

Summary
Alcohol abuse is one of the most common causes of pancreatitis. The risk of developing alcohol-induced pancreatitis is related to the amount and duration of drinking. However, only a small portion of heavy drinkers develop disease, indicating that other factors (genetic, environmental or dietary) contribute to disease initiation. Epidemiologic studies suggest roles for cigarette smoking and dietary factors in the development of alcoholic pancreatitis. The mechanisms underlying alcoholic pancreatitis are starting to be understood. Studies from animal models are revealing that alcohol sensitizes the pancreas to key pathobiologic processes that are involved in pancreatitis. Current studies are focused on the mechanisms responsible for the sensitizing effect of alcohol; recent findings reveal disordering of key cellular organelles including endoplasmic reticulum, mitochondria, and lysosomes. As our understanding of alcohol’s effects continue to advance to the level of molecular mechanisms, insights into potential therapeutic strategies will emerge providing opportunities for clinical benefit.
Factors involved in development of alcoholic pancreatitis
Alcohol abuse is associated with a spectrum of pancreatic disease clinical manifestations, from acute self-limiting pancreatitis to chronic unremitting pancreatitis leading to exocrine and endocrine pancreatic insufficiency (Lankisch et al. 2002;Pandol et al. 2007). The risk of developing alcohol-induced pancreatitis increases with the amount and duration of drinking. A minimum of 6–12 years of approximately 80 grams or more of alcohol per day is considered necessary for the development of clinically significant disease (Lin et al. 2000;Schenker & Montalvo 1998;Singer 2002;Strate et al. 2002;Yadav & Whitcomb 2010). However, less than 10% of heavy drinkers develop clinical pancreatitis suggesting that there are contributing genetic and environmental factors involved in disease expression. On the other hand, findings consistent with pancreatitis have been reported in up to 75% of autopsies performed on alcohol abusers (Dufour & Adamson 2003;Schenker & Montalvo 1998).
There are wide ranges in the reported incidence and prevalence of the disease between countries and sometimes within countries (Dufour & Adamson 2003;Go & Everheart 1994;Lankisch et al. 2002;Lin et al. 2000). However, there are patterns indicating that the incidence of alcoholic pancreatitis is more common among men, while pancreatitis caused by gallstones is more common among women (Lankisch et al. 2002;Pandol et al. 2007). Another pattern is related to ethnicity: the studies of discharge data from Los Angeles County and New York hospitals and one in Portugal demonstrated that for both men and women, black patients are more likely, compared to other ethnic groups, to be hospitalized for chronic pancreatitis than alcoholic cirrhosis (Dufour & Adamson 2003;Go & Everheart 1994;Tao et al. 2003). In the US, Native Americans and Alaskan natives have the highest rates of alcoholic cirrhosis of any ethnic/racial group but have rates of pancreatitis similar to those of whites (Lowenfels et al. 1999).
Both smoking and dietary factors may contribute to the risk of alcoholic pancreatitis. Although the issue is complicated by the interrelationship between smoking and drinking, recent studies have demonstrated that cigarette smoking is an independent risk factor for alcohol-related pancreatitis and that smoking accelerates the disease progression (Maisonneuve et al. 2005;Morton et al. 2004;Yadav & Whitcomb 2010). Further, one report (Yadav et al. 2009) provides evidence that there is possibly a synergistic association between alcohol and smoking in the development of pancreatitis. Different diets may also affect the development of the disease. Diets high in fat and protein may be associated with the development of alcoholic pancreatitis while saturated fats and vitamin E may decrease the toxic effect of alcohol (Dufour & Adamson 2003;Lankisch et al. 2002).
Overall hypothesis for the mechanism of alcoholic pancreatitis
The information provided above suggests that alcohol is a contributing factor to the initiation and development of both acute and chronic pancreatitis; and that alcohol alone may not cause pancreatitis unless accompanied by additional genetic and/or environmental factors. Thus, it is believed that alcohol “sensitizes” or “primes” the pancreas to pancreatitis. Additional factors, such as cigarette smoking, genetic factors (e.g., ethnicity) and/or diet, then act to initiate pancreatitis in the “alcohol-sensitized” pancreas. An extension of this hypothesis is that the pancreas has adaptation systems that protect it from insults caused by alcohol abuse and that pancreatic disease occurs when these adaptive systems are either disordered or insufficiently robust. In this scenario, genetic and/or environmental factors could increase the likelihood of disease development in alcohol abusers by altering key adaptive responses in the pancreas.
Clinical-pathological characteristics of alcoholic pancreatitis
The pathobiologic responses of alcoholic pancreatitis include acute and chronic inflammation, loss of parenchymal tissue, and fibrosis (Figure 1). These processes lead to irreversible and debilitating exocrine and endocrine insufficiency and a severe pain syndrome. The clinical course of a patient with alcoholic pancreatitis is usually one of repeated episodes of acute pancreatitis presenting as abdominal pain and difficulty in maintaining nutrition because the pain is increased or nausea and vomiting ensue with food or liquid intake (Dufour & Adamson 2003;Pandol et al. 2007;Sand & Nordback 2009). The acute episodes of pancreatitis cause both local vascular dysfunction in the pancreas leading to worsening of the disease because of ischemia; and a systemic inflammatory response that leads to pulmonary, renal and vascular failure (Pandol et al. 2007). Many patients also have pain and nutritional disorders between these more severe acute episodes, often despite cessation of drinking, suggesting that the disease progresses once initiated. In contrast to alcoholic pancreatitis, patients with biliary pancreatitis have complete resolution of pain and progression of disease once the inciting gallstone disease is dealt with. The mechanisms underlying the progressive nature of alcoholic pancreatitis are unknown. Adding to the morbidity and mortality of chronic pancreatitis is a markedly increased risk in pancreatic cancer that these patients have (Lowenfels et al. 1993;Maisonneuve & Lowenfels 2002).
Figure 1. The mechanisms of alcoholic pancreatitis.
