 ± 0.02 | |
| KI (mM) | kinact (hr−1) | ||
| 5-F-6-N3-UMP (12)17 | 18.2 ± 3.6 × 10−3 | 41 ± 3 | ND |
| 6-N3-UMP (14) | 6.3 ± 1.1 × 10−4 | 612 | ND |
| 2′-F-6-N3-UMP (34) | 1.2 ± 0.1 | 4.0 ± 0.2 | ND |
| 2′-F-6-CN-UMP (35) | 30 ± 6 | 2.0 ± 0.2 | ND |
The irreversible inhibitors of ODCase, 33–35, were further analyzed to confirm the covalent binding to the enzyme. Thus, the ODCase enzyme was incubated with each inhibitor, and the sample was analyzed using mass spectrometry. Compounds 33 and 34, the 6-iodo and 6-azido derivatives, showed a covalent complex after elimination of the 6-substitution, as expected (data not shown). This pattern was very similar to the corresponding 6-substituted UMP derivatives.15,16,17 However, compound 35, carrying a 6-cyano moiety, surprisingly produced a covalent complex; the mass implied that the mechanism for the time-dependent loss of activity was due to covalent bond formation (Figure 6A). In the control experiment, the mass for the enzyme was clearly seen at 27,344 a.m.u. (inset in Figure 6A). When the enzyme-ligand complex with 35 was analyzed, along with the native ODCase at 27,344 a.m.u., an additional and significant peak at 27,668 a.m.u. was observed, corresponding to the molecular weight of the enzyme and 33 after removal of the cyano group. This unequivocally confirmed covalent bond formation between the enzyme and ligand when ODCase was challenged with 33. This process was slow, as seen in the enzyme kinetics, with a kinact of 4.0 ± 0.2 h−1. This was unexpected because none of the other 6-cyano-UMP derivatives showed a covalent complex with ODCase, but they exhibited other unnatural biochemical transformations in the presence of ODCase. For example, 6-CN-UMP (11) in the presence of ODCase was transformed into 6-hydroxy-UMP or BMP (3), a chemically unknown transformation.22,23 There was no evidence for covalent bond formation in the case of 11 with the wild-type ODCase, but a hydrolysis (of C6–C7 bond) facilitated by ODCase leading to 3 occurred (Figure 6B). Substitution of the 2′-hydroxyl moiety with a fluorine moiety probably influenced the microenvironment and nucleophilicity of the active site residue, Lys72 (Mt ODCase numbering, equivalent to Lys125 residue in human ODCase), upon binding of ligand 35 to the active site of ODCase. Instead of a water molecule, as seen for 11, Lys72 displaced the C6 cyano group, leading to covalent bond formation similar to those of 6-iodo and 6-azido derivatives 33 and 34. These experiments were repeated using human ODCase, and results identical to those of Mt ODCase were observed.
Figure 6.

(A) Mass spectra for the control (inset) and 6-cyano-2′-deoxy-2′-fluoro-UMP (35) treated ODCases. (B) The biochemical transformation of 6-cyano- and 6-cyano-2′-deoxy-2′-fluoro-UMP derivatives by ODCase. In the former case, a hydrolytic process was observed, while in the latter case a covalent complex was observed in the active site of ODCase.
6-Iodo derivative 33 was co-crystallized with the human ODCase, and its three-dimensional structure was determined to understand the interactions of this class of compounds with the catalytic site of ODCase (Figure 4A). 2′-Fluoro-6-iodo-UMP (33) assumed a similar conformation, and a similar hydrogen-bonding network was formed, except at the 2′ position of the ribosyl moiety, when compared with the complex of its ribosyl derivative 10 (Figures 4A vs 4B). This is the first time that a 2′-fluoronucleotide was co-crystallized with a biological target. Gonin et al. reported the three-dimensional structure of the complex of a nucleoside diphosphate kinase (NDK) with a 2′,3′-dideoxy-3′-fluoro nucleotide, which was the only other fluororibosyl nucleotide co-crystallized with its target enzyme (Figure 4C).44 The substitution of the 3′-hydroxyl moiety in 2′-deoxy-UDP with a fluorine atom led to the loss of catalytic efficiency of NDK.44 However, 2′,3′-dideoxy-UDP, lacking the 3′-fluoro moiety, was a better substrate to NDK than the fluorinated analog.
