

Abstract
Normal differentiated hepatocytes primarily metabolize methionine, via homocysteine synthesis, through the transsulfuration pathway. In addition to glutathione, this pathway produces α-ketobutyrate that is further metabolized in the mitochondria. It is only under low methionine conditions that differentiated hepatocytes predominantly regenerate methionine from homocysteine. In contrast, proliferating hepatocytes and liver cancer cells regenerate methionine from homocysteine regardless of the availability of methionine. Here we propose that this less efficient metabolism of methionine in proliferating hepatocytes and cancer cells is an adaptation to the “Warburg effect” that is, to the well known phenomenon that cancer cells rely on aerobic glycolysis instead of oxidative phosphorylation to generate energy. The observation that knockout mice with impaired S-adenosylmethionine (SAMe) synthesis (the first step in methionine metabolism) or catabolism spontaneously develop fatty liver and hepatocellular carcinoma, together with the observation that SAMe administration induces apoptosis in hepatoma cells and prevents liver cancer support this hypothesis.
Keywords: S-Adenosylmethionine, methionine adenosyltransferase, glycine N-methyltransferase, oxidative methionine metabolism, aerobic methionine metabolism
Introduction
The body of a normal, adult, lean individual has an energy reserve of about 138,000 kcal, the majority of them (about 135,000 kcal) stored in the white adipose tissue as triglycerides (TGL) (reviewed in Cahill, 1976). In addition, a small amount of TGL (about 450 kcal) that play a critical role in maintaining metabolic homeostasis for the whole organism is stored in the liver. Current estimates indicate that about 20% to 40% of the general adult population living in Western countries has fatty liver (defined as more than 5% of fat by weight; reviewed in Adams, 2007). In these individuals, the liver loses its characteristic reddish color and get instead a yellowish tint due to the excessive accumulation of TGL. Fatty liver is mainly associated with increased consumption of alcohol and obesity although it is also frequently observed in individuals suffering from essential nutrients deficiency. Fatty liver is a progressive disease that goes from the simple accumulation of fat in the liver (steatosis) to steatosis with inflammation, necrosis and fibrosis (steatohepatitis; reviewed in Adams, 2007). In turn, steatohepatitis may progress to cirrhosis and hepatocellular carcinoma (HCC) suggesting that this association of cancer with fatty liver is the result of a disturbed state of energy metabolism homeostasis.
In 1932 Charles Best’s observed that choline prevented the deposition of fat in the liver, a phenomenon known as “lipotropism” (reviewed in 3). Best also realized that the rate of oxygen uptake by the slices of livers from rats on a diet low in choline was appreciably less than that of the normal liver slices. In a subsequent study carried out in 1937, Tucker and Eckstein discovered the lipotropic action of methionine (reviewed in 3). Then, in 1940 du Vigneaud and his colleagues administered deuterium labeled methionine to rats fed a diet low in methionine and choline and found a massive accumulation of the isotope in choline (reviewed in Poirier, 2002). This process, referred as “transmethylation”, undoubtedly established the synthesis of choline from methionine. Later work by Stetten established that ethanolamine (we know now that in the form of phosphatidylethanolamine) receives methyl groups from methionine to form choline (reviewed in Poirier, 2002); and in 1953 Cantoni showed that in order to transfer its methyl group, methionine needs first to be converted to an active sulfonium ion by reacting with ATP to form S-adenosylmethionine (SAMe) (reviewed in Poirier, 2002). In 1983, Poirier demonstrated that a severe methyl-deficient diet also caused liver cancer (reviewed in 3); and more recently, Lu and Mato demonstrated that knockout mice deficient in hepatic SAMe synthesis spontaneously developed fatty liver and HCC (reviewed in 4,5). An explanation for these observations has remained elusive since the connection of methionine metabolism with lipid utilization as well as with hepatocyte proliferation is, at first glance, not apparent.