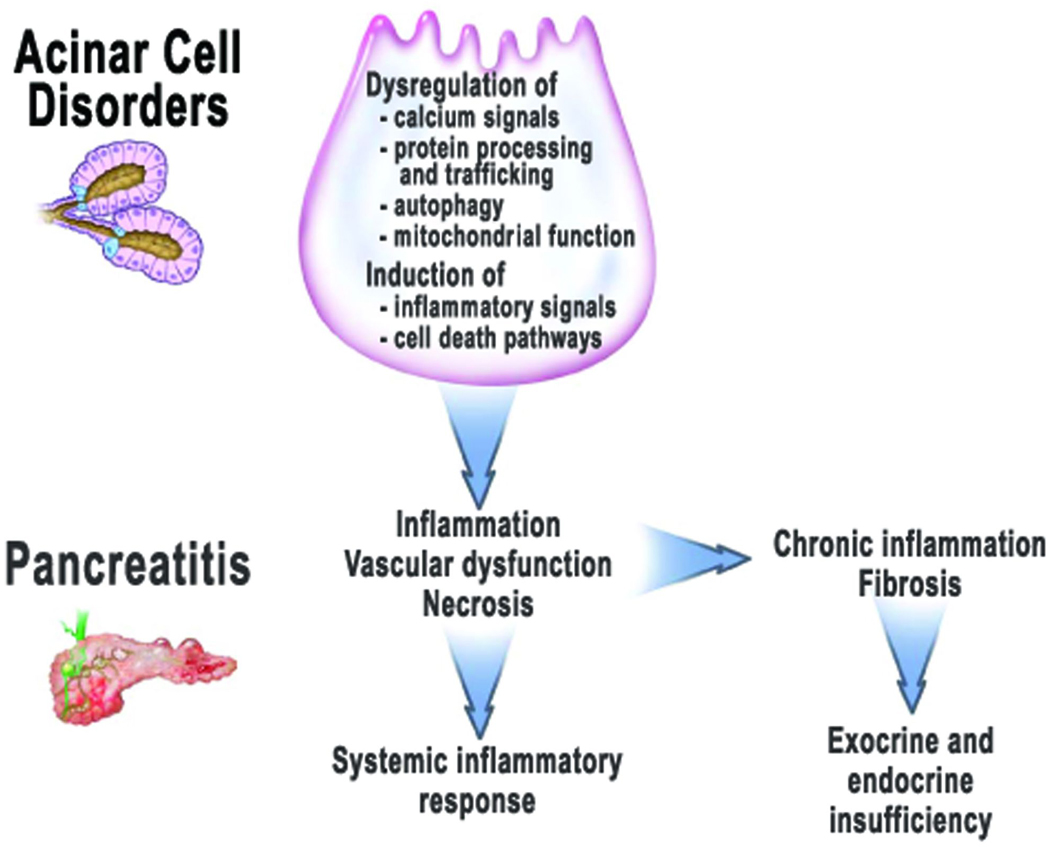
Intracellular events in the pancreatic acinar cell associated with alcohol abuse include dysregulation of calcium signals, protein processing and trafficking, the autophagic, lysosomal and mitochondrial functions; and the induction of inflammatory signals and cell death pathways. These disorders “sensitize” the pancreas so that is in not capable of adapting to stressful stimuli, thus leading to pancreatitis with key pathologic responses of organ inflammation, vascular dysfunction, and parenchymal necrosis. Severe acute pancreatitis can lead to systemic inflammatory response, multiple organ failure, and death. Chronic pancreatitis results from repeated episodes of acute pancreatitis leading to persistent inflammation and fibrosis. The insults of acute and chronic pancreatitis lead to loss of normal function so that exocrine and endocrine pancreatic insufficiency develops.
The current paradigm called necrosis-fibrosis sequence provides a framework to consider the pathologic process in the clinical course of chronic pancreatitis (Ammann et al. 1996;Comfort et al. 1946;Oruc & Whitcomb 2004). This hypothesis is based on morphology of surgical specimens showing that early in the disease the predominant lesions in the pancreas are necrosis, acute inflammation and mild fibrosis. In contrast, specimens obtained at autopsy several years after the onset of disease show severe fibrosis and calcification with no necrosis.
Thus, investigations into the pathogenesis of alcoholic pancreatitis must include strategies to elucidate the effects of alcohol on the pathobiologic processes of inflammation (acute and chronic), parenchymal cell necrosis, and fibrosis. In addition, experimental approaches should include investigations into the effects of smoking, dietary and genetic factors on the development of alcoholic pancreatitis. In the following discussion, we present a summary of our current understanding of the mechanism of the disease to bring into focus research directions that need to be pursued in order to identify the molecular events that initiate and perpetuate alcoholic pancreatitis. We envision that only when these molecular events are identified will there be an ability to develop therapeutic strategies for this disease.
Alcohol and the inflammatory response of pancreatitis
In recent years, the inflammatory response and its initiation in the acinar cell of the pancreas has emerged as a key pathologic process in the mechanism of pancreatitis (Bhatia et al. 2000;Norman 1998;Pandol et al. 2007). The findings from animal and in vitro models of acute pancreatitis indicate that the inflammatory response, once initiated, contributes to the pathologic, intra-acinar activation of the digestive enzyme trypsinogen and the parenchymal cell necrosis (Gukovskaya et al. 2002b;Sandoval et al. 1996). The acinar cell mechanisms of inflammation involve activation of pro-inflammatory signals, such as the key transcription factors nuclear factor (NF)-κB and activator protein-1, and p38 MAP kinase, resulting in the production of a multitude of inflammatory mediators, such as cytokines, chemokines, and adhesion molecules (Pandol et al. 2007).
Studies using both in vivo and in vitro experimental models (Gukovsky et al. 2003;Pandol et al. 1999;Satoh et al. 2006) have demonstrated that alcohol “sensitizes” the pancreas to the inflammatory response through a mechanism involving NF-κB activation by protein kinase C epsilon. This sensitization of the pancreas by alcohol enables physiologic neurohumoral stimulation of the acinar cell (by CCK or carbachol) to induce pancreatitis responses (Lugea A et al. 2010;Pandol et al. 1999).
As indicated earlier, in contrast to biliary pancreatitis, alcoholic pancreatitis does not completely resolve. Although the mechanism of this incomplete resolution has not been established, experimental findings suggest that immune dysregulation may be involved (Deng et al. 2005;Gukovsky et al. 2008). Dysregulation of the immunoinflammatory response by alcohol may impair pancreas recovery/regeneration from the episode of acute pancreatitis, providing an explanation for why alcohol uniquely promotes the transition from acute to chronic pancreatic injury (Gukovsky et al. 2008;Oruc & Whitcomb 2004). The lack of resolution of pancreatic injury is also associated with activation and proliferation of pancreatic stellate cells that mediate the fibrosing response (see below).
Alcohol and the cell death response of pancreatitis
There are several potential mechanistic routes involved in alcohol’s effects on acinar cell death in pancreatitis. Both major types of cell death, apoptosis and necrosis, are observed in models of pancreatitis, as well as in human disease (Gukovskaya & Pandol 2004;Pandol et al. 2007). Apoptosis is characterized by activation of a cascade of the serine proteases, caspases, leading to cell shrinkage and nuclear chromatin condensation. A key event upstream of the caspase activation cascade is mitochondrial dysfunction (permeabilization), resulting in the release of pro-apoptotic factors, such as cytochrome c, into the cytosol. Importantly, apoptosis preserves the integrity of the plasma membrane and is thus associated with little or no tissue inflammation. Necrosis is characterized by swelling of intracellular organelles and rupture of the plasma membrane, leading to spillage of cellular contents into the extracellular space and, therefore, an inflammatory response. Clinically, parenchymal necrosis is a major determinant of poor outcome in patients with pancreatitis (Pandol et al. 2007). Of note, several studies on experimental models (Gukovskaya et al. 1996;Gukovskaya & Pandol 2004;Kaiser et al. 1995;Mareninova et al. 2006) indicate that the severity of pancreatitis is directly related to the amount of necrosis, whereas it correlates inversely with the amount of apoptosis. Thus, shifting the acinar cell death pattern away from necrosis towards apoptosis may have a therapeutic value.