Figure 4.
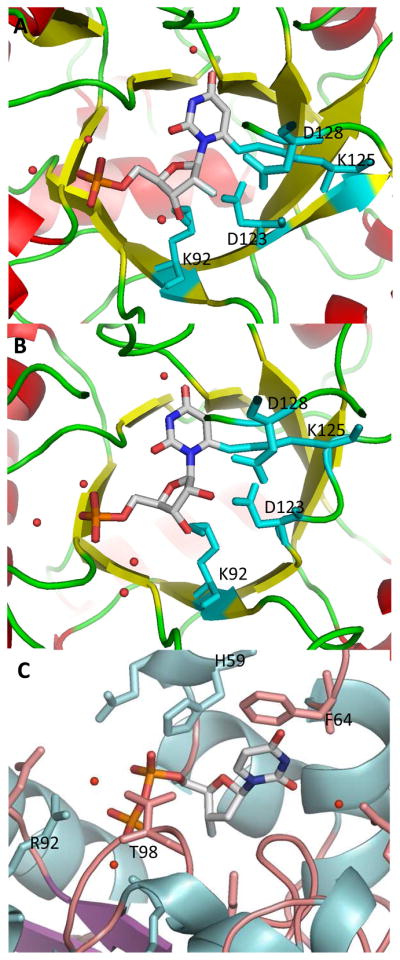
(A) Co-crystal structure of human ODCase covalently modified by 2′-fluoro-6-iodo-UMP (33). (B) X-ray crystal structure of human ODCase covalently modified by 6-iodo-UMP (10) (pdb code: 3BGJ). (C) X-ray crystal structure of nucleoside diphosphate kinase bound by 3′-fluoro-UDP (pdb code: 1B99). Active site regions are shown with the ligand rendered according to atom type. The secondary structures of the proteins are shown, and only the important active site residues are shown in a capped stick representation. Water molecules are shown as red spheres.
In the co-crystal structure of 33 bound to ODCase, a covalent bond was observed between the C6 of 33 and Lys-125 in the active site of human ODCase (Figure 4A). Two strong hydrogen bonds with the residues Asp128 and Thr132 in the active site of ODCase were compromised in the case of 33 due to the weaker hydrogen bonding potential of fluorine as well as unfavorable interactions (Figure 5). An unfavorable interaction of the carboxyl moiety of Asp128 and the 2′-fluoro moiety was observed in the complex between 33 and ODCase, leading to hydrogen-bonding interactions between Asp128 and Lys125- Nε. Such an interaction was absent in the co-crystal structure with 10 because the 2′-hydroxyl moiety was occupied by Asp128 via a classical hydrogen bond.
Figure 5.

Hydrogen-bonding networks of 2′-fluoro-6-iodo-UMP (33, Panel A) and of 6-iodo-UMP (10, Panel B) bound in the active site of human ODCase.
The 2′-fluoro-6-substituted uridine derivatives were evaluated for their antimalarial and anticancer activities. None of the synthesized nucleosides exhibited significant antimalarial activities against Plasmodium falciparum (3D7) or any significant anticancer activities in OCI-AML-1 cell lines (acute myeloid leukemia) in the cell-based assays (Table 4). Although the inhibition constants against the isolated enzyme ODCase revealed reasonable potency for the 2′-fluoro-6-substituted mononucleotide derivatives, the lack of cellular activities may have been due to the poor activation of these modified nucleosides into the corresponding mononucleotide forms. It is also interesting to note that 2′-fluoro-2′-deoxyuridine possessing a 5-(2-iodovinyl) substituent and 2′-deoxy-2′-fluoro-5-iodouridine undergo very low uptake in normal cells, but very high uptake in thymidine kinase postive cells.45 Both compounds exhibit weak antiviral activities. This suggests a 2′-ribo-2′-deoxyuridine moiety should be considered carefully for potential cellular and in vivo therapeutic activities.