Proliferating hepatocytes and liver cancer cells show a less efficient methionine metabolism than normal differentiated hepatocytes
SAMe is synthesized by methionine adenosyltransferase (MAT). In mammals there are three isoforms of MAT (MATI, MATII and MATIII) that are encoded by two genes (MAT1A and MAT2A) (reviewed in Mato and Lu, 2007; Mato et al., 2008). MATI and MATIII are tetrameric and dimeric forms, respectively, of the same subunit (α1) encoded by MAT1A, whereas the MATII isoform is a tetramer of a different subunit (α2) encoded by MAT2A. A third gene, MAT2β encodes for a β subunit that regulates the activity of MATII (lowering the Km and Ki for methionine and SAMe respectively) but not of MATI or MATIII (reviewed in Mato and Lu, 2007; Mato et al., 2008). Adult differentiated liver expresses MAT1A, whereas extrahepatic tissues and fetal liver expresses MAT2A. MAT1A expression is silenced in HCC. It is an intriguing question why there are three different MAT isoforms in the liver. The predominant liver form, MATIII, has higher Km for its substrates, is activated by methionine and has higher Vmax, in contrast with the other two isoenzymes (reviewed in Mato and Lu, 2007; Mato et al., 2008). Under normal conditions MATI would, as MATII outside the liver, synthesize most SAMe, which fuels the methylation reactions required by the hepatic cells to be converted into homocysteine, via S-adenosylhomocysteine (SAH), and used primarily for the regeneration of methionine (Figure 1). We refer to this process as conservative methionine metabolism. However, after an increase in methionine concentration, i.e. after a protein-rich meal, conversion to the high activity MATIII would occur and methionine excess will be eliminated (Figure 1). This will lead to an accumulation of SAMe and to the activation of glycine N-methyltransferase (GNMT), the main enzyme involved in hepatic SAMe catabolism (reviewed in Mato et al., 2008). Consequently, the excess of SAMe will be eliminated and converted to homocysteine. Once formed, homocysteine will be used primarily for the synthesis of cysteine and α-ketobutyrate as result of its transsulfuration, since betaine homocysteine methyltransferase (BHMT) and methionine synthase (MS), the two enzymes that regenerate methionine in the liver, have low Km for homocysteine, and elevated SAMe inhibits BHMT activity and expression as well as the synthesis of methyl-tetrahydrofolate, the co-substrate of MS (reviewed in Mato et al., 2008). The transsulfuration pathway involves two enzymes: cystathionine β-synthase (CBS), that is activated by SAMe, and γ-cystathionase. Cysteine is then utilized for the synthesis of glutathione as well as other sulfur containing molecules such as taurine, while α-ketobutyrate penetrates the mitochondria where it is further metabolized. This coordinated modulation by SAMe of the flux through the remethylation and transsulfuration pathways maximizes the production of cysteine and α-ketobutyrate after a methionine load minimizing the regeneration of this amino acid (Figure 1). We refer to this process as catabolic methionine metabolism.
Figure 1.
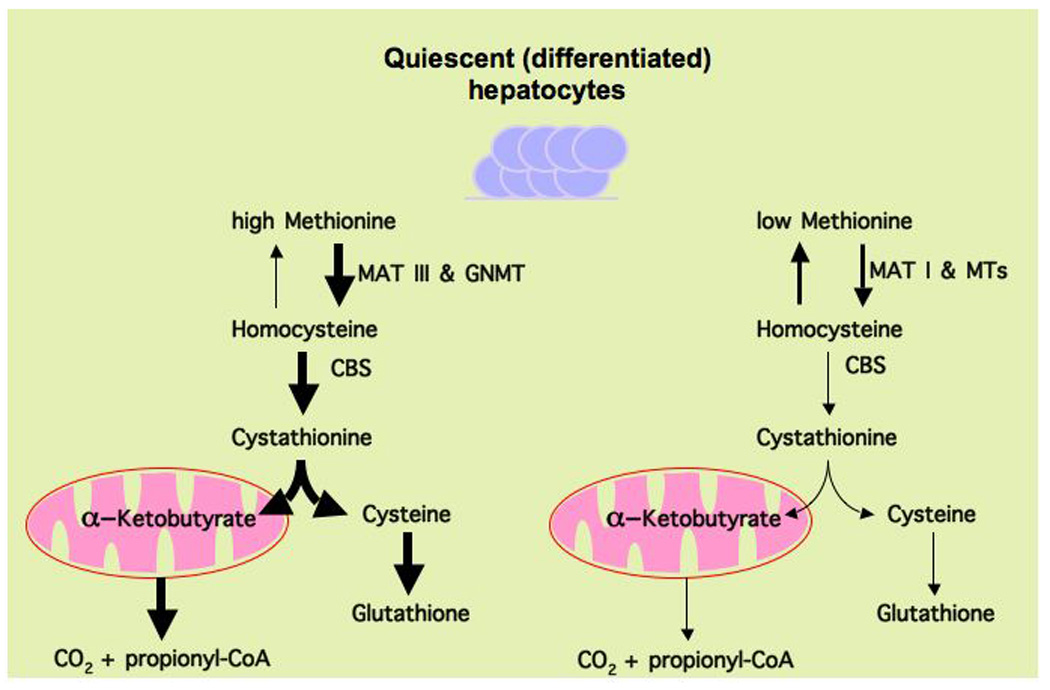
