

Abstract
Inbreeding coefficients and consanguineous mating types are usually inferred from population surveys or pedigree studies. Here, we present a method to estimate them from dense genome-wide single-nucleotide polymorphism genotypes and apply it to 940 unrelated individuals from the Human Genome Diversity Panel (HGDP-CEPH). Inbreeding is observed in almost all populations of the panel, and the highest inbreeding levels and frequencies of inbred individuals are found in populations of the Middle East, Central South Asia and the Americas. In these regions, first cousin (1C) marriages are the most frequent, but we also observed marriages between double first cousins (2 × 1C) and between avuncular (AV) pairs. Interestingly, if 2 × 1C marriages are preferred to AV marriages in Central South Asia and the Middle East, the contrary is found in the Americas. There are thus some regional trends but there are also some important differences between populations within a region. Individual results can be found on the CEPH website at ftp://ftp.cephb.fr/hgdp_hbd/.
Keywords: genome-based IBD, inbreeding, homozygosity by descent, mating-type inference, HGDP, linkage disequilibrium
Introduction
In many human populations, mating between relatives is relatively frequent and encouraged for social and/or economic reasons. Measuring inbreeding levels around the world has been the subject of many studies dating back to the 1950s (reviewed in Bittles and Black1). Estimates of these levels were often obtained by determining the prevalence in populations of different types of reported marriages between relatives (usually up to second cousins (2C)). When pedigree information is available, it is possible to evaluate individual inbreeding coefficients by counting the number of meioses in the different inbreeding loops. However, these estimates are dependent on the accuracy of genealogy data and can be quite unreliable.
With the availability of dense, genome-wide marker maps it has become possible to estimate individual inbreeding coefficients by inferring from observed homozygosity the proportion of the individual genome that is identical by descent (IBD) or equivalently autozygous. We will refer hereafter to homozygosity due to IBD as homozygosity by descent (HBD). This approach provides a genome-based alternative to genealogy2, 3, 4 and has been used so far in homozygosity mapping studies5, 6, 7 or to study levels of inbreeding in isolated human populations.8, 9 Here, we propose to use the approach in population surveys to infer mating-type habits.
To do so, we have extended the FEstim method,2 which provides more reliable inbreeding coefficient estimates than other available methods.9 We propose to estimate by a maximum-likelihood method the proportion of some specific mating types (first cousin (1C), double first cousin (2 × 1C), avuncular (AV), 2C etc) on the basis of the distribution of HBD segments over the genome of individuals from the studied populations. Indeed, the number and length of HBD segments in an individual genome depend on the relatedness of his/her parents and can thus be used to assess parental mating-type preferences. FEstim requires that the markers in the map be in minimal linkage disequilibrium (LD), as, otherwise, inflation in inbreeding estimates has been demonstrated.9 To avoid this bias, we developed an original strategy consisting of generating multiple sparse genome maps. This strategy has the advantage of not requiring any LD computation on the sample and of minimizing loss of information, as compared with a strategy that consists of using a single map of markers in minimal LD (as can be carried out with PLINK4 or MASEL,10 for instance).
Materials and methods
The HGDP-CEPH panel
We applied the method to the individuals from the Human Genome Diversity Panel (HGDP-CEPH)11 sampled from 52 populations from seven geographic regions in all inhabited continents. The panel is managed and maintained at the Fondation Jean Dausset-CEPH. Genotypes for 6 44 258 (Illumina650Y) autosomal single-nucleotide polymorphisms (SNPs)12 are available for 1043 individuals (available on the HGDP-CEPH web page). This group of individuals contain first- and second-degree relative pairs.13 After excluding one member of each relative pair, 940 HGDP-CEPH individuals could be used for this study (details in Supplementary Table 1). SNPs that had less than 95% genotype calls (1344 SNPs) and were monomorphic across all populations (51 SNPs) on the 940 individuals were removed, leaving 642 863 SNPs for the analysis. Finally, one Tujia individual (HGDP01097) was removed because almost all his chromosome 1 genotypes were found to be homozygous. This was confirmed by microsatellite and other SNP genome scan data available in the HGDP-CEPH genome database and is presumably a cell-line artifact.