Both the mitochondria-dependent and -independent pathways, and the caspase activation cascade have been shown to participate in acinar cell death during pancreatitis (Gukovskaya & Pandol 2004;Mareninova et al. 2006). Inhibition of apoptosis with specific caspase inhibitors increased necrosis and worsened experimental pancreatitis (Mareninova et al. 2006). Conversely, promoting apoptosis through inhibition of endogenous proteins that negatively regulate caspase activation (IAPs), resulted in decreased necrosis (Mareninova et al. 2006). These findings indicate that caspases protect from acinar cell necrosis in pancreatitis and that it is possible to modulate disease severity by shifting the necrosis/apoptosis ratio. Acinar cell death is also regulated by Bcl-xL and Bcl-2, the “prosurvival” members of the Bcl-2 family proteins (Sung et al. 2009).
Clinical data (Papachristou et al. 2006) indicate that chronic alcohol consumption is a significant risk factor for parenchymal necrosis in pancreatitis. Further, data on experimental models suggest that ethanol feeding promotes a shift from apoptosis to necrosis in pancreas (Fortunato et al. 2006;Fortunato et al. 2009;Gukovskaya et al. 2006;Wang et al. 2006). However, little is known about the effects of alcohol on specific pathways regulating acinar cell death in pancreatitis. One mechanism whereby alcohol may promote necrosis during pancreatitis is through mitochondrial dysfunction (Fortunato et al. 2006;Odinokova et al. 2008); another, by down-regulating the expression of caspases in pancreas (Wang et al. 2006).
Organellar damage in pancreatitis
As stated earlier, in addition to alcohol’s effects on inflammation, it can cause acinar cell damage through facilitating the inappropriate, intra-pancreatic activation of digestive enzymes, i.e., trypsinogen (Gorelick 2003;Gukovskaya et al. 2002b;Katz et al. 1996;Lu et al. 2002). The intra-acinar trypsinogen activation is considered a key initiating event of pancreatitis (Pandol et al. 2007;Saluja et al. 2007) and can directly lead to acinar cell necrosis (Ji et al. 2009). Although the mechanism of this pathologic event is not fully understood, recent evidence from models of nonalcoholic acute pancreatitis demonstrates that it is mediated by disordering of the autophagic and lysosomal pathways (Gukovsky & Gukovskaya 2010;Mareninova et al. 2009a). Autophagy (more precisely, macroautophagy) is the main cellular degradative, lysosome-driven pathway (Levine & Kroemer 2008); the recent findings (Mareninova et al. 2009a) indicate that flux through this pathway is impaired in pancreatitis due to inefficient lysosomal degradation caused by defective processing of key lysosomal proteases, cathepsins. The impaired autophagy mediates not only the pathologic, intra-pancreatic trypsinogen activation but also accumulation of large vacuoles in acinar cells, another hallmark response of pancreatiis (Gukovsky & Gukovskaya 2010;Mareninova et al. 2009a).
Of note, during the last 15 years the focus of research into the mechanism of pancreatitis, both nonalcoholic and alcoholic, was mainly on signaling pathways (Pandol et al. 2007;Saluja et al. 2007), whereas the role of organellar damage has been under-appreciated and under-explored. Impairment of autophagic/lysosomal functions is one example of organellar dysfunction caused by pancreatitis. Mitochondrial dysfunction is another early event in experimental acute pancreatitis, mediating its pathologic responses (Mareninova et al. 2009b;Mukherjee et al. 2008;Odinokova et al. 2009;Sung et al. 2009). In particular, pancreatitis stimulates mitochondrial permeabilization through opening of the permeability transition pore (PTP), a multi-protein nonselective channel, resulting in loss of the mitochondrial membrane potential. Properties of the PTP in pancreatic mitochondria are different from the “classical”, i.e., liver mitochondria, PTP (Odinokova et al. 2009). Recent findings (Mareninova et al. 2009b) reveal that PTP inactivation (by genetic ablation of a key PTP subunit) greatly ameliorates the pathologic responses of pancreatitis, including trypsinogen activation, the inflammatory and cell death responses. The pro-survival Bcl-xL and Bcl-2 proteins protect acinar cells from mitochondrial depolarization and hence necrosis, likely by counteracting the PTP opening (Sung et al. 2009).
Endoplasmic reticulum (ER) is another key organelle the function of which is perturbed in acute pancreatitis (Kubisch & Logsdon 2008). Proper ER functioning, responsible for protein synthesis and correct folding, is especially important for the pancreatic acinar cell, which has the highest rate of protein synthesis among all tissues in adult organism. Short term perturbations of ER function are normally resolved by the adaptive Unfolded Protein Response (UPR), which triggers several signaling pathways aimed to up-regulate the transcription of ER chaperones and foldases to augment the folding and export capacity of the ER, and activate the removal of misfolded or aberrant proteins from the ER (Ron & Walter 2007). Failure of UPR to resolve persistent or severe ER stress can lead to inflammation and cell death (Zhang & Kaufman 2008).
Investigation of alcohol’s effects on organellar damage in pancreatitis is only starting. Alcohol treatments both in vivo and in vitro lead to augmentation of the pathologic, intra-acinar cell activation of digestive enzymes (Cosen-Binker et al. 2008;Gorelick 2003;Katz et al. 1996;Lu et al. 2002;Lugea et al. 2010). Recent data indicate that alcohol treatments perturb the autophagic and lysosomal functions in the acinar cell (Fortunato et al. 2009;Mareninova et al. 2009b;Wang et al. 2006), as well as the processes of exocytosis (Cosen-Binker et al. 2008). Exposure of acinar cells to alcohol leads to PTP opening similar to that observed in pancreatitis (Odinokova et al. 2008;Shalbueva et al. 2009;Yuan et al. 2009). Finally, recent studies (Lugea et al. 2009;Lugea et al. 2010) indicate that alcohol feeding activates the UPR in pancreas with upregulation of the transcription factor XBP1, a key regulator of ER function and development in the pancreatic acinar cell [as well as other secretory cells (Lee et al. 2005)]. Prevention of XBP1 upregulation by heterozygous deletion of the XBP1 gene resulted in pathologic findings in acinar cells. Thus, while alcohol feeding did not induce pancreatic damage in wild type mice, XBP1 deficient mice fed ethanol displayed pathologic features such as acinar cell ER stress, disordered autophagy, and parenchymal necrosis (Lugea et al. 2009). These results suggest that activation of UPR in the acinar cell, and XBP1 in particular, represents an adaptive mechanism against alcohol’s toxicity, and when the UPR response is prevented or defective, pancreatic pathology ensues.
