

Abstract
Angiogenesis alleviates hypoxic stress in ischemic tissues or during tumor progression. In addition to endothelial cell proliferation and migration, the angiogenic process requires bone marrow–derived cell (BMDC) recruitment to sites of neovascularization. However, the mechanism of communication between hypoxic tissues and the BM remains unknown. Using 2 models of hypoxia-induced angiogenesis (ischemic hindlimb surgery and subcutaneous tumor growth), we show that platelet infusion promotes BMDC mobilization into the circulation, BMDC recruitment into growing neovasculature, tumor vascularization, and blood flow restoration in ischemic limbs, whereas platelet depletion inhibits these effects. Thus, platelets are required for BMDC recruitment into ischemia-induced vasculature. Secretion of platelet α-granules, but neither dense granules nor platelet aggregation is crucial for BMDC homing and subsequent angiogenesis, as determined using VAMP-8−/−, Pearl, and integrin Beta 3−/− platelets. Finally, platelets sequester tumor-derived promoters of angiogenesis and BMDC mobilization, which are counterbalanced by the antiangiogenic factor thrombospondin-1. A lack of thrombospondin-1 in platelets leads to an imbalance in proangiogenic and antiangiogenic factors and accelerates tumor growth and vascularization. Our data demonstrate that platelets stimulate BMDC homing in a VAMP-8–dependent manner, revealing a previously unknown role for platelets as key mediators between hypoxic tissues and the bone marrow during angiogenesis.
Introduction
Angiogenesis, the process of new vessel formation, is necessary for tissue repair after ischemia, myocardial infarction, or during wound healing. Angiogenesis also occurs in various pathologies, including neoplastic growth and retinopathies. Angiogenesis is a systemic process, not limited to existing vasculature expansion, but also involving the recruitment of other cell types, including immune cells, bone marrow–derived cells (BMDCs), and fibroblasts. Because the angiogenesis begins with vascular leakage, the role of blood cells in this process is significant but remains undetermined.
We and others have demonstrated the importance of recruited BMDCs in tumor growth and wound healing.1–3 Recruited BMDCs cluster at future metastatic sites before the arrival of tumor cells.4 In tumors, BMDCs localize to the perivascular area1,2 and are critical for tumor vessel maturation through cytokine and growth factor secretion.5,6 In addition, proangiogenic BMDCs accumulate around developing collateral arteries in regenerating myocardial tissues and in ischemic hindlimbs, promoting revascularization.7–10 Although the importance of BMDCs in hypoxia-induced angiogenesis is clear, the question of how ischemic tissue communicates with the BM remains unanswered. Although it is logical that the signal for BM to release proangiogenic cells is transmitted by the circulation, there is a need to protect this signal from reaching other parts of the organism and to ensure the specificity of communication. Thus, circulating blood cells, such as platelets, could play a role in protecting the signal between hypoxic tissues and the BM.
Several studies have suggested that circulating platelets have a strong proangiogenic function,11–14 the mechanisms of which are poorly understood. Platelets contain 3 secretory vesicles: α-granules, dense granules, and lysosomes, which each store a different set of proteins. Importantly, platelets are capable of selectively uptaking and releasing proangiogenic and antiangiogenic factors.15,16 Proangiogenic factors predominate in platelets, whereas at least 3 angiogenesis inhibitors are present: platelet factor-4, endostatin, and thrombospondin-1 (TSP-1).12,17 Thus, we theorize that platelets serve as a regulated storage compartment for growth factors and cytokines. Importantly, platelets are recruited to sites of vascular injury and are functionally associated with leukocytes and other blood cells14,18–21; however, the exact functions of platelets in angiogenesis are enigmatic. Therefore, we hypothesize that, during neovascularization, in addition to hemostasis,11,13 platelets play a novel role in BMDC-induced angiogenesis. In this study, using 2 distinct models of hypoxia-induced angiogenesis (tumor growth and hindlimb ischemia), we demonstrate that platelets facilitate the release of proangiogenic cells from the BM and their subsequent recruitment into hypoxic tissues, which promotes neoangiogenesis in vivo.
Methods
Animal studies
Six- to 10-week-old, sex- and age-matched C57BL/6 (WT), WT/GFP, TSP-1−/−, and integrin Beta 3−/− mice were purchased from The Jackson Laboratory. Matched Pearl (HPS2−/−) mice were obtained from Dr Lisa Young (University of Cincinnati), whereas VAMP-8−/− (endobrevin) mice were provided by S.W.W. All transgenic animals were on a C57BL/6 background. Separately, matched NOD/SCID mice were obtained from The Jackson Laboratory. All animal procedures were performed in accordance with an approved institutional protocol according to the guidelines of the Institutional Animal Care and Use Committee of the Cleveland Clinic.
Unilateral hindlimb ischemia model
A unilateral 1.0-cm incision was made in the left thigh of anesthetized mice to expose the femoral artery. An upper ligation was placed 0.5 cm proximal to the femoral artery bifurcation, with 2 additional ligatures placed on the deep bridge and collateral artery 3.0 mm from the bifurcation. The femoral artery was severed between the ligatures to terminate blood flow (BF).
Doppler imaging
A Moor LDI2-VR Laser Doppler was used for noncontact measurement of BF. Each mouse was scanned before and after ischemia, with additional biweekly monitoring, until 14 days postoperatively from a 20-cm distance at a low speed (10 m/s per pixel). Uniform regions of interest were applied to the operated hindlimb, and BF units were recorded and compared with readings obtained before surgical intervention.
Tumor implantation
WT mice were injected subcutaneously with 2 × 106 B16-F10 murine melanoma cells (8 mice/group). Tumors were harvested by detaching the surrounding connective tissue 9 days after implantation, weighed, and processed for immunohistochemistry. In addition, WT mice were injected subcutaneously with 4 × 105 RM1 murine prostate cancer cells, and tumors were harvested 12 days after implantation (5 mice/group). Separately, NOD/SCID mice were injected subcutaneously with 4 × 105 LNCaP-C4-2 human prostate cancer cells and tumors were harvested 28 days after implantation (4 mice/group).
Platelet isolation and injection
Blood was collected from WT, transgenic, or NOD/SCID mice through the vena cava into 0.1 volume of acid-citrate-dextrose buffer containing 1 μg/mL prostaglandin E1. Platelets were separated from the platelet-rich plasma of blood by centrifugation, washed by gel filtration, and resuspended in buffer (137mM NaCl, 4mM KCl, 0.5mM MgCl2, 0.5mM Na2HPO4, 11.1mM dextrose, 0.1% bovine serum albumin, 10mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, pH 7.4), as previously described.22 Platelets (∼ 3 × 109) from 2 donor mice in resuspension buffer were injected into a single recipient WT mouse via the tail vein. Control animals were injected with resuspension buffer. Platelet infusion was repeated every 5 days until experimental termination.
Platelet depletion
Mice received tail vein injection of rat antimouse GPIbα or control rat IgG (2 μg/g, Emfret Analytics) antibodies. Injections were repeated every 3 days to maintain low platelet counts. Tail vein blood samples were used for platelet counts before intervention and after each injection of GPIbα antibody via complete blood count analysis with differential using an Advia 120 laser hematology system (Siemens Healthcare Diagnostics) in the Center for Cardiovascular Research at the Lerner Research Institute.