Minimal LD map
To produce a sparse map with minimal LD, an SNP was randomly chosen on each chromosome, and subsequent SNPs were then selected every 0.5 cM in both directions from the initial marker. To avoid the systematic selection of the same SNPs after a gap (intermarker distance >0.5 cM), a random SNP was selected beyond the gap, and the map-building process was continued. This process was repeated to produce M maps. The genetic distances used here are the ones provided by Illumina and are based on the deCODE map.14
We generated 100 sparse maps that each contained about 6500 SNPs (∼1% of the original markers). This is similar to the number of SNPs present in the Illumina Linkage-12 panel (but note that we could not use the SNPs from the Illumina Linkage panel for comparison, as most of them are not included in the Illumina650Y chip). The 100 maps captured 34% of the markers from the original map of 642 863 SNPs (only one SNP located between two gaps on chr8 was common to all the maps). When gaps were not treated as described above (41 gaps were found), the different maps had much more overlap and only 11% of the markers from the original map could be captured (data not shown).
Genomic inbreeding coefficient estimation
FEstim2 is a maximum likelihood method that uses a hidden Markov chain to model the dependencies along the genome between the (observed) marker genotypes and the (unobserved) HBD status. In addition to the inbreeding coefficient, a parameter A is estimated, where AF is the instantaneous rate of change per unit map length (here cM) from no HBD to HBD. Both HBD and non-HBD segment lengths are assumed to be distributed exponentially with mean lengths 1/(A(1−F)) and 1/(AF), respectively. The inbreeding coefficient Fm and parameter Am were estimated for each map m=1 to M. The median values over the M maps were reported as F and A, respectively, for each individual.
To test whether F was significantly different from zero, we performed a likelihood-ratio test contrasting the maximum likelihood and the likelihood of being outbred. P-values (based on a χ2 test with two degrees of freedom) were obtained for each map and the median values over the M maps were reported.
Marker allele frequencies, required to estimate F, were determined separately for each of the 52 populations. They provided slightly more accurate F estimates than allele frequencies determined at the regional level (seven geographic regions). This is especially true for sub-Saharan Africa and the Americas (Supplementary Figure 1), where populations are more differentiated from each other than in other regions.
A few individuals had extreme A value estimates (much larger than 1 on most of the 100 maps) and were removed from subsequent analyses. Values of A must be strictly positive and are usually observed to be <1. Values of A>1 are possible, but would mean that the average HBD segment length is <1 cM, which is unlikely to be detected with an SNP density of 1 per 0.5 cM. Two individuals (one Bedouin HGDP00621 and one Mozabite HGDP01270) had A>1. Interestingly, we found from principal component analysis (data not shown) that these two individuals (probably the same individuals found by Jakobsson et al15 in their analysis) were closer to the sub-Sahara African than to the Northern-African–Middle-East populations, to which they were supposed to belong. It is thus likely that F and A were not correctly estimated for these two individuals because of undetected levels of admixture.
As the sparse genome maps used here were based on intermarker distances of about 0.5 cM, we checked the data for deletions greater than 0.5 Mb that might interfere with our inbreeding estimations by artificially increasing the estimates. Itsara et al16 reported CNV calls for the HGDP-CEPH samples based on rigorous analysis of SNP intensity data and direct validation with CGH oligonucleotide arrays tested on a small subsample. Only three individuals had a deletion larger than 0.5 Mb, 3.2 Mb for HGDP00894, 1.2 Mb for HGDP01156 and 1.6 Mb for HGDP00780. These individuals had estimates of F=0. Therefore, it is unlikely that large deletions have interfered with our results.
To test the significance of the differences of the genomic inbreeding estimates (F) among populations and among geographical regions, we performed a variance components analysis. Two linear-mixed models for F were compared: one including region and population information as random effects, and the other including region information only. We used the lme function from the nlme package (version 3.1-96) in R software (version 2.10.1) and, to deal with non-normality of F, we used log(F) for values of F>0, and log(T), where T is one-half of the lowest non-zero F value, for F=0. The Akaike information criterion (AIC)17 as implemented in the nlme package was used to select the best model.