Table 4.
Antimalarial and anticancer activities of 2′-fluoro-6-substituted nucleosides (expressed as IC50 in μM).
In summary, we determined the structure-activity relationships of 2′-deoxy-2′-fluoro-UMP derivatives as potential ODCase inhibitors and revealed novel chemistry of these fluorinated nucleotides with the target enzyme. While these nucleosides do meet the general criteria defined for the ODCase pharmacophore,14 issues of species of origin of the specific ODCase and the activation of the fluorinated nucleosides into the corresponding mononucleotides affect their ultimate therapeutic activities. The influence of 2′-fluoro moiety on modulation of the reactivity at the C6 center of the nucleotide could be used in the design of novel inhibitors of ODCase and perhaps other enzymes that accept nucleotides as ligands.
Supplementary Material
Figure 3.

Enzyme inhibition kinetics for 6-cyano derivative 35 (Panel A), 6-amido derivative 36 (Panel B), and 5-cyano derivative 37 (Panel C) against Mt ODCase.
Acknowledgments
The authors thank Mr. Terence To for help with the expression and purification of ODCase enzymes. This work was supported by the Canadian Institutes of Health Research (EFP & LPK) and University Health Network. LPK, EFP, KCK and IC thank the financial support from ISTPCanada. EFP acknowledges support through the Canada Research Chairs program. We thank the staff at BioCARS, sector 14 beamlines, at the Advanced Photon Source, Argonne National Laboratories, for their generous time commitment and support. Use of the Advanced Photon Source was supported by the Basic Energy Sciences, Office of Science, United States Department of Energy, under contract W-31-109-Eng-38. Use of the BioCARS sector 14 was supported by the National Center for Research Resources, National Institutes of Health, under grant RR07707. We also gratefully acknowledge the help we received from the staff of the Canadian Macromolecular Crystallography Facility at the Canadian Light Source, which is supported by NSERC, NRC, CIHR, and the University of Saskatchewan.
Abbreviations
- Mt
Methanobacterium thermoautotrophicum
- ODCase
Orotidine-5′-monophosphate decarboxylase
- UMP
Uridine-5′-monophosphate
- OMP
Orotidine-5′-monophosphate
- ITC
Isothermal calorimeter
- BMP
Barbiturate-5′-monophosphate
Footnotes
Supplementary Information. Purity data for compounds 27–38.
References
- 1.Cui W, DeWitt JG, Miller SM, Wu W. No metal cofactor in orotidine 5′-monophosphate decarboxylase. Biochem Biophys Res Comm. 1999;259:133–135. doi: 10.1006/bbrc.1999.0737. [DOI] [PubMed] [Google Scholar]
- 2.Shostak K, Jones ME. Orotidylate decarboxylase: insights into the catalytic mechanism from substrate specificity studies. Biochemistry. 1992;31:12155–12161. doi: 10.1021/bi00163a026. [DOI] [PubMed] [Google Scholar]
- 3.Reichard P. The enzymic synthesis of pyrimidines. Adv Enzymol Mol Biol. 1959;21:263–294. [Google Scholar]
- 4.Donovan WP, Kushner SR. Purification and characterization of orotidine-5′-phosphate decarboxylase from Escherichia coli K-12. J Bacteriol. 1983;156:620–624. doi: 10.1128/jb.156.2.620-624.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pragobpol S, Gero AM, Lee CS, O’Sullivan WJ. Orotate phosphoribosyltransferase and orotidylate decarboxylase from Crithidia luciliae: subcellular location of the enzymes and a study of substrate channeling. Arch Biochem Biophys. 1984;230:285–293. doi: 10.1016/0003-9861(84)90109-7. [DOI] [PubMed] [Google Scholar]