Flow cytometric analysis
Blood samples were collected in 0.1M ethylenediaminetetraacetic acid from the tail vein. Red blood cells were removed using a Mouse Erythrocyte Lysing Kit (R&D Systems). Lymphocytes (1 × 106) were blocked with murine Fc (1:50), incubated with a phycoerythrin-conjugated CXCR4 antibody (1:50; R&D Systems) and fixed in 1% formalin. Fluorescence values of stained lymphocytes analyzed using a BD FACS Canto II (BD Biosciences) running the FACS Diva software Version 6.0 were normalized to an unstained control sample.
Bone marrow transplantation
Six- to 10-week-old WT C57BL/6 mice were lethally irradiated (9 Gy) and reconstituted through tail vein injection with 1 × 107 BM cells isolated from green fluorescent protein-positive (GFP+) donor femurs. Mice were analyzed 8 weeks after bone marrow transplantation.
Histology and immunohistochemistry
Tissues were embedded in OCT and snap frozen in liquid nitrogen or fixed in 10% buffered formalin followed by paraffin embedding. Frozen sections were used for CD31 staining (1:100, Fitzgerald Industries). The Vectastain Elite ABC kit was used for chromogenic visualization (Vector Laboratories). Paraffin sections were used for laminin (1:1000; Sigma-Aldrich), von Willebrand factor (1:2000; Dako North America), and GFP (1:500, AnaSpec) immunohistochemistry. Secondary antibodies were used at 1:200 dilutions.
Image acquisition
Images were captured using a Leica DMR 2500 Inverted fluorescence microscope with a QImaging Retiga EXi camera controlled by the QImaging QCapturePro v6.0 software. The objectives used were 10×/0.30 NA, 20×/0.05 NA, or 40×/1.25 NA oil. Leica Type N Immersion Liquid was used for the 40× oil images. Stained areas were quantified for 5 fields per section by scanning the entire image with ImagePro Plus software (Version 5.1; Media Cybernetics).
Platelet content analysis
Isolated platelets from NOD/SCID mice without or bearing LNCaP-C4-2 tumors were lysed in RIPA buffer containing 1mM protease inhibitors. The resulting lysates were assayed with a custom-designed Quantibody human-specific protein array from RayBiotech to detect tumor-derived proteins according to the manufacturer's protocol. The fluorescently labeled array was analyzed using an Axon 4000B (Molecular Devices) scanner in the LRI Genomics Core. Four replicate values were extracted using the Axon Gene Pix Pro Version 4.1 software and analyzed using the RayBiotech Custom Raybio Q-Analyzer Version 1.0 software, which uses standardized dilutions of each protein to create standard curves used in determining the concentration in picograms per milliliter of each protein in the samples.
Statistical analysis
Data were represented as the mean ± SEM and analyzed using a 2-tailed Student t test or 1-way analysis of variance with Newman-Keuls posttest using the GraphPad Prism Version 4.03 software.
Results
Platelets induce BMDC mobilization in response to distal hypoxia
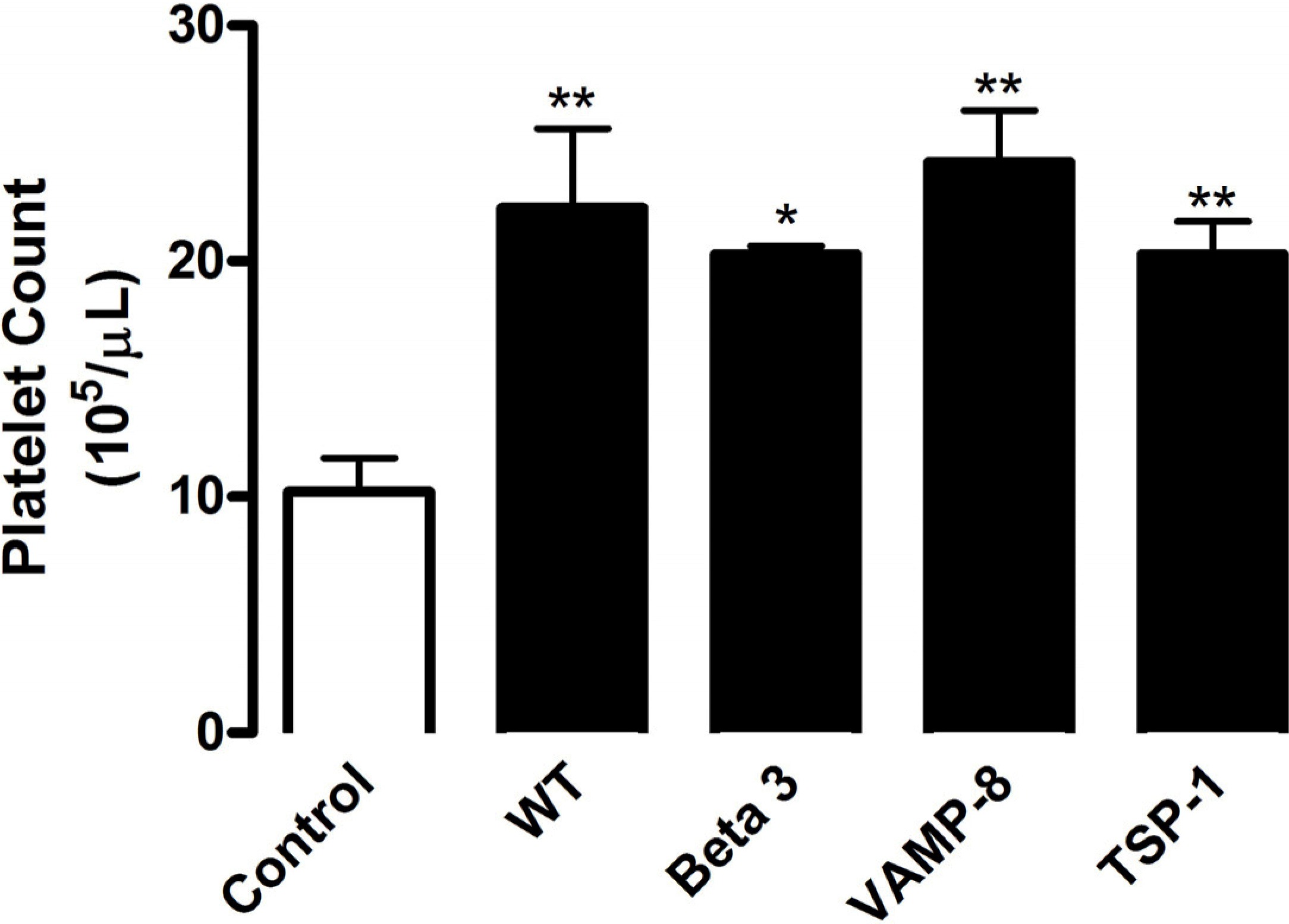
We and others have shown that tumors summon BMDCs to sites of angiogenesis1–3; however, the mechanism behind this recruitment remains unknown. In this study, we used 2 models of hypoxia-induced angiogenesis (subcutaneous tumor growth and hindlimb ischemia) to elucidate the mechanism of BMDC recruitment to neovascularization sites. We used CXCR4/CD184 expression, a G-protein linked 7 transmembrane domain chemokine receptor found on a majority of BM progenitor cells, as a marker to quantify the presence of BMDCs in the circulation by flow cytometry. As shown in Figure 1A, ischemia, represented either by hindlimb muscle with impaired blood supply or by a growing tumor, resulted in mobilization of CXCR4+ cells into the circulation. Hindlimb ischemia stimulated 2.05-fold higher CXCR4+ circulating cell levels compared with samples collected before intervention (Figure 1A). Likewise, similar increases were observed on tumor implantation using 3 tumor models: B16-F10 murine melanoma (1.84-fold), RM1 murine prostate cancer (1.63-fold), and LNCaP-C4-2 human prostate cancer (1.23-fold) compared with levels before tumor implantation (Figure 1A). The lower mobilization of CXCR4+ cells induced by LNCaP-C4-2 tumors is probably the result of the use of immunocompromised mice. These data indicate that distant hypoxic sites communicate with the BM to stimulate BMDC release, which might support neovascularization. Because B16-F10 implantation induced the greatest change in CXCR4+ levels and a percentage (10.04% ± 3.09%) consistent with the consequences of hindlimb ischemia (7.53% ± 2.87%), we focused on these 2 in vivo models to assess the role of platelets.