Schematic representation of the differences between conservative and catabolic methionine metabolism in normal non-proliferating hepatocytes. In the presence of high methionine, quiescent (differentiated) hepatocytes rely primarily on methionine adenosyltransferase III (MATIII), which is activated by methionine, to generate SAMe and maintain methionine homeostasis. Excess SAMe is then converted to homocysteine, by the combined action of glycine N-methyltransferase (GNMT) and S-adenosylhomocysteine hydrolase, to avoid aberrant methylation reactions such as irregular methylation of histones and DNA. While GNMT is allosterically activated by SAMe, S-adenosylhomocysteine hydrolase is a reversible enzyme. Once formed, the excess homocysteine is used for the synthesis of cysteine and α-ketobutyrate as result of its transsulfuration (cycling homocysteine back to methionine is inhibited by SAMe). The transsulfuration pathway involves two enzymes that require pyridoxal phosphate as cofactor: cystathionine β-synthase (CBS), which is also allosterically activated by SAMe, and γ-cystathionase. Cysteine is then utilized for the synthesis of glutathione as well as other sulfur containing molecules such as taurine, while α-ketobutyrate is further metabolized in the mitochondria. We refer to this process as catabolic methionine metabolism. When methionine concentration is limiting MATI synthesizes most SAMe, which feeds the multiple methyl transferase (MTs) reactions required by the hepatic cells, to be then converted into homocysteine, and used primarily for the regeneration of methionine. This recycling of homocysteine when methionine is limiting allows normal methylation reactions to continue although results in reduced α-ketobutyrate and glutathione synthesis when compared with oxidative methionine metabolism. We refer to this process as conservative methionine metabolism.
In contrast to normal non-proliferating (differentiated) hepatocytes, which rely primarily on MAT I/III to generate SAMe and maintain methionine homeostasis, embryonic and proliferating adult hepatocytes as well as liver cancer cells instead rely on MATII to synthesize SAMe (reviewed in Mato and Lu, 2007; Mato et al., 2008). Liver cancer cells often have very low levels of GNMT, CBS and γ-cystathionase expression and increased expression of MAT2β, which as mentioned above lowers the Km for methionine and the Ki for SAMe of MATII (reviewed in Mato and Lu, 2007; Mato et al., 2008). Consequently, proliferating hepatocytes and hepatoma cells tend to utilize methionine for protein synthesis regardless of whether methionine is present in high or low amounts and to divert most homocysteine away from the transsulfuration pathway by regenerating methionine (Figure 2). MAT2A/MAT2β expressing hepatoma cells have lower SAMe levels than cells expressing MAT1A, which also favors the regeneration of methionine. Conservation of methionine for protein synthesis is not only a metabolic adaptation of normal proliferating hepatocytes and liver cancer cells but is also a carcinogenic mechanism, as indicated by the observation that MAT1A and GNMT knockout (KO) mice spontaneously develop HCC (reviewed in Mato et al., 2008). Preservation of methionine during proliferation raises the question about how cells obtain cysteine, a metabolite of the transsulfuration pathway that is also needed for protein synthesis. In proliferating hepatocytes (i.e. during liver regeneration after partial hepatectomy), the transsulfuration pathway may be active enough to provide sufficient cysteine for protein synthesis while in hepatoma cells, where the transsulfuration pathway can be markedly diminished, this amino acid may be obtained from blood.