- 6.Gero AM, O’Sullivan WJ. Purines and pyrimidines in malarial parasites. Blood Cells. 1990;16:485–498. [PubMed] [Google Scholar]
- 7.Reyes P, Guganig ME. Studies on a pyrimidine phosphoribosyltransferase from murine leukemia P1534J. Partial purification, substrate specificity, and evidence for its existence as a bifunctional complex with orotidine 5-phosphate decarboxylase. J Biol Chem. 1975;250:5097–5108. [PubMed] [Google Scholar]
- 8.McClard RW, Black MJ, Livingstone LR, Jones ME. Isolation and initial characterization of the single polypeptide that synthesizes uridine 5′-monophosphate from orotate in Ehrlich ascites carcinoma. Purification by tandem affinity chromatography of uridine-5′-monophosphate synthase. Biochemistry. 1980;19:4699–4706. doi: 10.1021/bi00561a024. [DOI] [PubMed] [Google Scholar]
- 9.Meza-Avina ME, Wei L, Buhendwa MG, Poduch E, Bello AM, Pai EF, Kotra LP. Inhibition of orotidine 5′-monophosphate decarboxylase and its therapeutic potential. Mini-Rev Med Chem. 2008;8:239–247. doi: 10.2174/138955708783744065. [DOI] [PubMed] [Google Scholar]
- 10.Levine HL, Brody RS, Westheimer FH. Inhibition of orotidine-5′ phosphate decarboxylase by 1-(5′-phospho-®-D-ribofuranosyl)barbituric acid, 6-azauridine-5′-phosphate, and uridine 5′-phosphate. Biochemistry. 1980;19:4993–4999. doi: 10.1021/bi00563a010. [DOI] [PubMed] [Google Scholar]
- 11.Scott HV, Gero AM, O’Sullivan WJ. In vitro inhibition of Plasmodium falciparum by Pyrazofurin, an inhibitor of pyridine biosynthesis de novo. Mol Biochem Parasitol. 1986;18:3–15. doi: 10.1016/0166-6851(86)90045-9. [DOI] [PubMed] [Google Scholar]
- 12.Christopherson RI, Lyons SD, Wilson PK. Inhibitors of de novo nucleotide biosynthesis as drugs. Acc Chem Res. 2002;35:961–971. doi: 10.1021/ar0000509. [DOI] [PubMed] [Google Scholar]
- 13.Najarian T, Traut TW. Nifedipine and nimodipine competitively inhibit uridine kinase and orotidine-phosphate decarboxylase: theoretical relevance to poor outcome in stroke. Neurorehabil Neural Repair. 2000;14:237–2341. doi: 10.1177/154596830001400310. [DOI] [PubMed] [Google Scholar]
- 14.Meza-Avina ME, Wei L, Liu Y, Poduch E, Bello AM, Mishra RK, Pai EF, Kotra LP. Structural determinants for inhibitory ligands of orotidine-5′-monophosphate decarboxylase. Bioorg Med Chem. 2010 doi: 10.1016/j.bmc.2010.04.017. in press. ( http://dx.doi.org/10.1016/j.bmc.2010.04.017) [DOI] [PMC free article] [PubMed]
- 15.Poduch E, Bello AM, Tang S, Fujihashi M, Pai EF, Kotra LP. Design of inhibitors of orotidine monophosphate decarboxylase using bioisosteric replacement and determination of inhibition kinetics. J Med Chem. 2006;49:4937–4945. doi: 10.1021/jm060202r. [DOI] [PubMed] [Google Scholar]
- 16.Bello AM, Poduch E, Liu Y, Wei L, Crandall I, Wang X, Dyanand C, Kain KC, Pai EF, Kotra LP. Structure-activity relationships of c6-uridine derivatives targeting plasmodia orotidine monophosphate decarboxylase. J Med Chem. 2008;51:439–448. doi: 10.1021/jm7010673. [DOI] [PubMed] [Google Scholar]