Figure 1.

Platelets promote release of BMDCs during angiogenesis. (A) To induce ischemia, ligation of the femoral artery was performed and the experiment was terminated 14 days later (n = 4). In a separate group of mice, tumor cells were implanted subcutaneously: B16-F10 (2 × 106 cells, n = 8), RM1 (4 × 105 cells, n = 5), or LNCaP-C4–2 (4 × 105 cells, n = 4), and excised after 9 days (B16-F10), 12 days (RM1), or 28 days (LNCaP-C4–2). Blood samples were collected from mice before tumor implantation or ischemic surgery (Initial, white columns) and on experimental termination (Final, black columns). (B-D) After tumor implantation or hindlimb ischemia surgery, 3 × 109 platelets were infused into mice every 5 days by tail vein injection. Control mice were injected with phosphate-buffered saline (PBS). Separately, mice were treated intravenously with 2 μg/g body weight rat anti–mouse GPIbα to deplete platelets or rat IgG as a control. Injections were repeated every 3 days. (B) Whole blood was collected before (Initial, white columns) and 9 days after (Final, black columns) B16-F10 tumor implantation (n = 6). (C-D) Whole blood was collected before ischemic surgery (Initial, white columns), from IgG or PBS-treated mice (Ischemia, gray columns), and from mice after platelet (PLT) infusion or depletion (black columns) (n = 6). Red blood cells were lysed, and the sample was labeled with CXCR4 antibody conjugated with phycoerythrin and analyzed by flow cytometry. Values are mean percentage of CXCR4+ cells ± SEM. *P < .05, **P < .01, and ***P < .005 by Student t test (A-B) or 1-way analysis of variance (C-D) versus initial samples.
We hypothesized that platelets function as communicators between hypoxic tissues and the BM. Accordingly, we altered platelet levels in our 2 neovascularization models using complement approaches: platelet depletion and platelet infusion. Circulating platelets were reduced using a GPIbα antibody by greater than 90% compared with control IgG (data not shown), consistent with previous reports.1,12,13 Infusion of approximately 3 × 109 gel-filtered platelets into mice increased the platelet count by 2.70- to 3.00-fold compared with control mice (data not shown). Using B16-F10 tumor implantation and flow cytometry, we determined that the levels of CXCR4+ cells in the circulation increased with platelet infusion by 3.94-fold and decreased with platelet depletion by 1.63-fold compared with mice before implantation (Figure 1B). Likewise, hindlimb ischemia alone stimulated CXCR4+ mobilization 1.80-fold compared with before ischemia. Platelet infusion increased circulating CXCR4+ cells 2.00-fold (Figure 1C), whereas platelet depletion diminished cell levels 2.67-fold (Figure 1D) compared with ischemia alone. Thus, the presence of platelets promotes BMDC release in both models of angiogenesis.
Platelet levels regulate BMDC recruitment to sites of hypoxic stress
Having demonstrated that platelets control BMDC mobilization, we then determined the effects of altering platelet levels on BMDC recruitment to sites of hypoxia-induced angiogenesis. To visualize BMDCs in hypoxic tissues, we replaced the BM of WT mice with that from GFP mice via bone marrow transplantation, implanted tumors or performed ischemia surgery, and then depleted or infused mice with platelets. Tumors from mice with depleted or infused platelets were stained with smooth muscle actin to visualize the vasculature (Figure 2A). GFP+ cells were recruited to the tumor periphery and alongside tumor vasculature. Platelet infusion resulted in 1.91-fold higher infiltration of GFP+ BMDCs adjacent to tumor vasculature, whereas platelet depletion resulted in a 1.87-fold decrease compared with tumors from control mice (Figure 2B). Thus, platelet depletion inhibited GFP+ BMDC recruitment, whereas platelet infusion elevated BMDC levels in hypoxic tumor tissue. Similar results were obtained using the hindlimb ischemia model. Two weeks after femoral artery ligation surgery, muscles were immunostained for GFP+ BMDCs (Figure 2C arrows). The number of GFP+ cells in muscles increased 2.48-fold after platelet infusion and decreased 2.17-fold after platelet depletion compared with muscles from the control groups (Figure 2D). Taken together, these data underscore the importance of platelets in BMDC recruitment to sites of hypoxia-induced neovascularization.
Figure 2.

Platelet presence alters BMDC recruitment to hypoxic tissues. Lethally irradiated WT mice were reconstituted with GFP BM. After tumor implantation (A-B) or hindlimb ischemia surgery (C-D), 3 × 109 platelets were infused into mice every 5 days by tail vein injection (PLT Infusion, Treated). Control mice were injected with PBS (PLT Infusion, Control). Separately, mice were treated intravenously with 2 μg/g body weight rat antimouse GPIbα to deplete platelets (PLT Depletion, Treated) or rat IgG (PLT Depletion, Control). Injections were repeated every 3 days. (A-B) Subcutaneous B16-F10 tumors were removed after 9 days of growth. Tumors were sectioned and stained for blood vessels using a smooth muscle actin antibody (red) and 4,6-diamidino-2-phenylindole (blue). GFP+ BMDCs were counted per field. (C-D) Muscles were sectioned after 14 days of ischemia. GFP+ BMDCs were visualized by immunostaining (brown) and counted per field. Arrows indicate GFP+ cells. Scale bars represent 50 μm. Staining was quantified as mean GFP+ cells per field ± SEM (n = 5). *P < .05 vs control (Student t test). ***P < .005 vs control (Student t test).
