

Abstract
Florida has the highest degree of endemicity for eastern equine encephalitis virus (EEEV) of any state in the United States and is the only state with year-round transmission of EEEV. To further understand the viral population dynamics in Florida, the genome sequence of six EEEV isolates from central Florida were determined. These data were used to identify the most polymorphic regions of the EEEV genome from viruses isolated in Florida. The sequence of these polymorphic regions was then determined for 18 additional Florida isolates collected in four geographically distinct regions over a 20-year period. Phylogenetic analyses of these data suggested a rough temporal association of the Florida isolates, but no clustering by region or by source of the isolate. Some clustering of northeastern isolates with Florida isolates was seen, providing support for the hypothesis that Florida serves as a reservoir for the periodic introduction of EEEV into the northeastern United States.
Introduction
Eastern equine encephalitis virus (EEEV; family Togaviridae, genus Alphavirus) is the most virulent of the arthropod-borne viruses (arboviruses) in the United States. The virus is found primarily along the Atlantic Seaboard and the Gulf Coast states, although it is also found as far west as the Great Lakes region. Additional lineages of the virus are found in many parts of Central and South America.1,2 In the northeastern United States, the primary vector responsible for maintaining the enzootic cycle of the virus is the ornithophilic mosquito Culiseta melanura,3,4 although other mosquito species may be responsible for enzootic maintenance in the south central United States.5,6 Enzootic cycles are often located in hardwood swamp habitats, where vector and avian hosts are found. There are also numerous species of bridge vectors with catholic feeding patterns important in epizootic transmission of the virus to humans, horses, and other mammals, which are generally considered dead end hosts for the virus.7,8
In the United States, Florida is the state with the most reported neuroinvasive human cases of EEEV.9 In Florida, unlike in the rest of the United States, EEEV has been found to circulate throughout the year.10 Because of this stable transmission cycle of EEEV in Florida, some investigators have proposed that Florida may serve as a reservoir from which EEEV is introduced periodically into Connecticut, New Hampshire, and New York in the northeastern United States, areas in which virus is endemic,11–14 through migration of infected birds.
Phylogenetic analyses of EEEV have been performed to study the overall evolutionary history of North American strains, and to study transmission, localized perpetuation, and movement of the virus in selected regions of the northeastern United States.11,14 However an in depth study of the transmission and evolutionary history of EEEV in Florida has not been reported.
The Florida Department of Health Bureau of Laboratories (BOL), in Tampa has a long history of statewide arbovirus surveillance, including EEEV, St. Louis encephalitis virus, highlands J virus, and more recently West Nile virus (WNV). The BOL coordinates an extensive sentinel chicken program throughout most of Florida, screens veterinary and wild bird serum samples for arboviruses, and tests mosquito pools from local mosquito control districts. As a result of these efforts, numerous isolations/detections of EEEV have been made by the BOL from many counties across the state dating from the late 1980s.
To study the transmission and evolutionary history of EEEV in Florida, 24 EEEV isolates were chosen for gene sequencing and phylogenetic analysis. Strains were chosen from four geographically distinct regions of the state and from different years. These selection criteria enabled an examination of the level of the genetic diversity existing between geographic regions of the state and over a temporal scale of two decades. These data were compared with similar data collected from EEEV isolates from other states to test the hypothesis that Florida might serve as a reservoir for the introduction of EEEV to other regions of the United States.
Materials and Methods
Virus isolation.
The EEEV isolates from Florida were provided by the Florida Department of Health BOL in Tampa. Collection dates of these specimens ranged from 1986 through 2008. Isolates were derived from nine counties and from a variety of sources including avian, mammalian, and insect hosts. All virus isolates from Florida were previously cultured in either cell culture or suckling mouse brain and had a history of one or two such passages. Samples from Alabama were collected in 2003 at an EEEV-endemic site located in the Tuskegee National Forest in east central Alabama.6,11–14 Collection details on all isolates used in this study are shown in Table 1.