Figure 2.
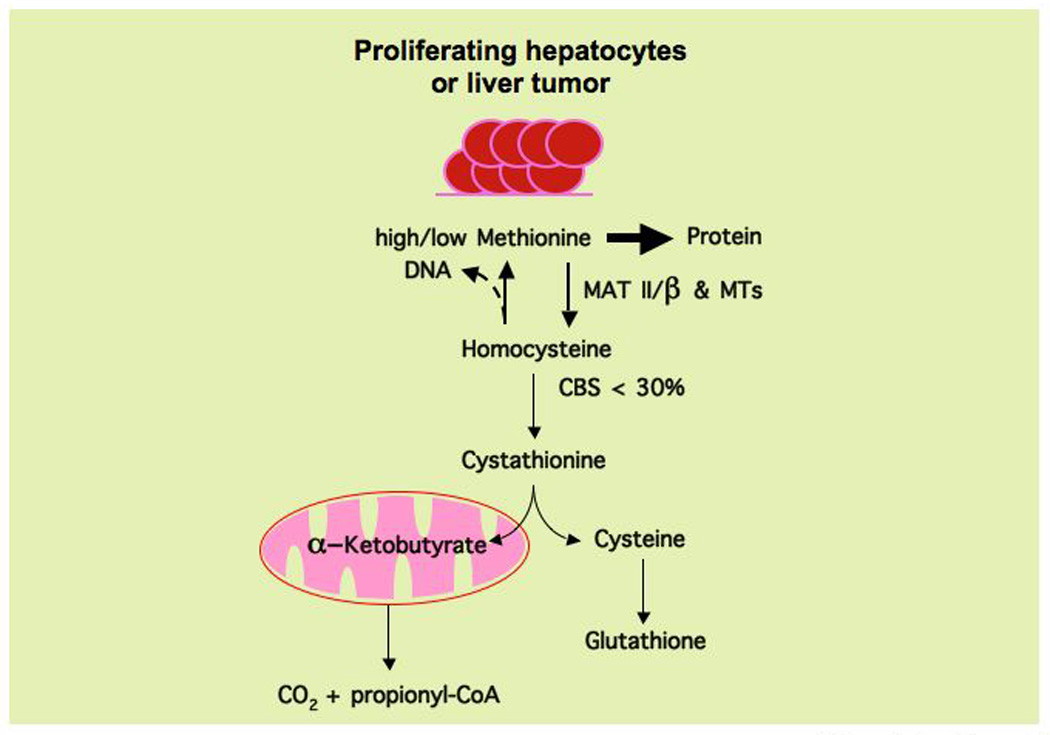
Proliferating hepatocytes and liver cancer cells recycle most homocysteine to methionine (conservative methionine metabolism) regardless of whether this amino acid is present in high quantities or is limiting. Proliferating hepatocytes and liver cancer cells rely on methionine adenosyltransferase II (MATII, which is inhibited by SAMe) and the β-subunit (which lowers the Km and Ki of MATII for methionine and SAMe respectively) to synthesize SAMe, which is used in multiple methyl transferase (MTs) reactions required by the proliferating cells to be converted into homocysteine, via S-adenosylhomocysteine, and used primarily for the regeneration of methionine. In liver tumors, the expression of CBS is less then 30% of that found in normal hepatic tissue. This recycling of homocysteine maximizes the utilization of methionine for protein synthesis and results in minimal α-ketobutyrate and glutathione synthesis. Homocysteine recycling is coupled to the synthesis of thymidine monophosphate (through the folate cycle), which is subsequently phosphorylated to thymidine triphosphate for use in DNA synthesis.