Platelet-induced BMDC recruitment stimulates angiogenesis
Angiogenesis is induced either by platelet presence in or by BMDC recruitment to perivascular regions of hypoxic tissues.1,2,6–8,14,23 We hypothesized that these 2 processes may be linked and demonstrated that platelet levels altered BMDC homing to hypoxic tissues. We then used platelet depletion in combination with tumor implantation to assess the importance of platelets in tissue neovascularization. Vessels were visualized using laminin, a blood vessel basement membrane protein, or CD31, an endothelial marker, staining in tumor sections (Figure 3A,C arrows). At early stages of vascularization (4 days after tumor implantation), the number of laminin-positive vessels in tumor sections of control mice was 2 or 3 per field, whereas no well-defined vascular structures were observed in platelet-depleted mice (Figure 3A-B). At the same time, platelet infusion stimulated blood vessel formation by 30% (data not shown). Nine days after tumor implantation, there were still substantial differences (2.68-fold decrease on platelet depletion) in vascular area as judged by CD31 staining (Figure 3C-D). Visually, the length of blood vessels was diminished in tumors with platelet depletion (Figure 3C). Tumors in control mice contained approximately 6 vessels/mm2 with well-defined laminin staining (Figure 3A,C). Conversely, tumors from mice undergoing platelet depletion displayed a significantly lower vessel density and minimal laminin staining indicating a delay in angiogenesis and vascular maturation.
Figure 3.

Blood vessel density is diminished on platelet depletion. (A-D) Subcutaneous B16-F10 tumors were implanted in mice undergoing platelet (PLT) depletion or injected with control rat IgG. After 9 days of growth, tumors were excised and sectioned. (A-B) Tumor sections were stained for laminin to visualize blood vessels. Laminin-positive vessels were quantified and represented as mean vessel number per section ± SEM (n = 6). (C-D) Frozen tumor sections were stained for CD31+ vessels (red) and 4,6-diamidino-2-phenylindole (blue). CD31+ blood vessels were quantified and represented as mean percentage of area ± SEM (n = 24). Arrows indicate vessels within the tissues. (E-F) Platelets were depleted or infused in mice directly after hindlimb ischemia surgery. Muscles from mice injected with PBS (PLT Infusion, Control) or 3 × 109 platelets (PLT Infusion, Treated) or rat IgG (PLT Depletion, Control) or rat antimouse GPIbα (PLT Depletion, Treated) were sectioned 14 days after surgery and immunostained for von Willebrand factor-positive blood vessels. Vessels were quantified and represented as mean vessel number per field ± SEM (n = 5). Scale bars represent 50 μm. *P < .05 vs control (Student t test). ***P < .005 vs control (Student t test).
Next, we assessed angiogenesis in our ischemic model on platelet infusion and depletion. Vessels within the muscles 2 weeks after ischemic surgery were stained with von Willebrand factor (Figure 3E). In both control and platelet-infused mice, no significant differences were noted in the diameter or location of blood vessels (Figure 3E). However, on platelet depletion, less than one vessel/field was present (Figure 3E). Quantitative analysis demonstrated that platelet infusion induced vascular density by 1.28-fold, whereas platelet depletion inhibited vessel formation by 2.19-fold compared with their respective controls (Figure 3F). Taken together, our data demonstrated that platelet infusion stimulated blood vessel formation in hypoxic tissues, whereas platelet depletion inhibited this process. Together with our results on the BMDC release and homing, these data show that platelets promote angiogenesis by stimulating the mobilization and recruitment of proangiogenic BMDCs, which are necessary for neovascularization of hypoxic sites.6–8
Platelet infusion stimulates BF in ischemic limbs
Our data demonstrate that platelets control hypoxia-induced neovascularization. To determine whether platelets mediate the formation of functional vasculature, BF was measured by Doppler imaging before and after hindlimb ischemia surgery. Before surgery, mice in each group displayed similar BF levels (Figure 4). After surgery, mice were infused with or depleted of platelets. BF levels in the nonoperated limbs did not change significantly throughout the experiment (data not shown). Directly after surgical intervention, BF diminished to approximately 30% of the initial levels for both control groups, platelet-infused, and platelet-depleted animals (Figure 4A). With platelet infusion, BF was increased 2.04-fold at 2 days and 1.45-fold at 2 weeks compared with control animals (Figure 4A left). By 2 days postoperatively, BF in the platelet-infused animals had returned to 75% of the initial reading and was at 91% by 2 weeks (Figure 4A left). Control mice recovered approximately 70% of their initial BF by 2 weeks (Figure 4A). Conversely, platelet depletion inhibited BF recovery and suppressed angiogenesis postoperatively (Figure 4B). Platelet-depleted BF was 0.43-fold of control after 2 days and was 0.47-fold at 2 weeks (Figure 4A right). Two weeks after surgery, BF in platelet-depleted animals had only recovered 25% of initial BF (Figure 4A right). Thus, platelet infusion promoted BF recovery after surgery, whereas platelet depletion reduced it. These data correlated directly with the changes noted in blood vessel formation and maturation. Taken together, our results indicate that platelets are crucial to angiogenesis during tumor expansion and postischemia recovery.
Figure 4.

Platelet levels alter BF in ischemic limbs. Platelets were depleted or infused in mice directly after hindlimb ischemia surgery. BF was monitored by Doppler for 2 weeks postoperatively in mice injected with PBS (PBS Control) or 3 × 109 platelets (PLT Infusion) or rat IgG (IgG Control) or rat anti–mouse GPIbα (PLT Depletion). (A) Doppler values are represented as mean BF ± SEM for control (dashed line) and treated (solid line) animals (n = 5). *P < .05 vs control (Student t test). **P < .01 vs control (Student t test). ***P < .005 vs control (Student t test). (B) Representative Doppler images over the experimental time course are depicted. The color scale represents the BF values.
Platelet α-granule secretion regulates BMDC recruitment
Having established a role for platelets in BMDC recruitment and neovascularization, we assessed which platelet characteristic was essential for their proangiogenic function by performing platelet infusion from knockout mice with various platelet defects. Platelets perform 2 main functions in hemostasis: platelet aggregation and secretion. Aggregation mediated by platelet fibrinogen receptor αIIbβ3 integrin is an essential part of thrombus formation. Beta 3 integrin null platelets lack aggregation responses, thereby resembling human Glanzmann thrombasthenia patients.24 Platelet secretion has a prohemostatic function involving release of the contents from 3 types of granules: α-granules, dense granules, and lysosomes. Pearl mice, which serve as a model of Hermansky-Pudlak syndrome, are characterized by deficiencies in dense granule and lysosome secretion.25,26 Vesicle-associated membrane protein-8 (VAMP-8/endobrevin) null mice have a defect in the secretion machinery of α-granules (70% reduced secretion), with minor defects in dense granules and lysosomes. VAMP-8 deficiency affects membrane fusion without any effect on granule biosynthesis or platelet signaling.27 Platelets isolated from these knockout mice were transfused into chimeric WT/GFP mice followed by tumor implantation. Platelet levels in transfused mice were approximately 2.5-fold higher compared with mice without platelet infusion regardless of genotype (supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). Several characteristics were monitored, including BMDC recruitment into vasculature, tumor weight, and tumor vascularization (Figure 5). As anticipated, infusion of WT platelets promoted a 2.43-fold increase in BMDC recruitment (Figure 5B), which resulted in augmentation of tumor growth by 1.73-fold compared with control mice (Figure 5A). Accordingly, vascular area correlated with tumor progression because platelet infusion promoted a 2.00-fold increase in vascular density (Figure 5C). Infusion of aggregation-deficient Beta 3 integrin null or dense granule secretion-impaired Pearl platelets produced increases in BMDC recruitment and in tumor weight over control comparable that of WT platelets (3.14- and 2.39-fold for BMDCs and 1.76 and 1.67 for tumor weight, respectively; Figure 5A-B). Therefore, platelet aggregation and dense granule secretion are not crucial for the proangiogenic role of platelets and for BMDC recruitment. In contrast, VAMP-8-deficient platelet infusion diminished BMDC recruitment 2.88-fold compared with WT infusion (Figure 5B). Indeed, the numbers of recruited BMDCs were not significantly different between control mice without platelet infusion and mice infused with VAMP-8 null platelets (Figure 5B). Thus, lack of α-granule secretion completely abolished the effect of infused platelets on the recruitment of proangiogenic BMDCs. Likewise, respective differences were observed in tumor weight because infusion of VAMP-8 null platelets reduced tumor weight 1.52-fold compared with WT platelet infusion (Figure 5A). Consistent with a key role for BMDCs in angiogenesis, infusion of VAMP-8 null platelets did not promote tumor vascularization and there was no difference between control mice and mice infused with VAMP-8 null platelets (Figure 5C). Thus, VAMP-8 deficiency in platelets abolished the effects of platelet infusion on BMDC recruitment, angiogenesis, and tumor growth. Therefore, our data suggest that α-granule secretion is required for platelet-induced BMDC recruitment and the function of these cells in tumor angiogenesis and growth.