- 17.Bello AM, Konforte D, Poduch E, Furlonger C, Wei L, Lewis M, Pai EF, Paige CJ, Kotra LP. Structure-activity relationships of orotidine-5′-monophosphate decarboxylase inhibitors as anticancer agents. J Med Chem. 2009;52:1648–1658. doi: 10.1021/jm801224t. [DOI] [PubMed] [Google Scholar]
- 18.Crowther GJ, Napuli AJ, Gilligan JH, Gagaring K, Borboa R, Francek C, Chen Z, Dagostino EF, Stockmyer JB, Wang Y, et al. Identification of inhibitors for putative malaria drug targets among novel antimalarial compounds. Mol Biochem Parasitol. 2011;175:21–29. doi: 10.1016/j.molbiopara.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wittmann JG, Heinrich D, Gasow K, Frey A, Diederichsen U, Rudolph MG. Structures of the human orotidine-5′-monophosphate decarboxylase support a covalent mechanism and provide a framework for drug design. Structure. 2008;16:82–92. doi: 10.1016/j.str.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 20.Poduch E, Wei L, Pai EF, Kotra LP. Structural diversity and plasticity associated with nucleotides targeting orotidine monophosphate decarboxylase. J Med Chem. 2008;51(3):432–438. doi: 10.1021/jm700968x. [DOI] [PubMed] [Google Scholar]
- 21.Bello AM, Poduch E, Fujihashi M, Amani M, Li Y, Crandall I, Hui R, Lee PI, Kain KC, Pai EF, Kotra LP. A potent covalent inhibitor of orotidine 5′-monophosphate decarboxylase with antimalarial activity. J Med Chem. 2007;50:915–921. doi: 10.1021/jm060827p. [DOI] [PubMed] [Google Scholar]
- 22.Fujihashi M, Bello AM, Poduch E, Wei L, Annedi SC, Pai EF, Kotra LP. An unprecedented twist to odcase catalytic activity. J Am Chem Soc. 2005;127:15048–15050. doi: 10.1021/ja054865u. [DOI] [PubMed] [Google Scholar]
- 23.Fujihashi M, Wei L, Kotra LP, Pai EF. Structural characterization of the molecular events during a slow substrate-product transition in Orotinde-5′-monophosphate decarboxylase. J Mol Biol. 2009;387:1199–1210. doi: 10.1016/j.jmb.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pankiewicz KW. Fluorinated nucleosides. Carbohydrate Res. 2000;327:87–105. doi: 10.1016/s0008-6215(00)00089-6. [DOI] [PubMed] [Google Scholar]
- 25.Isanbor C, O’Hagan D. Fluorine in medicinal chemistry: A review of anti-cancer agents. J Fluorine Chem. 2006;127:303–319. [Google Scholar]
- 26.Begue JP, Bonnet-Delpon D. Recent advances (1995–2005) in fluorinated pharmaceuticals based on natural-products. J Fluorine Chem. 2006;127:992–1012. [Google Scholar]
- 27.Meng W-D, Qing F-L. Fluorinated nucleosides as antiviral and antitumor agents. Curr Top Med Chem. 2006;6:1499–1528. doi: 10.2174/156802606777951082. [DOI] [PubMed] [Google Scholar]
- 28.Barton-Burke M. Gemcitabine: a pharmacologic and clinical overview. Cancer Nurs. 1999;22:176–183. doi: 10.1097/00002820-199904000-00011. [DOI] [PubMed] [Google Scholar]
- 29.Jeong LS, Tosh DK, Choi WJ, Lee SK, Kang YJ, Choi S, Lee JH, Lee H, Lee HW, Kim HO. Discovery of a new template for anticancer agents: 2′-deoxy-2′-fluoro-4′-selenoarabinofuranosyl-cytosine (2′-F-4′-seleno-ara-C) J Med Chme. 2009;52:5303–5306. doi: 10.1021/jm900852b. [DOI] [PubMed] [Google Scholar]
- 30.Asselah T, Lada O, Moucari R, Marcellin P. Clevudine: a promising therapy for the treatment of chronic hepatitis B. Expert Opin Investig Drugs. 2008;17:1963–1974. doi: 10.1517/13543780802535760. [DOI] [PubMed] [Google Scholar]