Effects of alcohol metabolites in the acinar cell
Alcohol’s unique metabolism in the pancreas leads to products that have direct effects on pancreatic acinar cell organellar functions (e.g., mitochondria) that can lead to necrosis. The liver is the major alcohol metabolizing organ where ethanol is converted first to acetaldehyde and then acetate by enzymatic oxidative steps (Cederbaum et al. 2009). In contrast, the pancreas contributes only a small portion to overall oxidative metabolism of ethanol and mostly converts ethanol to fatty acid ethyl esters, which then rapidly metabolize to fatty acids (Gukovskaya et al. 2002a;Haber et al. 1998;Haber et al. 2004;Laposata & Lange 1986;Pfutzer et al. 2002). The importance of the non-oxidative metabolism of ethanol in cell death pathways comes from recent findings (Criddle et al. 2004;Criddle et al. 2006;Gerasimenko et al. 2009) that fatty acid ethyl esters cause acinar cell ATP depletion by a process that first involves activation of inositol trisphosphate receptors on the intracellular (e.g., ER) calcium stores, resulting in the release of the large amount of stored Ca2+ into the cytosol. The increase in cytosolic Ca2+ leads to calcium overload of mitochondria, resulting in their depolarization and loss of ability to produce ATP. The end result is necrosis of the acinar cell.
Alcohol and the fibrosing response of pancreatitis
Considerable progress has been made in our understanding of the fibrosing response of chronic pancreatic as a result of the identification and characterization of the pancreatic stellate cell (PaSC) (Apte et al. 1998;Bachem et al. 1998). In normal pancreas, PaSCs are quiescent, with long cytoplasmic processes encircling the base of the acinus (Omary et al. 2007). Similar to hepatic stellate cells, in the quiescent state PaSCs store significant amounts of vitamin A as cytoplasmic lipid droplets. Also similar to hepatic stellate cells, PaSCs are “activated” during pancreatic injury leading to their transformation to a myofibroblastic phenotype, with loss of vitamin A stores, proliferation, and production of high amounts of extracellular matrix (ECM) proteins, i.e., collagen (Omary et al. 2007).
Evidence supporting a key role for activation of PaSCs in the pathogenesis of alcoholic pancreatitis comes from studies using pancreatic tissue from patients with chronic pancreatitis, as well as from animal models (Casini et al. 2000;Haber et al. 1999). These studies show the presence of activated PaSCs in areas of fibrosis, using markers of stellate cell activation (α-smooth muscle actin by immunostaining) and collagen gene transcription (by in situ hybridization). Activated PaSCs participate in tissue repair processes; although their precise role remains to be elucidated, these cells likely contribute to the formation of a provisional matrix at the site of injury that allows new parenchymal cell proliferation and migration, and tissue regeneration. Abnormal, persistent activation of PaSCs and sustained ECM deposition after tissue damage may play a key role in the development of the fibrosing response of chronic pancreatitis (Lugea et al. 2006).
The mechanisms of PaSC activation and ECM protein production involve cytokines generated during pancreatitis by acinar cells as well as by the inflammatory cells and the stellate cells themselves (Omary et al. 2007). Furthermore, alcohol, its metabolites, and reactive oxygen species generated by alcohol-stimulated NADPH oxidase all contribute to the PaSC activation process (Apte et al. 2000;Hu et al. 2007;Masamune et al. 2002;Omary et al. 2007).
In addition to stimulating PaSC activation, alcohol and its metabolites can modulate pancreatic ECM turnover by altering the activity of two major systems of ECM degradation, the matrix metalloproteinases and the plasminogen system (Apte et al. 2000;Apte et al. 2006;Lugea et al. 2003). Both proteolytic systems play an important role in tissue remodeling during pancreatitis and in the recovery after pancreas damage (Lugea et al. 2006;Shek et al. 2002). Although several cell types produce components of these ECM degradation systems, activated PaSCs are an important source of several metalloproteinases and their inhibitors, as well as of plasminogen activators and the plasminogen activator inhibitor 1 (Lugea et al. 2008;Phillips et al. 2003). Studies on cultured PaSCs suggest that ethanol facilitates fibrosis via alterations in the plasminogen system that inhibit ECM degradation and thus increase ECM deposition (Lugea et al. 2008).
Effects of alcohol on molecular processes and future directions of research
As illustrated in the graphic in figure 1, it is generally accepted that disorders of the processes in the acinar cell initiate both acute and chronic pancreatitis. As indicated throughout this review, information about the effects of alcohol consumption on pancreatic pathologic processes is emerging. However, our understanding of the molecular mechanisms underlying these processes is limited. The main unresolved issues are: Why doesn’t alcohol consumption by man, as well as animals, lead invariably to pancreatitis? What is the mechanism of alcohol’s “sensitizing” effect on the pancreas? How does alcohol affect adaptive responses, such as the UPR or autophagy, in the pancreatic acinar cell?
As discussed above, recent data indicate that dysfunction of organelles, such as the ER, mitochondria, and lysosomes, is critical in the pathogenesis of pancreatitis. Investigation of the mechanisms underlying alcohol’s effects on organellar dysfunction in pancreatitis has only recently started. For example, there is evidence that alcohol feeding activates the UPR response in the ER, and that up-regulation of the key transcription factor XBP1 represents an adaptive mechanism against alcohol’s toxicity to pancreas (Lugea et al. 2009;Lugea et al. 2010). Mitochondrial damage, through PTP opening, leads to acinar cell death and other pathologic responses of pancreatitis (Mareninova et al. 2009b;Mukherjee et al. 2008;Odinokova et al. 2009;Sung et al. 2009). In this regard, one molecular mechanism underlying alcohol’s effects could be the induction by fatty acid ethyl esters of Ca2+ release from intracellular stores (Criddle et al. 2004;Criddle et al. 2006;Gerasimenko et al. 2009), leading, in turn, to mitochondrial calcium overload and PTP opening (Baumgartner et al. 2009;Odinokova et al. 2009). Alcohol-induced changes could also make the mitochondria more sensitive to small increases in Ca2+ that occur during physiologic neurohumoral stimulation (Shalbueva et al. 2009;Yuan et al. 2009). Finally, recent data (Mareninova et al. 2010) indicate that alcohol feeding differently affects the autophagic and lysosomal functions in the acinar cell.
There is some evidence that dysregulation of proton transport in the acinar cell may be an important factor in pancreatitis. A major class of cellular proton transporters is vacuolar ATPases; these have been shown to mediate protease activation in the acinar cell (Waterford et al. 2005). Preliminary studies (FSG, unpublished) suggest that short-term ethanol exposure can activate vacuolar ATPase and that this event is linked to trypsinogen activation. Further studies are needed to determine the mechanism of this effect and whether it also occurs with chronic ethanol exposure.
Other examples of alcohol’s disordering effects on molecular pathways in the acinar cell include the recent finding (Cosen-Binker et al. 2007) that alcohol causes re-direction of digestive enzyme secretion from the apical (duct-facing) surface to the basolateral surface of the acinar cell through a protein kinase C alpha medicated effect on key SNARE proteins responsible for exocytosis. This effect likely leads to inappropriate digestive enzyme activation resulting in pancreatic injury. Another recent finding (Mee et al. 2010) is that alcohol treatments markedly suppress transport of folate into the acinar cell, which could potentially cause a whole array of cellular defects considering the important role of folate as a methyl-donor.