Figure 5.

Platelet α-granules are necessary for tumor growth, BMDC recruitment, and angiogenesis. Lethally irradiated WT mice were reconstituted with BM from GFP mice. Mice were implanted with B16-F10 tumors and injected with platelets from WT, integrin Beta 3−/−, Pearl, or VAMP-8−/− mice (black columns). Control represents mice bearing tumors without platelet infusion (white column). (A) After 9 days tumors were excised, weighed, and represented as mean weight ± SEM (n = 10). (B) Tumors were sectioned and GFP+ BMDCs were quantified in tumor sections. Values are represented as mean number GFP+ cells per field ± SEM (n = 5). (C) Tumor sections were stained for laminin to visualize vessel density. Vessels were quantified and represented as mean percentage of area ± SEM (n = 5). *P < .05 vs control or WT (Student t test). **P < .01 vs control or WT (Student t test).
Platelet α-granules sequester tumor-derived factors
Our data demonstrate that platelet α-granule secretion is crucial for communication between hypoxic tissues and the BM, which results in mobilization of proangiogenic BMDCs. We used a tumor xenograft model to determine what factors produced by tumor were sequestered by platelet α-granules. Platelets were isolated mice with or without tumors, lysed, and assayed using an enzyme-linked immunosorbent assay-based, human-specific protein array to determine the levels of tumor-derived proteins within platelets. Several factors associated with BMDC mobilization were found to be tumor-produced and present within platelet lysates (Figure 6). Among these were granulocyte colony-stimulating factor, macrophage colony-stimulating factor, matrix metalloproteinase-9, and granulocyte-macrophage colony-stimulating factor, which are known to be associated with the release of stem cells from the BM niche into the circulation8,28–30 and ranged from 25 to 335 pg/mL in platelets (Figure 6A). While in the BM and circulation, thrombopoietin and IL-6 support stem cell survival and expansion29,31 and were found at the highest levels within platelets (thrombopoietin, 13 798.81 ± 3292.31 pg/mL; IL-6, 8500.57 ± 2356.11 pg/mL; Figure 6B). Further, IL-6–induced CXCR4 expression on BMDCs potentiates their migration and recruitment.31 SDF-1α and vascular endothelial growth factor (VEGF) regulate BMDC adhesion and homing toward hypoxic sites2,32 and were present in platelet lysates (SDF-1α, 5783.01 ± 532.61 pg/mL; VEGF, 1068.25 ± 629.11 pg/mL; Figure 6B). Release of SDF-1α in the vasculature is of particular interest as it might induce a localized SDF-1α enrichment, resulting in BMDC mobilization toward the circulation. Taken together, these results show that platelet α-granules serve as reservoirs for factors produced by hypoxic tissues; many of which promote BMDC mobilization.
Figure 6.

Platelets contain proangiogenic and BMDC mobilization factors. NOD/SCID mice were injected subcutaneously with human LNCaP-C4–2 cells. After 28 days of tumor growth, platelets were isolated from the whole blood of control (white columns) and tumor-bearing (black columns) mice and lysed. A species-specific array for human proteins was used to determine the concentrations of tumor-derived cytokines, represented as mean values ± SEM (n = 4). (A) Tumor-derived thrombopoietin (TPO), interleukin-6, (IL-6), stromal-derived factor 1a (SDF-1a), and vascular endothelial growth factor (VEGF) are highly concentrated in platelets. (B) Tumor-derived granulocyte cololny-stimulating factor (G-CSF), monocyte colony-stimulating factor (M-CSF), matrixmetalloproteinase-9 (MMP-9), and granulocyte-macrophage colony-stimulating factor (GM-CSF) are present at lower levels in platelets. No tumor-derived proteins were found in the platelets of control mice, demonstrating the specificity of the array. *P < .05 vs control (Student t test). ***P < .005 vs control (Student t test).
Platelet TSP-1 diminishes tumor growth
As shown in Figure 6, platelet α-granules contained high concentrations of angiogenic and BMDC-stimulating factors, but only sequester a few antiangiogenic proteins, mainly represented by TSP-1. Therefore, platelet TSP-1 might effectively balance the activity of various proangiogenic factors. Accordingly, using our tumor model, we infused mice with WT or TSP-1 null platelets resulting in a 2.5-fold increase in platelet numbers (supplemental Figure 1). Tumors from mice infused with TSP-1–deficient platelets were larger than those from mice infused with WT platelets, indicating that the lack of only platelet TSP-1 was sufficient to shift the balance toward increased tumorigenesis (Figure 7A). This was consistent with previous reports emphasizing the suppressive role of TSP-1 in tumor growth.17,33 Importantly, TSP-1 deficiency in infused platelets resulted in increased CXCR4+ BMDC mobilization (Figure 7B). TSP-1 null platelet infusion stimulated 1.37-fold higher circulating BMDC levels compared with WT infusion and 2.31-fold compared with control (Figure 7B). Thus, the absence of TSP-1 in platelet α-granules stimulated tumor growth and enhanced BMDC mobilization. Therefore, the primary antiangiogenic factor TSP-1 in platelets can counteract the effects of the plethora of proangiogenic and BMDC mobilization factors within platelets.
Figure 7.

Tumor growth is enhanced in the absence of platelet α-granule–derived TSP-1. Lethally irradiated WT mice were reconstituted with BM from GFP mice. Mice were implanted with B16-F10 tumors and injected with platelets from WT or TSP-1−/− mice (black columns). Control represents mice bearing tumors without platelet infusion (white columns). (A) After 9 days tumors were excised, weighed, and represented as mean weight ± SEM (n = 10). (B) Whole blood samples were collected from mice after tumor implantation. Red blood cells were lysed, and the sample was labeled with CXCR4 antibody conjugated with phycoerythrin and analyzed by flow cytometry. Levels of CXCR4+ cells in the circulation are represented as mean percentage ± SEM (n = 4). *P < .05 vs control or WT (Student t test). **P < .01 vs control or WT (Student t test). ***P < .005 vs control or WT (Student t test).