- 31.Shi J, Du J, Ma T, Pankiewicz KW, Patterson SE, Tharnish PM, McBrayer TR, Stuyver LJ, Otto MJ, Chu CK, Schinazi RF, Watanabe KA. Synthesis and anti-viral activity of a series of D- and L-2′-deoxy-fluororibonucleosides in the subgenomic HCV replicon system. Bioorg Med Chem. 2005;13:1641–1652. doi: 10.1016/j.bmc.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 34.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likehood method. Acta Crystallogr, Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan K. Model building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 36.Van Rompay AR, Johansson M, Karlsson A. Substrate specificity and phosphorylation of antiviral and anticancer nucleoside analogues by human deoxyribonucleoside kinases and ribonucleoside kinases. Pharmacol Ther. 2003;100:119–139. doi: 10.1016/j.pharmthera.2003.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan X, Chu CK, Boudinot FD. Development and optimization of anti-HIV nucleoside analogs and prodrugs: A review of their cellular pharmacology, structure-activity relationships and pharmacokinetics. Adv Drug Delivery Rev. 1999;39:117–151. doi: 10.1016/s0169-409x(99)00023-x. [DOI] [PubMed] [Google Scholar]
- 38.Plavec J, Koole LH, Sandstrom A, Chattopadhyaya J. Structural studies of anti-HIV 3′-α-fluorothymidine & 3′-α-azidothymidine by 500 MHz 1H NMR spectroscopy and molecular mechanics (MM2) calculations. Tetrahedron. 1991;47:7363–7376. [Google Scholar]
- 39.Barchi JJ, Jr, Jeong LS, Siddiqui MA, Marquez VE. Conformational analysis of the complete series of 2′ and 3′ monofluorinated dideoxyuridines. J Biochem Biophys Methods. 1997;34:11–29. doi: 10.1016/s0165-022x(96)00032-2. [DOI] [PubMed] [Google Scholar]
- 40.Mikhailopulo I, Pricota TI, Sivets GG, Altona C. 2′-Chloro-2′,3′-dideoxy-3′-fluoro-D-ribonucleosides: Synthesis, stereospecificity, some chemical transformations, and conformational analysis. J Org Chem. 2003;68:5897–5908. doi: 10.1021/jo0340859. [DOI] [PubMed] [Google Scholar]
- 41.Shimuzu M, Tanaka H, Hayakawa H, Miyasaka T. Dynamic aspects in the LDA lithiathion of an arabinofuranosyl derivative of 4-ethoxy-2-pyrimidinone: Regioselective entry to both C-5 and C-6 substitutions. Tetrahedron Lett. 1990;31:1295–1298. [Google Scholar]
- 42.Ueda T, Inoue H, Matsuda A. Synthesis and reaction of some 6-substituted pyrimidine nucleosides. Ann NY Acad Sci. 1975;255:121–130. doi: 10.1111/j.1749-6632.1975.tb29218.x. [DOI] [PubMed] [Google Scholar]
- 43.Torrence PF, Bhooshan B, Descamps J, De Clercq E. Improved synthesis and in vitro antiviral activities of 5-cyanouridine and 5-cyano-2′-deoxyuridine. J Med Chem. 1977;20:974 – 976. doi: 10.1021/jm00217a026. [DOI] [PubMed] [Google Scholar]
- 44.Gonin P, Xu Y, Milon L, Dabernat S, Morr M, Kumar R, Lacombe ML, Janin J, Lascu I. Catalytic mechanism of nucleoside diphosphate kinase investigated using nucleotide analogs, viscosity effects, and X-ray crystallography. Biochemistry. 1999;38:7265–7272. doi: 10.1021/bi982990v. [DOI] [PubMed] [Google Scholar]
- 45.Morin KW, Atrazheva ED, Knaus EE, Wiebe LI. Synthesis and cellular uptake of 2′-substituted analogs of (E)-5-(2-[125I]iodovinyl)-2′-deoxyuridine in tumor cells transduced with the herpes simplex type-1 thymidine kinase gene. Evaluation as probed for monitoring gene therapy. J Med Chem. 1997;40:2184–2190. doi: 10.1021/jm9606406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.