The discussion here indicates that alcohol causes disordering of several key pathways in the acinar cell, as determined in experimental systems. Little is known about the relative contributions of these defects to the acinar cell pathology that occurs during human disease. Further, it is very possible that different pathways predominate as a function of type of drinking (e.g., binge drinking) or synergistic risk factors (e.g., genetics, diet, or smoking). Deciphering the key mechanistic steps in alcohol’s effects on pancreas should provide insights into potential molecular targets to treat or mitigate the severity of alcoholic pancreatitis.
Acknowledgments
Southern California Research Center for Alcoholic Liver and Pancreatic Diseases (P50AA11999); UCLA Center for Excellence in Pancreatic Diseases (P01AT003960); NIH/NIAAA (U56 AA0114643, R21AA016010, R21AA015781, R21AA017276); NIH/NCI (R01CA119025); NIH/NIDDK (R01DK059936, R01DK54021); and the AGA Foundation Designated Research Scholar Award in Pancreatitis.
References
- Ammann RW, Heitz PU, Kloppel G. Course of alcoholic chronic pancreatitis: a prospective clinicomorphological long-term study. Gastroenterology. 1996;vol. 111(no. 1):224–231. doi: 10.1053/gast.1996.v111.pm8698203. [DOI] [PubMed] [Google Scholar]
- Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;vol. 43(no. 1):128–133. doi: 10.1136/gut.43.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte MV, Phillips PA, Fahmy RG, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA, Pirola RC, Naidoo D, Wilson JS. Does alcohol directly stimulate pancreatic fibrogenesis? Studies with rat pancreatic stellate cells. Gastroenterology. 2000;vol. 118(no. 4):780–794. doi: 10.1016/s0016-5085(00)70148-x. [DOI] [PubMed] [Google Scholar]
- Apte MV, Pirola RC, Wilson JS. Battle-scarred pancreas: role of alcohol and pancreatic stellate cells in pancreatic fibrosis. J Gastroenterol.Hepatol. 2006;vol. 21 Suppl 3:S97–S101. doi: 10.1111/j.1440-1746.2006.04587.x. [DOI] [PubMed] [Google Scholar]
- Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, Siech M, Beger H, Grunert A, Adler G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;vol. 115(no. 2):421–432. doi: 10.1016/s0016-5085(98)70209-4. [DOI] [PubMed] [Google Scholar]
- Baumgartner HK, Gerasimenko JV, Thorne C, Ferdek P, Pozzan T, Tepikin AV, Petersen OH, Sutton R, Watson AJ, Gerasimenko OV. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J Biol.Chem. 2009;vol. 284(no. 31):20796–20803. doi: 10.1074/jbc.M109.025353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia M, Brady M, Shokuhi S, Christmas S, Neoptolemos JP, Slavin J. Inflammatory mediators in acute pancreatitis. J Pathol. 2000;vol. 190(no. 2):117–125. doi: 10.1002/(SICI)1096-9896(200002)190:2<117::AID-PATH494>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Casini A, Galli A, Pignalosa P, Frulloni L, Grappone C, Milani S, Pederzoli P, Cavallini G, Surrenti C. Collagen type I synthesized by pancreatic periacinar stellate cells (PSC) co-localizes with lipid peroxidation-derived aldehydes in chronic alcoholic pancreatitis. J.Pathol. 2000;vol. 192(no. 1):81–89. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH675>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch.Toxicol. 2009;vol. 83(no. 6):519–548. doi: 10.1007/s00204-009-0432-0. [DOI] [PubMed] [Google Scholar]
- Comfort M, Gambill E, Baggenstoss A. Chronic relapsing pancreatitis: a study of 29 cases without associated disease of the biliary or gastro-intestinal tract. Gastroenterology. 1946;vol. 6:239–285. [PubMed] [Google Scholar]
- Cosen-Binker LI, Binker MG, Wang CC, Hong W, Gaisano HY. VAMP8 is the v-SNARE that mediates basolateral exocytosis in a mouse model of alcoholic pancreatitis. J.Clin.Invest. 2008;vol. 118(no. 7):2535–2551. doi: 10.1172/JCI34672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosen-Binker LI, Lam PP, Binker MG, Reeve J, Pandol S, Gaisano HY. Alcohol/cholecystokinin-evoked pancreatic acinar basolateral exocytosis is mediated by protein kinase C alpha phosphorylation of Munc18c. J.Biol.Chem. 2007;vol. 282(no. 17):13047–13058. doi: 10.1074/jbc.M611132200. [DOI] [PubMed] [Google Scholar]
- Criddle DN, Murphy J, Fistetto G, Barrow S, Tepikin AV, Neoptolemos JP, Sutton R, Petersen OH. Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology. 2006;vol. 130(no. 3):781–793. doi: 10.1053/j.gastro.2005.12.031. [DOI] [PubMed] [Google Scholar]
- Criddle DN, Raraty MG, Neoptolemos JP, Tepikin AV, Petersen OH, Sutton R. Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proc.Natl.Acad.Sci.U.S.A. 2004;vol. 101(no. 29):10738–10743. doi: 10.1073/pnas.0403431101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Wang L, Elm MS, Gabazadeh D, Diorio GJ, Eagon PK, Whitcomb DC. Chronic alcohol consumption accelerates fibrosis in response to cerulein-induced pancreatitis in rats. Am.J.Pathol. 2005;vol. 166(no. 1):93–106. doi: 10.1016/S0002-9440(10)62235-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour MC, Adamson MD. The epidemiology of alcohol-induced pancreatitis. Pancreas. 2003;vol. 27(no. 4):286–290. doi: 10.1097/00006676-200311000-00002. [DOI] [PubMed] [Google Scholar]
- Fortunato F, Burgers H, Bergmann F, Rieger P, Buchler MW, Kroemer G, Werner J. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology. 2009;vol. 137(no. 1):350–360. doi: 10.1053/j.gastro.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Fortunato F, Deng X, Gates LK, McClain CJ, Bimmler D, Graf R, Whitcomb DC. Pancreatic response to endotoxin after chronic alcohol exposure: switch from apoptosis to necrosis? Am.J Physiol Gastrointest Liver Physiol. 2006;vol. 290(no. 2):G232–G241. doi: 10.1152/ajpgi.00040.2005. [DOI] [PubMed] [Google Scholar]
- Gerasimenko JV, Lur G, Sherwood MW, Ebisui E, Tepikin AV, Mikoshiba K, Gerasimenko OV, Petersen OH. Pancreatic protease activation by alcohol metabolite depends on Ca2+ release via acid store IP3 receptors. Proc.Natl.Acad.Sci.U.S.A. 2009;vol. 106(no. 26):10758–10763. doi: 10.1073/pnas.0904818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go V, Everheart J. Digestive Diseases in the United States: Epidemiology and Impact. Washington, DC: US Government Office; 1994:693–712. NIH Publication no 94–1447.