Inference of population mating types (α) and individual parental mating types (P)
We assume that a population is a mixture of offspring from several mating types. Here, we considered four different consanguineous mating type groups: 2C, 1C, AV or 2 × 1C matings. We want to classify the individuals into each mating type group to estimate the proportions of the (parental) mating types in the population. Note that an individual is not usually classified into a single mating-type group, but rather has a probability for each group. The population proportion of a mating type can then be thought of as the sum of the individual probabilities for this mating type. To estimate the proportion of parental mating k for a given map M (αk,m), the following likelihood was maximized in each population (of size n):
 |
where Lk,m(i) is the likelihood of individual i being the offspring of mating type k and L0,m(i) is the likelihood for individual i to have unrelated parents (that is, individual i is outbred). We computed each likelihood Lk,m(i) as in Leutenegger et al,2 but instead of estimating F and A, we used fixed values calculated from the genealogy of the mating type. For the genealogy-based inbreeding coefficient, we used the usual Wright's path counting method.18 For the genealogy-based value of A, we used simulations as in Leutenegger et al2 The fixed values were as follows: for k=2C, (F, A)=(0.015625, 0.080); for 1C, (F, A)=(0.0625, 0.063); for AV, (F, A)=(0.125, 0.057); for 2 × 1C, (F, A)=(0.125, 0.068). Maximization of (1) was performed with ConstrOptim function from the stats package (version 2.10.1) in R software (version 2.10.1) with multiple starting points to avoid local maxima.
For each individual i, we used Bayes formula to determine the posterior probability Lk,m(i) of mating type 0:
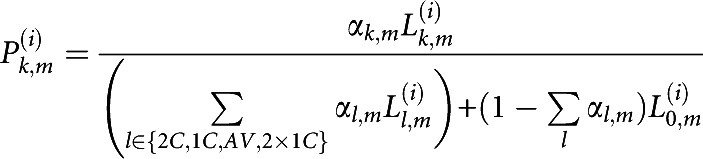 |
The median of αk,m and Pk,m(i) over the M maps (noted αk and Pk(i), respectively) were considered and plotted using Distruct software.19
Simulation study to validate the mating-type inference
Genotype data at 5000 SNPs (corresponding roughly to the number of SNPs in one sparse map) were simulated over the genome for 2C, 1C, AV and 2 × 1C offspring using the Genedrop program of MORGAN2.8 (available from Pangaea website). We also generated genotype data over the genome for outbred individuals. For each of these mating types, we performed 1000 replicates of a population of 20 individuals (equivalent to an average-sized population in the HGDP-CEPH panel). At each replicate, we estimated α and P as presented above. In addition, we estimated the true values of (F, A) from the true HBD data (accessible through the haplotype labels of the founder individuals of the genealogy).
Results
From the simulation study, we found that when individuals were 1C, 2C and outbred offspring, the mating types were usually correctly inferred (average proportion of individuals correctly inferred over replicates (95% variation interval)): α1C=0.94 (0.75; 1), α2C=0.94 (0.73; 1) and α0=1 (0.98; 1), respectively. In the case of 2 × 1C and AV, these numbers were 0.67 (0.40; 1) and 0.78 (0.39; 1), respectively. Incorrect inferences in these two latter cases were most often from 2 × 1C offspring to AV offspring and vice versa, as could be expected from the fact that they have the same genealogy-based inbreeding coefficient (0.125), but different distributions of the HBD regions along their genomes: offspring of 2 × 1C matings tend to have multiple, shorter HBD segments compared with AV mating offspring who have longer HBD segments. This is well illustrated in Supplementary Figure 2A. The true inbreeding coefficient is plotted against the true mean HBD segment length, with the ellipses representing the boundaries containing 95% of replicates for each mating type. In Supplementary Figure 2B, we plotted each simulated 2 × 1C offspring. Whenever the posterior probability of a mating type was higher than 0.7, the individual was considered as an offspring of this mating type and coloured accordingly. One can see that the simulated 2 × 1C offspring who are inferred as AV offspring (green dots) do resemble AV offspring (dashed line ellipse) more than 2 × 1C offspring (dotted line ellipse). The reverse can be observed for simulated AV offspring in Supplementary Figure 2C.