Most cancer and normal proliferating cells metabolize glucose to lactate, rather than to mitochondrial oxidative phosphorylation and ATP production. Otto Warburg first realized the existence of this association of metabolism and growth in 1924 (reviewed in vander Heiden et al., 2009), after observing that cancer cells metabolize glucose into lactate even in the presence of enough oxygen to support mitochondrial oxidative phosphorylation, a phenomenon referred as “aerobic glycolysis.” This less efficient way to generate ATP has the advantage of enhancing the incorporation of nutrients into the building blocks (lipids, proteins, DNA and RNA) needed to make a new cell (reviewed in vander Heiden et al., 2009). From this perspective, it seems that minimizing the amount of methionine that is oxidized in the mitochondria, via α–ketobutyrate, and maximizing the amount of methionine that is available for protein synthesis as well as the amount of homocysteine that is used for the regeneration of methionine, may be considered as another example of the same phenomenon, the diversion of nutrients for the synthesis of new cells.
Deletion of MAT1A, GNMT or CBS in mice leads to fatty liver and, in the case of MAT1A and GNMT, also to HCC (reviewed in Mato et al., 2008). Disruption of methionine catabolism in these three mice models, however, affected hepatic SAMe differently. While loss of MAT1A resulted in a decrease in hepatic SAMe, deletion of GNMT caused a marked increase in SAMe liver, and CBS loss induced a moderate increase in SAMe and an increase in SAH, an inhibitor of methylation reactions (reviewed in Mato et al., 2008). How do these results support a unifying hypothesis regarding synchrony between methionine metabolism and lipid metabolism and hepatocyte growth? One possibility is that any disruption of the catabolism of methionine may be key for the development of fatty liver disease and HCC. Alternatively, SAMe may regulate different pathways with opposite effects on lipid metabolism and cell growth. For instance, SAMe increases the stability of prohibitin 1 (PHB1) (Santamaría et al., 2003), a protein essential to maintain normal mitochondrial function and consequently fatty acid oxidation, but inhibits AMP-activated protein kinase (AMPK) (Martínez-Chantar et al., 2006), a cellular energy sensor that is activated when the AMP levels are elevated stimulating fatty acid oxidation. Hence, in MAT1A KO mice hepatic AMPK is activated but mitochondrial function is impaired, which may explain at least in part the accumulation of liver triglycerides in this model (Martínez-Chantar et al., 2006; Santamaría et al., 2003). Another example of this dual effect of SAMe is its effect on the expression of liver tumor suppressors. PHB1 is a liver tumor suppressor, as demonstrated recently by the finding that liver-specific PHB1-KO mice develop HCC (Ko et al., in press). Although from this perspective it could be concluded that high SAMe would prevent hepatocarcinogenesis, in GNMT-KO mice excess SAMe causes hypermethylation and silencing of SOCS2 and RASSF1A (Martínez-Chantar et al., 2008), two well-known tumor suppressor genes implicated in liver cancer. Obviously, other factors, such as inflammation, may also be necessary for neoplastic transformation in these mice models.
How does the methionine-sparing concept relate to choline deficient diets and cancer? Reduced betaine, resulting from a choline deficient diet, diminishes methionine regeneration and, accordingly with the methionine-sparing concept, SAMe synthesis (reviewed in Poirier, 2002). This reduction in total homocysteine flux, caused by the decrease of the BHMT pathway, cannot be compensated by MS, which has low Km for homocysteine, or by CBS, which needs high SAMe to be fully active. This situation, where the synthesis of SAMe and the transsulfuration pathway are both maintained at low level, is similar to that observed in MAT1A KO mice and may explain, at least in part, the development of fatty liver and the carcinogenic effect of choline deficient diets.
Does changing the metabolism of hepatocytes through manipulation of methionine metabolism holds promise for improving HCC prognosis?
An explanation to Best’s effect is that a less efficient methionine metabolism where SAMe level and transsulfuration activity are reduced enhances the capacity of hepatocytes and hepatoma cells to utilize nutrients for anabolic processes to support growth. In principle, this dependency of HCC on methionine metabolism may be utilized for cancer treatment. Consistent with this hypothesis, hepatoma cells transfected with MAT1A grow slower than control tumor cells (Lu and Mato, 2008), and the addition of SAMe to hepatoma cells, but not to normal hepatocytes, induces apoptosis (reviewed in Li et al., 2010). In vivo, SAMe treatment is also effective in preventing the establishment of HCC although is ineffective as a treatment of already established liver tumors in a rodent model (possibly due to the adaptation of the surrounding normal liver tissue to eliminate excess exogenous SAMe) (Lu et al., 2009).