- Gorelick FS. Alcohol and zymogen activation in the pancreatic acinar cell. Pancreas. 2003;vol. 27(no. 4):305–310. doi: 10.1097/00006676-200311000-00006. [DOI] [PubMed] [Google Scholar]
- Gukovskaya AS, Mareninova OA, Odinokova IV, Sung KF, Lugea A, Fischer L, Wang YL, Gukovsky I, Pandol SJ. Cell death in pancreatitis: effects of alcohol. J Gastroenterol.Hepatol. 2006;vol. 21 Suppl 3:S10–S13. doi: 10.1111/j.1440-1746.2006.04571.x. [DOI] [PubMed] [Google Scholar]
- Gukovskaya AS, Mouria M, Gukovsky I, Reyes CN, Kasho VN, Faller LD, Pandol SJ. Ethanol metabolism and transcription factor activation in pancreatic acinar cells in rats. Gastroenterology. 2002a;vol. 122(no. 1):106–118. doi: 10.1053/gast.2002.30302. [DOI] [PubMed] [Google Scholar]
- Gukovskaya AS, Pandol SJ. Cell death pathways in pancreatitis and pancreatic cancer. Pancreatology. 2004;vol. 4(no. 6):567–586. doi: 10.1159/000082182. [DOI] [PubMed] [Google Scholar]
- Gukovskaya AS, Perkins P, Zaninovic V, Sandoval D, Rutherford R, Fitzsimmons T, Pandol SJ, Poucell-Hatton S. Mechanisms of cell death after pancreatic duct obstruction in the opossum and the rat. Gastroenterology. 1996;vol. 110(no. 3):875–884. doi: 10.1053/gast.1996.v110.pm8608898. [DOI] [PubMed] [Google Scholar]
- Gukovskaya AS, Vaquero E, Zaninovic V, Gorelick FS, Lusis AJ, Brennan ML, Holland S, Pandol SJ. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology. 2002b;vol. 122(no. 4):974–984. doi: 10.1053/gast.2002.32409. [DOI] [PubMed] [Google Scholar]
- Gukovsky I, Gukovskaya AS. Impaired autophagy underlies key pathological responses of acute pancreatitis. Autophagy. 2010;vol. 6(no. 3):428–429. doi: 10.4161/auto.6.3.11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gukovsky I, Lugea A, Shahsahebi M, Cheng JH, Hong PP, Jung YJ, Deng QG, French BA, Lungo W, French SW, Tsukamoto H, Pandol SJ. A rat model reproducing key pathological responses of alcoholic chronic pancreatitis. Am.J.Physiol Gastrointest.Liver Physiol. 2008;vol. 294(no. 1):G68–G79. doi: 10.1152/ajpgi.00006.2007. [DOI] [PubMed] [Google Scholar]
- Gukovsky I, Reyes CN, Vaquero EC, Gukovskaya AS, Pandol SJ. Curcumin ameliorates ethanol and nonethanol experimental pancreatitis. Am.J.Physiol Gastrointest.Liver Physiol. 2003;vol. 284(no. 1):G85–G95. doi: 10.1152/ajpgi.00138.2002. [DOI] [PubMed] [Google Scholar]
- Haber PS, Apte MV, Applegate TL, Norton ID, Korsten MA, Pirola RC, Wilson JS. Metabolism of ethanol by rat pancreatic acinar cells. J.Lab Clin.Med. 1998;vol. 132(no. 4):294–302. doi: 10.1016/s0022-2143(98)90042-7. [DOI] [PubMed] [Google Scholar]
- Haber PS, Apte MV, Moran C, Applegate TL, Pirola RC, Korsten MA, McCaughan GW, Wilson JS. Non-oxidative metabolism of ethanol by rat pancreatic acini. Pancreatology. 2004;vol. 4(no. 2):82–89. doi: 10.1159/000077608. [DOI] [PubMed] [Google Scholar]
- Haber PS, Keogh GW, Apte MV, Moran CS, Stewart NL, Crawford DH, Pirola RC, McCaughan GW, Ramm GA, Wilson JS. Activation of pancreatic stellate cells in human and experimental pancreatic fibrosis. Am.J.Pathol. 1999;vol. 155(no. 4):1087–1095. doi: 10.1016/S0002-9440(10)65211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Wang YL, Edderkaoui M, Lugea A, Apte MV, Pandol SJ. Ethanol augments PDGF-induced NADPH oxidase activity and proliferation in rat pancreatic stellate cells. Pancreatology. 2007;vol. 7(no. 4):332–340. doi: 10.1159/000105499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji B, Gaiser S, Chen X, Ernst SA, Logsdon CD. Intracellular trypsin induces pancreatic acinar cell death but not NF-kappaB activation. J.Biol.Chem. 2009;vol. 284(no. 26):17488–17498. doi: 10.1074/jbc.M109.005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser AM, Saluja AK, Sengupta A, Saluja M, Steer ML. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am.J.Physiol. 1995;vol. 269(no. 5 Pt 1):C1295–C1304. doi: 10.1152/ajpcell.1995.269.5.C1295. [DOI] [PubMed] [Google Scholar]
- Katz M, Carangelo R, Miller LJ, Gorelick F. Effect of ethanol on cholecystokinin-stimulated zymogen conversion in pancreatic acinar cells. Am.J.Physiol. 1996;vol. 270(no. 1 Pt 1):G171–G175. doi: 10.1152/ajpgi.1996.270.1.G171. [DOI] [PubMed] [Google Scholar]
- Kubisch CH, Logsdon CD. Endoplasmic reticulum stress and the pancreatic acinar cell. Expert.Rev Gastroenterol.Hepatol. 2008;vol. 2(no. 2):249–260. doi: 10.1586/17474124.2.2.249. [DOI] [PubMed] [Google Scholar]
- Lankisch PG, Assmus C, Maisonneuve P, Lowenfels AB. Epidemiology of pancreatic diseases in Luneburg County. A study in a defined German population. Pancreatology. 2002;vol. 2(no. 5):469–477. doi: 10.1159/000064713. [DOI] [PubMed] [Google Scholar]
- Laposata EA, Lange LG. Presence of nonoxidative ethanol metabolism in human organs commonly damaged by ethanol abuse. Science. 1986;vol. 231(no. 4737):497–499. doi: 10.1126/science.3941913. [DOI] [PubMed] [Google Scholar]
- Lee AH, Chu GC, Iwakoshi NN, Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005;vol. 24(no. 24):4368–4380. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;vol. 132(no. 1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Tamakoshi A, Matsuno S, Takeda K, Hayakawa T, Kitagawa M, Naruse S, Kawamura T, Wakai K, Aoki R, Kojima M, Ohno Y. Nationwide epidemiological survey of chronic pancreatitis in Japan. J.Gastroenterol. 2000;vol. 35(no. 2):136–141. doi: 10.1007/s005350050026. [DOI] [PubMed] [Google Scholar]
- Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andren-Sandberg A, Domellof L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N.Engl.J.Med. 1993;vol. 328(no. 20):1433–1437. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- Lowenfels AB, Maisonneuve P, Grover H, Gerber E, Korsten MA, Antunes MT, Marques A, Pitchumoni CS. Racial factors and the risk of chronic pancreatitis. Am.J.Gastroenterol. 1999;vol. 94(no. 3):790–794. doi: 10.1111/j.1572-0241.1999.00952.x. [DOI] [PubMed] [Google Scholar]