Overall, in the sample, 36% of the individuals (Figure 1) have an estimated inbreeding coefficient F significantly different from 0. These inbred individuals are found in all geographical regions, but the most inbred individuals are from the Americas, the Middle East and Central South Asia (details in Supplementary Table 2). F estimates show significant differences at the regional and population level, with the model including the population level providing a better fit to the data (AIC difference=66.8, Table 1). Indeed, within-population differences account for most of the variability of F (63%).
Figure 1.

F estimates for each individual by population sample and geographical region. Closed circles represent the median values over 100 (LD minimal) maps. Dots represent Fm estimates for each map m.
Table 1. Variance components (%) of the inbreeding coefficient estimates F.
| Between | Within regions | AIC | ||
|---|---|---|---|---|
| regions | Between populations | Within populations | ||
| Region+population | 26 | 11 | 63 | 3362.659 |
| Region | 26 | 74 | 3429.419 | |
Abbreviation: AIC, Akaike information criterion.
To illustrate the advantage of the proposed strategy of using several sparse maps, we show in Figure 1 the variability of the inbreeding estimates across all maps: nearly a quarter of the individuals with a median F at 0 have at least one map-specific Fm of 0.015 (expected for 2C offspring) or higher.
We then continue to characterize the nature of the inbreeding within each population by inferring the mating-type habits. We found that for 23% of the individuals from the sample the inferred parental mating type (posterior probability ≥0.7) was 2C, for 9% it was 1C, for 3% 2 × 1C and for 0.2% for AV. Finally, 55% of the individuals were inferred as offspring of unrelated parents. Note that there is a difference between this number and the number of individuals with an F not significantly different from 0 who were found to represent 64% of the sample. This could be related to the fact that, when inferring mating-type preferences, we take into account the population context contrary to what is done when testing to determine whether F is different from 0. An individual from a population in which most of the individuals are offspring of consanguineous matings is then given a high prior probability of being inbred; thus, even if his F is very close to 0, he might still be inferred as inbred.
Of interest is the difference in the distribution of consanguineous marriages. In Figure 2a and Supplementary Table 3, the inference of the most likely parental mating type (αk) is presented per population grouped by region. The most likely mating types are very different among populations, and in some instances between regions, for example, Middle East, Central South Asia and the Americas versus sub-Saharan Africa, Europe and East Asia. In the regions showing the highest inbreeding levels (Middle East, Central South Asia and the Americas), 2 × 1C matings were more frequent in Central South Asia and the Middle East than AV matings, whereas the contrary was usually observed in the Americas. The Surui, Pima, Karitiana and Kalash populations stand out from the others, as all or almost all individuals in these populations are found to have related parents.
Figure 2.

Inference of mating-type preferences. (a) Population mating-type frequencies α. (b) Parental mating-type probabilities P for each individual within the population. Matings between unrelated individuals are in white; second cousin, first cousin, double first cousin and avuncular matings are in increasing shades of grey.
In Figure 2b, one can see the parental mating-type posterior probabilities for each individual. For about 63% of the individuals, the data clearly point to one mating type (Pk(i)≥0.95). For the remaining individuals, the picture is a mixture of at most three mating types. This occurs mostly in Middle-Eastern, Central South-Asian and American individuals.
Discussion
Relying on genomic data, we have estimated the inbreeding levels and mating-type proportions in 52 populations from all inhabited continents. We found that consanguinity was present in almost all populations.
A global overview of consanguinity was recently published by Bittles and Black1 based on self-reported information: household surveys, obstetric inpatients and pedigree information. Compared with this study, we found the same general trend with high rates of inbreeding in North Africa, the Middle East and Central South Asia.
They estimated a worldwide rate of marriages between 2Cs or closer of 10.4%. It might be difficult to compare the Bittles and Black worldwide estimates with those of the HGDP-CEPH panel, which does not include the same populations and, in many cases, is concerned with more isolated ancestral populations. Moreover, consanguinity is found to be very different between populations, and discussing it globally is probably less interesting than focusing on specific regional and population patterns.
Even for the few populations in common between the Bittles and Black study and the HGDP-CEPH panel, the results are different. The reason might be the differing sampling locations and times. This is well illustrated by the Yoruba of Nigeria for whom Bittles and Black found 51% of consanguineous marriages in a rural sampling location in 1974 (reviewed in Scott-Emuakpor20), as compared to our 6% of consanguineous matings in an urban area in the 1990s (see population description on ALFRED website). In the case of the Bantu from South Africa, where we estimate 22% of consanguineous marriages and Bittles and Black report 6%, another explanation could be the self-reported consanguinity in the latter that is less likely to account for remote relationships than our genome-based method, which is not limited by the available genealogical depth.