Discussion
In this study, we use 2 models of hypoxia-induced neovascularization (tumor growth and hindlimb ischemia) to demonstrate the cellular mechanisms underlying the proangiogenic role of circulating platelets. We show that platelets facilitate communication between hypoxic tissues and the BM and mediate the release of proangiogenic BMDCs into the circulation. These cells are then recruited to angiogenic sites and contribute to augmented vascularization and improved blood supply. This novel mechanism is operational in both tumors and ischemic limbs. We further show that platelet α-granule secretion, but neither platelet aggregation nor dense granule secretion is responsible for BMDC mobilization and subsequent angiogenesis. We show that platelets sequester several tumor-secreted growth factors and cytokines that promote BMDC mobilization. These factors are counterbalanced by the main antiangiogenic agent, TSP-1, which is abundant in platelet α-granules. A loss of TSP-1 in only infused platelets resulted in dramatically increased BMDC mobilization and tumor growth. Thus, platelets serve as a key mediator between hypoxic tissues and the BM facilitating the release of proangiogenic BMDCs, which are recruited into the tissue in need and promote its blood supply, thereby contributing to the systemic nature of the angiogenic process.
In recent years, several reports suggest a role for blood components in angiogenesis, especially during tumorigenesis.11 Infusion of platelets together with leukocytes results in improved BF and vessel formation in several angiogenic models: corneal pockets, hindlimb ischemia, and arterial occlusion.14,34 Conversely, platelet depletion diminishes angiogenesis in models of retinal neovascularization,12 corneal pockets, and Matrigel implantation studies.13 In tumors, platelet depletion results in intratumoral hemorrhage because of vessel destabilization.11 Thus, previously published studies establish that platelets are proangiogenic in most models of neoangiogenesis. Our study shows that platelets benefit growing vasculature not directly but by facilitating the recruitment of another important cellular component of angiogenesis: BM-derived myeloid cells.
BMDC mobilization occurs through several stages. First, they are released from the BM niche and migrate to the sinusoidal vascular niche. Next, BMDCs cross the endothelium into the circulation and home to hypoxic tissues. Mobilization signals must travel from the site of injury to the BM without being used or degraded and, thus, must be protected in the circulation. Our data demonstrate that platelets fulfill this particular role. Monitoring BMDC release in our tumor and ischemia models, we observe that platelet injection enhances BMDC mobilization and recruitment into the sites of neovascularization, whereas platelet depletion results in the opposite effects. In our study, a significant increase in the number of CXCR4+ cells is recorded 2 days after ischemia (data not shown) and reaches a peak 2 weeks later, during the time of intense tissue regeneration. The presence of CXCR4+ cells in the circulating blood and GFP+ cells in the ischemic tissue indicates an accelerated healing process, propagated by repeated platelet injections. Several reports suggest an interaction between BMDCs and platelets. It is reported that BMDCs adhere to platelets under static and dynamic flow conditions and that platelets directly induce differentiation of “endothelial progenitors” into mature endothelial cells,35 which leads to vascular rescue and promotes tissue recovery.36 We observe BMDCs only in close proximity with blood vessels, with a negligible amount of GFP+ cells directly incorporating into the tumor endothelium, similar to observations reported by multiple groups.2,3,7,37,38 The increased presence of BMDCs in the dynamically reorganized vasculature after platelet injection indicates that platelets might not only promote release of BMDC into the circulation but that platelets also facilitate BMDC recruitment into ischemic tissues.
In this study, we use a general marker for BMDCs (CXCR4, a key receptor for SDF-1α) and do not attempt to differentiate between the diverse populations of recruited cells. CXCR4+ proangiogenic cells represent a heterogeneous population and have been described as endothelial progenitor cells, smooth muscle progenitors, mesenchymal stem cells, hematopoietic stem cells, fibroblasts, or tumor-associated myeloid cells.2,7,28,30,39–41 Based on our previously published studies, we think that the use of CXCR4 as a marker allows for tracking of the majority of recruited BMDCs.1 Regardless of the presence of additional markers, these BMDCs are crucial for angiogenesis and vascular maturation in various models.1,3,8 Reflecting the overall heterogeneity of these cells, multiple cytokines are reported to be essential for their recruitment; however, many studies emphasize the central function of SDF-1α.4,36,40 SDF-1α has an effect additive to that of VEGF on neovascularization in tumors42 and in ischemic limbs.43,44 Interestingly, SDF-1α accumulation in platelets is involved in angiogenesis regulation and vascular repair.45 Likewise, studies performed on SDF-1α and CXCR4 null mice emphasize that the SDF-1α/CXCR4 axis is crucial for BMDC mobilization into the bloodstream.46 Increased numbers of circulating BMDCs might be sufficient to promote vascularization.47 Homing of BMDCs to areas of neovascularization is proposed to be directed by a VEGF concentration gradient.2 Thus, VEGF is required for the recruitment and maintenance of BMDCs at sites of angiogenesis.2 In both experimental models used in this study, there is a substantial amount of VEGF produced either by ischemic muscle or by the growing tumor, which ensures the recruitment of circulating CXCR4+ cells and their subsequent proangiogenic function. Thus, together with published studies, our data show that the platelet-mediated release of CXCR4+ cells from BM is a crucial step during angiogenesis.
Circulating platelets sequester and store in their α-granules factors, which mediate BMDC recruitment.32,48 Therefore, our results demonstrating a crucial role for platelet α-granule secretion during BMDC recruitment are well supported by existing evidence. It was shown previously that infusion of degranulated platelets could not prevent intratumor hemorrhaging associated with vascular dysfunction after thrombocytopenia, whereas resting platelets could rescue tumors.11 It was surprising that neither the lack of dense granule secretion nor αIIbβ3 and platelet aggregation diminishes the proangiogenic function of platelets. Another important aspect of platelet activation is the release of microparticles, which can enhance angiogenesis. Further studies are required to determine the effects of platelet microparticle release on BMDC mobilization and recruitment. However, microparticle release is diminished in integrin Beta 3 null platelets (W.F. and T.V.B, manuscript in preparation); and because tumor growth is identical after WT and integrin Beta 3 null platelet infusion, we do not expect altered microparticle levels resulting from platelet infusion to play a role in tumor growth in our model. Thus, it appears that it is the platelet's ability to intake, transport, and release key factors from α-granules that is most important for angiogenesis. Importantly, platelets are highly selective in their uptake and secretion because they store and release proangiogenic and antiangiogenic proteins in an independent manner.15 Thus, platelets play an active role in the endocytosis of angiogenesis regulators. BMDC mobilization by platelets may be the result of the well-timed and controlled release of sequestered proteins within the BM niche. This makes platelets an efficient mediator between the BM and hypoxic tissues requiring new vasculature.