- Lu Z, Karne S, Kolodecik T, Gorelick FS. Alcohols enhance caerulein-induced zymogen activation in pancreatic acinar cells. Am.J.Physiol Gastrointest.Liver Physiol. 2002;vol. 282(no. 3):G501–G507. doi: 10.1152/ajpgi.00388.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugea A, Gong J, Nguyen J, Nieto J, French SW, Pandol SJ. Cholinergic mediation of alcohol-induced experimental pancreatitis. Alcohol Clin.Exp. Res. 2010 doi: 10.1111/j.1530-0277.2010.01264.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugea A, Gukovsky I, French SW, Gorelick FS, Pandol SJ. Role of XBP1 in the protective unfolded protein response to limit chronic ethanol-induced endoplasmic reticulum stress and damage in the pancreas. Gastroenterology. 2009;vol. 136 Suppl. 1:A-589. (Abstract) [Google Scholar]
- Lugea A, Gukovsky I, Gukovskaya AS, Pandol SJ. Nonoxidative ethanol metabolites alter extracellular matrix protein content in rat pancreas. Gastroenterology. 2003;vol. 125(no. 6):1845–1859. doi: 10.1053/j.gastro.2003.09.021. [DOI] [PubMed] [Google Scholar]
- Lugea A, Nan L, French SW, Bezerra JA, Gukovskaya AS, Pandol SJ. Pancreas recovery following cerulein-induced pancreatitis is impaired in plasminogen-deficient mice. Gastroenterology. 2006;vol. 131(no. 3):885–899. doi: 10.1053/j.gastro.2006.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugea A, Wu X, Dasari S, Pandol SJ. Plasminogen activator inhibitor-1 mediates fibrogenesis after cerulein-induced pancreatitis. Gastroenterology. 2008;vol. 134 Suppl.1:A-585. (Abstract) [Google Scholar]
- Maisonneuve P, Lowenfels AB. Chronic pancreatitis and pancreatic cancer. Dig.Dis. 2002;vol. 20(no. 1):32–37. doi: 10.1159/000063165. [DOI] [PubMed] [Google Scholar]
- Maisonneuve P, Lowenfels AB, Mullhaupt B, Cavallini G, Lankisch PG, Andersen JR, Dimagno EP, Andren-Sandberg A, Domellof L, Frulloni L, Ammann RW. Cigarette smoking accelerates progression of alcoholic chronic pancreatitis. Gut. 2005;vol. 54(no. 4):510–514. doi: 10.1136/gut.2004.039263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareninova OA, Yakubov I, French SW, Jia W, Lee MA, Pandol SJ, Gukovskaya AS, Gukovsky I. Ethanol feeding causes lysosomal dysfunction in exocrine pancreas similar to pancreatitis; but in contrast to pancreatitis, ethanol down-regulates autophagy. Gastroenterology. 2010:138. (Abstract) [Google Scholar]
- Mareninova OA, Hermann K, French SW, O'Konski MS, Pandol SJ, Webster P, Erickson AH, Katunuma N, Gorelick FS, Gukovsky I, Gukovskaya AS. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J.Clin.Invest. 2009a;vol. 119(no. 11):3340–3355. doi: 10.1172/JCI38674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareninova OA, Odinokova IV, Lee MA, Gukovsky I, Gukovskaya AS. Role of mitochondrial permeability transition pore in pancreatitis. Gastroenterology. 2009b;vol. 136 Suppl. 1:A-60. (Abstract) [Google Scholar]
- Mareninova OA, Sung KF, Hong P, Lugea A, Pandol SJ, Gukovsky I, Gukovskaya AS. Cell death in pancreatitis: caspases protect from necrotizing pancreatitis. J.Biol.Chem. 2006;vol. 281(no. 6):3370–3381. doi: 10.1074/jbc.M511276200. [DOI] [PubMed] [Google Scholar]
- Masamune A, Kikuta K, Satoh M, Satoh A, Shimosegawa T. Alcohol activates activator protein-1 and mitogen-activated protein kinases in rat pancreatic stellate cells. J.Pharmacol.Exp.Ther. 2002;vol. 302(no. 1):36–42. doi: 10.1124/jpet.302.1.36. [DOI] [PubMed] [Google Scholar]
- Mee L, Sekar VT, Ashokkumar B, Pandol SJ, Said H. Mechanism and regulation of folate uptake by pancreatic acinar cells: Effect of chronic alcohol consumption. Am.J.Physiol Gastrointest.Liver Physiol. 2010 doi: 10.1152/ajpgi.00068.2010. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton C, Klatsky AL, Udaltsova N. Smoking, coffee, and pancreatitis. Am.J.Gastroenterol. 2004;vol. 99(no. 4):731–738. doi: 10.1111/j.1572-0241.2004.04143.x. [DOI] [PubMed] [Google Scholar]
- Mukherjee R, Criddle DN, Gukovskaya A, Pandol S, Petersen OH, Sutton R. Mitochondrial injury in pancreatitis. Cell Calcium. 2008;vol. 44(no. 1):14–23. doi: 10.1016/j.ceca.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman J. The role of cytokines in the pathogenesis of acute pancreatitis. Am.J.Surg. 1998;vol. 175(no. 1):76–83. doi: 10.1016/s0002-9610(97)00240-7. [DOI] [PubMed] [Google Scholar]
- Odinokova IV, Sung KF, Mareninova OA, Hermann K, Evtodienko Y, Andreyev A, Gukovsky I, Gukovskaya AS. Mechanisms regulating cytochrome c release in pancreatic mitochondria. Gut. 2009;vol. 58(no. 3):431–442. doi: 10.1136/gut.2007.147207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odinokova IV, Sung KF, Mareninova OA, Hermann K, Gukovsky I, Gukovskaya AS. Mitochondrial mechanisms of death responses in pancreatitis. J Gastroenterol.Hepatol. 2008;vol. 23 Suppl 1:S25–S30. doi: 10.1111/j.1440-1746.2007.05271.x. [DOI] [PubMed] [Google Scholar]
- Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J.Clin.Invest. 2007;vol. 117(no. 1):50–59. doi: 10.1172/JCI30082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oruc N, Whitcomb DC. Theories, mechanisms, and models of alcoholic chronic pancreatitis. Gastroenterol.Clin.North Am. 2004;vol. 33(no. 4):733–7vi. doi: 10.1016/j.gtc.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Pandol SJ, Periskic S, Gukovsky I, Zaninovic V, Jung Y, Zong Y, Solomon TE, Gukovskaya AS, Tsukamoto H. Ethanol diet increases the sensitivity of rats to pancreatitis induced by cholecystokinin octapeptide. Gastroenterology. 1999;vol. 117(no. 3):706–716. doi: 10.1016/s0016-5085(99)70465-8. [DOI] [PubMed] [Google Scholar]