Among the HGDP-CEPH panel, populations not considered by Bittles and Black are the Surui and the Karitiana, both isolated and endogamous groups in Brazil, for whom we estimated that nearly all individuals are consanguineous. This can be explained by the small population sizes from which the individuals were sampled and the history of peopling of the Americas. The Surui individuals were sampled from a single village of 85 inhabitants in the 1980s (reviewed in Calafell et al21). The Karitiana individuals, also sampled in the 1980s, come from a population numbering less than 150 people and comprise essentially one family in a single village.22 In addition, the isolated Amazonian populations have been shown to have the lowest genetic diversity worldwide, likely because of serial founder effects along the colonization routes of the Americas.23
We found that our genome-based estimate was highly variable (see Figure 1 and ellipses in Supplementary Figure 2). Hence, it can be seen as unreliable depending on the goal of the study. The genome-based and genealogy-based approaches are in fact complementary. The former is probably best for homozygosity mapping and genetic studies and the latter for anthropological studies. Thus, when studying a population, it would be most informative to have both types of information.
Previous studies9, 24, 25 have highlighted the risk of overestimating inbreeding coefficients when the studied markers are in LD. The strategy developed here, which consists of generating several sparse maps, seems to be a reliable strategy that avoids losing as much information as when a single sparse map is considered. Recently, Browning and Browning26 proposed a new method for HBD detection that incorporates a comprehensive LD model. The method was developed for the study of outbred individuals of Northern European ancestry and does not allow estimating F. However, if the method were modified to estimate F, then it would be interesting to apply it to the HGDP-CEPH panel where consanguinity and diverse populations are present.
Inferring relationships from the genome has been used to check the relationship between two individuals with the goal of data cleaning or genealogy reconstruction.27, 28, 29 To our knowledge, this is the first study in which genome-wide genotypes of (singleton) individuals are used to assess mating-type preferences. The method we proposed can be used with any population-based sample genotyped with SNP chips to infer the frequency of consanguineous marriages in the population and estimate, for each individual, the probability of being inbred. No genealogical information is required, thus avoiding self-reported data or detailed pedigree studies. This method and the results obtained on the HGDP-CEPH panel will be useful in disease studies to evaluate the impact of consanguinity but also more generally to describe marriage patterns in human populations.
Acknowledgments
We thank Jean Maccario for helpful discussions on variance components and two anonymous reviewers for their constructive comments. SG is funded by the plateforme de génomique constitutionnelle (Faculté de médecine, Univ Paris-Diderot, Paris, France).
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Web resources
Data availability on the HGDP-CEPH webpage: http://www.cephb.fr/en/hgdp Consanguinity/Endogamy Resource: http://www.consang.net/index.php/Main_Page
ALFRED website: http://alfred.med.yale.edu/alfred/entity.asp?condition=populations Pangaea website: http://www.stat.washington.edu/thompson/Genepi/pangaea.shtml FEstim Software: on request from anne-louise.leutenegger@inserm.fr
Supplementary Material
References
- Bittles AH, Black ML. Evolution in health and medicine sackler colloquium: consanguinity, human evolution, and complex diseases. Proc Natl Acad Sci USA. 2009;107 (Suppl 1:1779–1786. doi: 10.1073/pnas.0906079106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leutenegger AL, Prum B, Genin E, et al. Estimation of the inbreeding coefficient through use of genomic data. Am J Hum Genet. 2003;73:516–523. doi: 10.1086/378207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carothers AD, Rudan I, Kolcic I, et al. Estimating human inbreeding coefficients: comparison of genealogical and marker heterozygosity approaches. Ann Hum Genet. 2006;70:666–676. doi: 10.1111/j.1469-1809.2006.00263.x. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leutenegger AL, Labalme A, Genin E, et al. Using genomic inbreeding coefficient estimates for homozygosity mapping of rare recessive traits: application to Taybi-Linder syndrome. Am J Hum Genet. 