We show that platelets specifically sequester several tumor-derived factors involved in BMDC mobilization and angiogenesis, including thrombopoietin, SDF-1α, VEGF, granulocyte colony-stimulating factor, macrophage colony-stimulating factor, matrix metalloproteinase-9, and granulocyte-macrophage colony-stimulating factor. Interestingly, these proteins were produced by the hypoxic tissue, the tumor itself. Proangiogenic factors predominate in the platelets of mice bearing tumors.16 Thus, we propose that the stimulatory effect on BMDC mobilization and angiogenesis is mediated not by one single factor, but rather by several factors in combination, thereby affecting multiple aspects of the multicellular process of angiogenesis.
Importantly, this plethora of angiogenic factors appears to be balanced by TSP-1, which is abundant in platelet α-granules and has several nonoverlapping antiangiogenic functions, including inhibition of matrix metalloproteinase-9 activity, which affects VEGF release from the extracellular matrix49 and ligation of endothelial CD36, leading to the reduction of endothelial cell migration.33,50 Although TSP-1 is up-regulated in the platelets of tumor-bearing mice, it serves as a powerful negative regulator during initial tumor angiogenesis.17 We show that the lack of TSP-1 in the proportion of infused platelets is sufficient to escalate tumor growth in vivo. Importantly, a lack of TSP-1 in platelets causes increased BMDC mobilization. This might be because of TSP-1's effects on VEGF release, which alters the VEGF gradient required to retain BMDCs in hypoxic tissues.
In conclusion, our study bridges several previous observations and provides a cellular mechanism underlying the role of platelets in angiogenesis. Platelets function as communicators between hypoxic tissues and the BM to stimulate BMDC mobilization, a key step for their recruitment and proangiogenic function. Platelets probably function in a similar fashion in other angiogenic situations, such as myocardial infarction repair and retinopathies. Control of platelet levels and their secretory function may aid in the treatment of various pathologies.
Supplementary Material
Acknowledgments
The authors thank Miroslava Tischenko and Steven Maximuk for technical assistance and Dr Pieter Faber for assistance with microarray analysis.
This study was supported by the National Institutes of Health (grants DK060933, CA126847, and HL071625; T.V.B.). B.A.K was supported by the National Institutes of Health/National Heart, Lung, and Blood Institute (training grant T32 HL007914).
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: W.F., M.M., B.A.K., and T.V.B. designed the study; S.W.W. provided reagents; W.F., M.M., B.A.K, G.H.M., and S.W.W. performed research; W.F., M.M., B.A.K., G.H.M., S.W.W., and T.V.B. analyzed data; and W.F., M.M., B.A.K., and T.V.B. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation of M.M. is Department of Plastic Surgery, Cleveland Clinic Foundation, Cleveland, OH. The current affiliation of G.H.M. is University Hospitals Harrington-McLaughlin Heart and Vascular Institute and Case Cardiovascular Research Institute, Case Western University School of Medicine, Cleveland, OH.
Correspondence: Tatiana V. Byzova, Cleveland Clinic Foundation, NB-50, 9500 Euclid Ave, Cleveland, OH 44195; e-mail: byzovat@ccf.org.
References
- 1.Feng W, McCabe NP, Mahabeleshwar GH, Somanath PR, Phillips DR, Byzova TV. The angiogenic response is dictated by β3 integrin on bone marrow-derived cells. J Cell Biol. 2008;183(6):1145–1157. doi: 10.1083/jcb.200802179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grunewald M, Avraham I, Dor Y, et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell. 2006;124(1):175–189. doi: 10.1016/j.cell.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 3.Lyden D, Hattori K, Dias S, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7(11):1194–1201. doi: 10.1038/nm1101-1194. [DOI] [PubMed] [Google Scholar]
- 4.Kaplan RN, Riba RD, Zacharoulis S, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shojaei F, Wu X, Malik AK, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25(8):911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 6.Nolan DJ, Ciarrocchi A, Mellick AS, et al. Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes Dev. 2007;21(12):1546–1558. doi: 10.1101/gad.436307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ziegelhoeffer T, Fernandez B, Kostin S, et al. Bone marrow-derived cells do not incorporate into the adult growing vasculature. Circ Res. 2004;94(2):230–238. doi: 10.1161/01.RES.0000110419.50982.1C. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi T, Kalka C, Masuda H, et al. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5(4):434–438. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 9.Zisa D, Shabbir A, Mastri M, Suzuki G, Lee T. Intramuscular VEGF repairs the failing heart: role of host-derived growth factors and mobilization of progenitor cells. Am J Physiol Regul Integr Comp Physiol. 2009;297(5):R1503–R1515. doi: 10.1152/ajpregu.00227.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Haider H, Ahmad N, Zhang D, Ashraf M. Evidence for ischemia induced host-derived bone marrow cell mobilization into cardiac allografts. J Mol Cell Cardiol. 2006;41(3):478–487. doi: 10.1016/j.yjmcc.2006.06.074. [DOI] [PubMed] [Google Scholar]
- 11.Ho-Tin-Noé B, Goerge T, Cifuni SM, Duerschmied D, Wagner DD. Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res. 2008;68(16):6851–6858. doi: 10.1158/0008-5472.CAN-08-0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rhee JS, Black M, Schubert U, et al. The functional role of blood platelet components in angiogenesis. Thromb Haemost. 2004;92(2):394–402. doi: 10.1160/TH03-04-0213. [DOI] [PubMed] [Google Scholar]
- 13.Kisucka J, Butterfield CE, Duda DG, et al. Platelets and platelet adhesion support angiogenesis while preventing excessive hemorrhage. Proc Natl Acad Sci U S A. 2006;103(4):855–860. doi: 10.1073/pnas.0510412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iba O, Matsubara H, Nozawa Y, et al. Angiogenesis by implantation of peripheral blood mononuclear cells and platelets into ischemic limbs. Circulation. 2002;106(15):2019–2025. doi: 10.1161/01.cir.0000031332.45480.79. [DOI] [PubMed] [Google Scholar]
- 15.Italiano JE, Jr, Richardson JL, Patel-Hett S, et al. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet α granules and differentially released. Blood. 2008;111(3):1227–1233. doi: 10.1182/blood-2007-09-113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klement GL, Yip T-T, Cassiola F, et al. Platelets actively sequester angiogenesis regulators. Blood. 2009;113(12):2835–2842. doi: 10.1182/blood-2008-06-159541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaslavsky A, Baek KH, Lynch RC, et al. Platelet-derived thrombospondin-1 (TSP-1) is a critical negative regulator and potential biomarker of angiogenesis. Blood. 2010;115(22):4605–4613. doi: 10.1182/blood-2009-09-242065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Massberg S, Konrad I, Schürzinger K, et al. Platelets secrete stromal cell-derived factor 1alpha and recruit bone marrow-derived progenitor cells to arterial thrombi in vivo. J Exp Med. 2006;203(5):1221–1233. doi: 10.1084/jem.20051772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karpatkin S, Pearlstein E, Ambrogio C, Coller BS. Role of adhesive proteins in platelet tumor interaction in vitro and metastasis formation in vivo. J Clin Invest. 1988;81:1012–1019. doi: 10.1172/JCI113411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng S, Shen J, Jiao Y, et al. Platelets and fibrinogen facilitate each other in protecting tumor cells from natural killer cytotoxicity. Cancer Sci. 2009;100(5):859–865. doi: 10.1111/j.1349-7006.2009.01115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nash GF, Turner LF, Scully MF, Kakkar AK. Platelets and cancer. Lancet Oncol. 2002;3:425–430. doi: 10.1016/s1470-2045(02)00789-1. [DOI] [PubMed] [Google Scholar]