Acknowledgments
Financial support
This work is supported by grants from NIH AT-1576 (to S.C.L. and J.M.M.), DK51719 (to S.C.L.), SAF 2008-04800, HEPADIP-EULSHM-CT-205 and ETORTEK-2008 (to J.M.M.); the Instituto de Salud Carlos III funds CIBERehd.
References
- Adams LA, Lindor KD. Nonalcoholic fatty liver disease. Ann Epidemiol. 2007;17:863–869. doi: 10.1016/j.annepidem.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Cahill GF. Starvation in man. J Clin Endocrinol Metab. 1976;5:397–415. doi: 10.1016/s0300-595x(76)80028-x. [DOI] [PubMed] [Google Scholar]
- Ko KS, Tomasi ML, Iglesias-Ara A, French BA, French SW, Ramani K, Lozano JJ, Oh P, He L, Stiles BL, Li TW, Yang H, Martínez-Chantar ML, Mato JM, Lu SC. Liver specific deletion of prohibitin 1 results in spontaneous liver injury, fibrosis and hepatocellular carcinoma in mice. Hepatology. doi: 10.1002/hep.23919. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ramani K, Sun Z, Zee C, Grant EG, Yang H, Xia M, Oh P, Ko K, Mato JM, Lu SC. Forced expression of methionine adenosylransferase 1A in human hepatoma cells suppresses in vivo tumorigenicity in mice. Am J Pathol. 2010;176:2456–2466. doi: 10.2353/ajpath.2010.090810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SC, Mato JM. S-adenosylmethionine in cell growth, apoptosis and liver cancer. J Gastroenterol Hepatol. 2008;1:S73–S77. doi: 10.1111/j.1440-1746.2007.05289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SC, Ramani K, Ou X, Lin M, Yu V, Ko K, Park R, Bottiglieri T, Tsukamoto H, Kanel G, French SW, Mato JM, Moats R, Grant E. S-Adenosylmethionine in the chemoprevention and treatment of hepatocellular carcinoma in a rat model. Hepatology. 2009;50:462–471. doi: 10.1002/hep.22990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Chantar ML, Vázquez-Chantada M, Ariz U, Martínez N, Varela M, Luka Z, Capdevila A, Rodríguez J, Aransay AM, Matthiesen R, Yang H, Calvisi DF, Esteller M, Fraga MF, Lu SC, Wagner C, Mato JM. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47:1191–1199. doi: 10.1002/hep.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Chantar ML, Vázquez-Chantada M, Garnacho M, Varela-Rey M, Dotor J, Santamaría M, Martínez-Cruz LA, Parada LA, Lu SC, Mato JM. S-adenosylmethionine regulates cytoplasmic HuR via AMP-activated kinase. Gastroenterology. 2006;131:223–232. doi: 10.1053/j.gastro.2006.04.019. [DOI] [PubMed] [Google Scholar]
- Mato JM, Lu SC. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology. 2007;45:1306–1312. doi: 10.1002/hep.21650. [DOI] [PubMed] [Google Scholar]
- Mato JM, Martínez-Chantar ML, Lu SC. Methionine metabolism and liver disease. Annu Rev Nutr. 2008;28:273–293. doi: 10.1146/annurev.nutr.28.061807.155438. [DOI] [PubMed] [Google Scholar]
- Poirier LA. The effects of diet, genetics and chemicals on toxicity and aberrant DNA methylation: an introduction. J Nutr. 2002;132:23368–23396. doi: 10.1093/jn/132.8.2336S. [DOI] [PubMed] [Google Scholar]
- Santamaría E, Avila MA, Latasa MU, Rubio A, Martín-Duce A, Lu SC, Mato JM, Corrales F. Functional proteomics of nonalcoholic steatohepatitis: mitochondrial proteins as targets of S-adenosylmethionine. Proc Natl Acad Sci U S A. 2003;100:3065–3070. doi: 10.1073/pnas.0536625100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vander Heiden MG, Cantley LC, Thompson CB. Science. 2008;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]