2006;79:62–66. doi: 10.1086/504640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Haynes C, Barany F, Ott J. Genome-wide autozygosity mapping in human populations. Genet Epidemiol. 2009;33:172–180. doi: 10.1002/gepi.20344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis D, Vine AE, Knight J. Study of regions of extended homozygosity provides a powerful method to explore haplotype structure of human populations. Ann Hum Genet. 2008;72:261–278. doi: 10.1111/j.1469-1809.2007.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuillan R, Leutenegger AL, Abdel-Rahman R, et al. Runs of homozygosity in European populations. Am J Hum Genet. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polasek O, Hayward C, Bellenguez C, et al. Comparative assessment of methods for estimating individual genome-wide homozygosity-by-descent from human genomic data. BMC Genomics. 2010;11:139. doi: 10.1186/1471-2164-11-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellenguez C, Ober C, Bourgain C. Linkage analysis with dense SNP maps in isolated populations. Hum Hered. 2009;68:87–97. doi: 10.1159/000212501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann HM, de Toma C, Cazes L, et al. A human genome diversity cell line panel. Science. 2002;296:261–262. doi: 10.1126/science.296.5566.261b. [DOI] [PubMed] [Google Scholar]
- Li JZ, Absher DM, Tang H, et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. doi: 10.1126/science.1153717. [DOI] [PubMed] [Google Scholar]
- Rosenberg NA. Standardized subsets of the HGDP-CEPH Human Genome Diversity Cell Line Panel, accounting for atypical and duplicated samples and pairs of close relatives. Ann Hum Genet. 2006;70:841–847. doi: 10.1111/j.1469-1809.2006.00285.x. [DOI] [PubMed] [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J, et al. A high-resolution recombination map of the human genome. Nat Genet. 2002;31:241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- Jakobsson M, Scholz SW, Scheet P, et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- Itsara A, Cooper GM, Baker C, et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike H. A new look at the statistical identification model. IEEE Trans Automat Contr. 1974;19:716–723. [Google Scholar]
- Wright S. Coefficient of inbreeding and relationship. Am Nat. 1922;56:330–338. [Google Scholar]
- Rosenberg NA. DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes. 2004;4:137–138. [Google Scholar]
- Scott-Emuakpor AB. The mutation load in an African population. I. An analysis of consanguineous marriages in Nigeria. Am J Hum Genet. 1974;26:674–682. [PMC free article] [PubMed] [Google Scholar]
- Calafell F, Shuster A, Speed WC, Kidd JR, Black FL, Kidd KK.Genealogy reconstruction from short tandem repeat genotypes in an Amazonian population Am J Phys Anthropol 1999108137–146. ( http://info.med.yale.edu/genetics/kkidd/302.pdf ). [DOI] [PubMed] [Google Scholar]
- Kidd JR, Pakstis AJ, Kidd KK.Global Levels of DNA Variation Proceedings from The Fourth International Symposium on Human Identification 1993 ( http://info.med.yale.edu/genetics/kkidd/302.pdf ).
- Wang S, Lewis CM, Jakobsson M, et al. Genetic variation and population structure in native Americans. PLoS Genet. 2007;3:e185. doi: 10.1371/journal.pgen.0030185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR. Estimation of pairwise identity by descent from dense genetic marker data in a population sample of haplotypes. Genetics. 2008;178:2123–2132. doi: 10.1534/genetics.107.084624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson EA.Analysis of data on related individuals through inference of identity by descent Seattle: Department of Statistics, University of Washington; Technical Report 5392008 [Google Scholar]
- Browning SR, Browning BL. High-resolution detection of identity by descent in unrelated individuals. Am J Hum Genet. 2010;86:526–539. doi: 10.1016/j.ajhg.2010.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein MP, Duren WL, Boehnke M. Improved inference of relationship for pairs of individuals. Am J Hum Genet. 2000;67:1219–1231. doi: 10.1016/s0002-9297(07)62952-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieberts SK, Wijsman EM, Thompson EA. Relationship inference from trios of individuals, in the presence of typing error. Am J Hum Genet. 2002;70:170–180. doi: 10.1086/338444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wilder K, McPeek MS. Enhanced pedigree error detection. Hum Hered. 2002;54:99–110. doi: 10.1159/000067666. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.