- 22.Byzova TV, Plow EF. Networking in the hematostatic system: integrin alphaIIbbeta3 binds prothrombin and influences its activation. J Biol Chem. 1997;272:27183–27188. doi: 10.1074/jbc.272.43.27183. [DOI] [PubMed] [Google Scholar]
- 23.Brill A, Elinav H, Varon D. Differential role of platelet granular mediators in angiogenesis. Cardiovasc Res. 2004;63(2):226–235. doi: 10.1016/j.cardiores.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 24.Hodivala-Dilke KM, McHugh KP, Tsakiris DA, et al. Beta3-integrin-deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Invest. 1999;103(2):229–238. doi: 10.1172/JCI5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhen L, Jiang S, Feng L, et al. Abnormal expression and subcellular distribution of subunit proteins of the AP-3 adaptor complex lead to platelet storage pool deficiency in the pearl mouse. Blood. 1999;94(1):146–155. [PubMed] [Google Scholar]
- 26.Young LR, Borchers MT, Allen HL, Gibbons RS, McCormack FX. Lung-restricted macrophage activation in the pearl mouse model of Hermansky-Pudlak syndrome. J Immunol. 2006;176(7):4361–4368. doi: 10.4049/jimmunol.176.7.4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ren Q, Barber HK, Crawford GL, et al. Endobrevin/VAMP-8 is the primary v-SNARE for the platelet release reaction. Mol Biol Cell. 2007;18(1):24–33. doi: 10.1091/mbc.E06-09-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petit I, Szyper-Kravitz M, Nagler A, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3(7):687–694. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- 29.Jin DK, Shido K, Kopp HG, et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med. 2006;12(5):557–567. doi: 10.1038/nm1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shiba Y, Takahashi M, Yoshioka T, et al. M-CSF accelerates neointimal formation in the early phase after vascular injury in mice: the critical role of the SDF-1-CXCR4 system. Arterioscler Thromb Vasc Biol. 2007;27(2):283–289. doi: 10.1161/01.ATV.0000250606.70669.14. [DOI] [PubMed] [Google Scholar]
- 31.Peled A, Petit I, Kollet O, et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science. 1999;283(5403):845–848. doi: 10.1126/science.283.5403.845. [DOI] [PubMed] [Google Scholar]
- 32.Stellos K, Langer H, Daub K, et al. Platelet-derived stromal cell-derived factor-1 regulates adhesion and promotes differentiation of human CD34+ cells to endothelial progenitor cells. Circulation. 2008;117(2):206–215. doi: 10.1161/CIRCULATIONAHA.107.714691. [DOI] [PubMed] [Google Scholar]
- 33.Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med. 2002;6(1):1–12. doi: 10.1111/j.1582-4934.2002.tb00307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobayashi T, Hamano K, Li TS, et al. Angiogenesis induced by the injection of peripheral leukocytes and platelets. J Surg Res. 2002;103(2):279–286. doi: 10.1006/jsre.2001.6309. [DOI] [PubMed] [Google Scholar]
- 35.Langer H, May AE, Daub K, et al. Adherent platelets recruit and induce differentiation of murine embryonic endothelial progenitor cells to mature endothelial cells in vitro. Circ Res. 2006;98(2):e2–e10. doi: 10.1161/01.RES.0000201285.87524.9e. [DOI] [PubMed] [Google Scholar]
- 36.Stellos K, Gawaz M. Platelet interaction with progenitor cells: potential implications for regenerative medicine. Thromb Haemost. 2007;98:922–929. doi: 10.1160/th07-02-0147. [DOI] [PubMed] [Google Scholar]
- 37.Purhonen S, Palm J, Rossi D, et al. Bone marrow-derived circulating endothelial precursors do not contribute to vascular endothelium and are not needed for tumor growth. Proc Natl Acad Sci U S A. 2008;105(18):6620–6625. doi: 10.1073/pnas.0710516105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shojaei F, Wu X, Qu X, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009;106(16):6742–6747. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahn GO, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. 2008;13(3):193–205. doi: 10.1016/j.ccr.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petit I, Jin D, Rafii S. The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol. 2007;28(7):299–307. doi: 10.1016/j.it.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pitchford SC, Furze RC, Jones CP, Wengner AM, Rankin SM. Differential mobilization of subsets of progenitor cells from the bone marrow. Cell Stem Cell. 2009;4(1):62–72. doi: 10.1016/j.stem.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 42.Manenti L, Paganoni P, Floriani I, et al. Expression levels of vascular endothelial growth factor, matrix metalloproteinases 2 and 9 and tissue inhibitor of metalloproteinases 1 and 2 in the plasma of patients with ovarian carcinoma. Eur J Cancer. 2003;39:1948–1956. doi: 10.1016/s0959-8049(03)00427-1. [DOI] [PubMed] [Google Scholar]
- 43.Salven P, Orpana A, Joensuu H. Leukocytes and platelets of patients with cancer contain high levels of vascular endothelial growth factor. Clin Cancer Res. 1999;5:487–491. [PubMed] [Google Scholar]
- 44.McDowell G, Temple I, Kirwan CC, et al. Alteration in platelet function in patients with early breast cancer. Anticancer Res. 2005;25:3963–3966. [PubMed] [Google Scholar]
- 45.Stellos K, Gawaz M. Platelets and stromal cell-derived factor-1 in progenitor cell recruitment. Semin Thromb Hemost. 2007;33(2):159–164. doi: 10.1055/s-2007-969029. [DOI] [PubMed] [Google Scholar]
- 46.Lapidot T, Kollet O. The essential roles of the chemokine SDF-1 and its receptor CXCR4 in human stem cell homin and repopulation of transplanted immune-deficient NOD/SCID and NOD/SCID/B2mnull mice. Leukemia. 2002;16:1992–2003. doi: 10.1038/sj.leu.2402684. [DOI] [PubMed] [Google Scholar]
- 47.Hattori K, Heissig B, Tashiro K, et al. Plasma elevation of stromal cell-derived factor-1 induces mobilization of mature and immature hematopoietic progenitor and stem cells. Blood. 2001;97(11):3354–3360. doi: 10.1182/blood.v97.11.3354. [DOI] [PubMed] [Google Scholar]
- 48.Rafii DC, Psaila B, Butler J, Jin DK, Lyden D. Regulation of vasculogenesis by platelet-mediated recruitment of bone marrow-derived cells. Arterioscler Thromb Vasc Biol. 2008;28:217–222. doi: 10.1161/ATVBAHA.107.151159. [DOI] [PubMed] [Google Scholar]
- 49.Vu TH, Shipley JM, Bergers G, et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93(3):411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roberts DD. Regulation of tumor growth and metastasis by thrombospondin-1. FASEB J. 1996;10(10):1183–1191. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}