- Pandol SJ, Saluja AK, Imrie CW, Banks PA. Acute pancreatitis: bench to the bedside. Gastroenterology. 2007;vol. 132(no. 3):1127–1151. doi: 10.1053/j.gastro.2007.01.055. erratum: ibid, vol. 133, p. 1056. [DOI] [PubMed] [Google Scholar]
- Papachristou GI, Papachristou DJ, Morinville VD, Slivka A, Whitcomb DC. Chronic alcohol consumption is a major risk factor for pancreatic necrosis in acute pancreatitis. Am.J Gastroenterol. 2006;vol. 101(no. 11):2605–2610. doi: 10.1111/j.1572-0241.2006.00795.x. [DOI] [PubMed] [Google Scholar]
- Pfutzer RH, Tadic SD, Li HS, Thompson BS, Zhang JY, Ford ME, Eagon PK, Whitcomb DC. Pancreatic cholesterol esterase, ES-10, and fatty acid ethyl ester synthase III gene expression are increased in the pancreas and liver but not in the brain or heart with long-term ethanol feeding in rats. Pancreas. 2002;vol. 25(no. 1):101–106. doi: 10.1097/00006676-200207000-00021. [DOI] [PubMed] [Google Scholar]
- Phillips PA, Wu MJ, Kumar RK, Doherty E, McCarroll JA, Park S, Pirola RC, Wilson JS, Apte MV. Cell migration: a novel aspect of pancreatic stellate cell biology. Gut. 2003;vol. 52(no. 5):677–682. doi: 10.1136/gut.52.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat.Rev Mol.Cell Biol. 2007;vol. 8(no. 7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Saluja AK, Lerch MM, Phillips PA, Dudeja V. Why does pancreatic overstimulation cause pancreatitis? Annu.Rev Physiol. 2007;vol. 69:249–269. doi: 10.1146/annurev.physiol.69.031905.161253. [DOI] [PubMed] [Google Scholar]
- Sand J, Nordback I. Acute pancreatitis: risk of recurrence and late consequences of the disease. Nat.Rev Gastroenterol.Hepatol. 2009;vol. 6(no. 8):470–477. doi: 10.1038/nrgastro.2009.106. [DOI] [PubMed] [Google Scholar]
- Sandoval D, Gukovskaya A, Reavey P, Gukovsky S, Sisk A, Braquet P, Pandol SJ, Poucell-Hatton S. The role of neutrophils and platelet-activating factor in mediating experimental pancreatitis. Gastroenterology. 1996;vol. 111(no. 4):1081–1091. doi: 10.1016/s0016-5085(96)70077-x. [DOI] [PubMed] [Google Scholar]
- Satoh A, Gukovskaya AS, Reeve JR, Jr, Shimosegawa T, Pandol SJ. Ethanol sensitizes NF-kappaB activation in pancreatic acinar cells through effects on protein kinase C-epsilon. Am.J.Physiol Gastrointest.Liver Physiol. 2006;vol. 291(no. 3):G432–G438. doi: 10.1152/ajpgi.00579.2005. [DOI] [PubMed] [Google Scholar]
- Schenker S, Montalvo R. Alcohol and the pancreas. Recent Dev.Alcohol. 1998;vol. 14:41–65. doi: 10.1007/0-306-47148-5_3. [DOI] [PubMed] [Google Scholar]
- Shalbueva N, Mareninova OA, Yuan JZ, Pandol SJ, Gukovskaya AS. Alcohol depolarizes pancreatic mitochondria through PTP-dependent mechanism. Pancreas. 2009;vol. 38(no. 8):1046. (Abstract) [Google Scholar]
- Shek FW, Benyon RC, Walker FM, McCrudden PR, Pender SL, Williams EJ, Johnson PA, Johnson CD, Bateman AC, Fine DR, Iredale JP. Expression of transforming growth factor-beta 1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am.J Pathol. 2002;vol. 160(no. 5):1787–1798. doi: 10.1016/s0002-9440(10)61125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer MV. Effect of ethanol and alcoholic beverages on the gastrointestinal tract in humans. Rom.J.Gastroenterol. 2002;vol. 11(no. 3):197–204. [PubMed] [Google Scholar]
- Strate T, Yekebas E, Knoefel WT, Bloechle C, Izbicki JR. Pathogenesis and the natural course of chronic pancreatitis. Eur.J.Gastroenterol. Hepatol. 2002;vol. 14(no. 9):929–934. doi: 10.1097/00042737-200209000-00002. [DOI] [PubMed] [Google Scholar]
- Sung KF, Odinokova IV, Mareninova OA, Rakonczay Z, Jr, Hegyi P, Pandol SJ, Gukovsky I, Gukovskaya AS. Prosurvival Bcl-2 proteins stabilize pancreatic mitochondria and protect against necrosis in experimental pancreatitis. Exp.Cell Res. 2009;vol. 315(no. 11):1975–1989. doi: 10.1016/j.yexcr.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao N, Sussman S, Nieto J, Tsukamoto H, Yuan JM. Demographic characteristics of hospitalized patients with alcoholic liver disease and pancreatitis in Los Angeles county. Alcohol Clin.Exp.Res. 2003;vol. 27(no. 11):1798–1804. doi: 10.1097/01.ALC.0000095862.30777.D9. [DOI] [PubMed] [Google Scholar]
- Wang YL, Hu R, Lugea A, Gukovsky I, Smoot D, Gukovskaya AS, Pandol SJ. Ethanol feeding alters death signaling in the pancreas. Pancreas. 2006;vol. 32(no. 4):351–359. doi: 10.1097/01.mpa.0000220859.93496.e1. [DOI] [PubMed] [Google Scholar]
- Waterford SD, Kolodecik TR, Thrower EC, Gorelick FS. Vacuolar ATPase regulates zymogen activation in pancreatic acini. J.Biol.Chem. 2005;vol. 280(no. 7):5430–5434. doi: 10.1074/jbc.M413513200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav D, Hawes RH, Brand RE, Anderson MA, Money ME, Banks PA, Bishop MD, Baillie J, Sherman S, DiSario J, Burton FR, Gardner TB, Amann ST, Gelrud A, Lawrence C, Elinoff B, Greer JB, O'Connell M, Barmada MM, Slivka A, Whitcomb DC. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch.Intern.Med. 2009;vol. 169(no. 11):1035–1045. doi: 10.1001/archinternmed.2009.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav D, Whitcomb DC. The role of alcohol and smoking in pancreatitis. Nat.Rev.Gastroenterol.Hepatol. 2010;vol. 7(no. 3):131–145. doi: 10.1038/nrgastro.2010.6. [DOI] [PubMed] [Google Scholar]
- Yuan JZ, Liu Y, Shalbueva N, Tan T, Gukovskaya AS, Pandol SJ. Protein kinase C epsilon mediates ethanol and carbachol-induced depolarization in pancreatic acinar cells. Pancreas. 2009;vol. 38(no. 8):1066. (Abstract) [Google Scholar]
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;vol. 454(no. 7203):455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]