Table 1.
Isolates of eastern equine encephalitis virus from Florida and Alabama from which sequence data were obtained
| Strain name | Passage history* | Isolation source | County | Collection date | Region† |
|---|---|---|---|---|---|
| 2001 aR1-27 | Vero | White-throated sparrow | Santa Rosa | 11/21/01 | 1 |
| 2002 aR1-56 | Vero | Finch | Santa Rosa | 11/1/02 | 1 |
| 2003 mR1-19 | Vero | Ochlerotatus infirmatus | Escambia | 6/10/03 | 1 |
| 2005 mR1-31 | Vero | Culiseta melanura | Escambia | 7/18/05 | 1 |
| 1986 eR2-10 | SM | Equine | Jefferson | 1/2/86 | 2 |
| 1991 aR2-11 | SM | Pheasant | Leon | 8/2/91 | 2 |
| 1994 aR2-32 | BGM | Pheasant | Leon | 8/3/94 | 2 |
| 2001 aR2-35 | Vero | Thrasher | Gadsden | 10/17/01 | 2 |
| 2003 eR2-38 | Vero | Equine | Jefferson | 7/10/03 | 2 |
| 2005 eR2-18 | Vero | Equine | Leon | 7/12/05 | 2 |
| 1992 aR3-1 | SM, BGM | Pheasant | Volusia | 6/10/92 | 3 |
| 1992 mR3-7 | SM, BGM | Culex erraticus | Volusia | 8/17/92 | 3 |
| 1992 aR3-52 | SM, BGM | White ibis | Orange | 6/25/92 | 3 |
| 1994 mR3-5 | SM | Culex nigrapalpus | Volusia | 3/27/94 | 3 |
| 2001 aR3-41 | Vero | Common grackle | Orange | 6/25/01 | 3 |
| 2003 eR3-3 | Vero | Equine | Volusia | 4/2/03 | 3 |
| 2003 eR3-40 | Vero | Equine | Seminole | 7/14/03 | 3 |
| 2005 mR3-4 | Vero | Culex nigripalpus | Volusia | 5/26/05 | 3 |
| 2005 mR3-39 | Vero | Coquillettidia perturbans | Volusia | 5/26/05 | 3 |
| 2008 mR3-6 | Vero | Culex salinarius | Volusia | 2/13/08 | 3 |
| 2001 aR4-12 | Vero | Mourning dove | Palm Beach | 10/17/01 | 4 |
| 2003 mAL-62 | Vero | Mosquito spp. | Macon | 7/25/03 | AL |
| 2003 mAL-63 | Vero | Culex erraticus | Macon | 7/31/03 | AL |
| 2003 mAL-64 | Vero | Culiseta melanura | Macon | 7/29/03 | AL |
VERO = Vero cell culture; SM = suckling mouse culture; BGM = buffalo green monkey cell culture.
Region 1 = Escambia and Santa Rosa Counties; Region 2 = Gadsden, Leon, and Jefferson Counties; Region 3 = Orange, Seminole, and Volusia Counties; Region 4 = Palm Beach County; AL = Alabama.
Alabama isolates, all of which were derived from mosquito pools, were positive for EEEV by reverse transcription–polymerase chain reaction (RT-PCR), but had not been confirmed by culture. Homogenates from these positive pools had been prepared in BA-1 tissue culture medium as described,5 and had been stored at –80°C. To culture these viruses, stored homogenates (approximately 1 mL) were thawed at 37°C and 1 mL of diluent (1× Hanks' minimal essential medium, 10% heat-inactivated fetal bovine serum, 200 U/mL penicillin, 200 μg/mL streptomycin, 2.5 μg/mL amphotericin B) was added. The sample was mixed for 1 minute, centrifuged at 4°C for 4 minutes at 13,000 × g, and the supernatant was filtered through a sterile 0.2-μm filter before inoculation into individual T-25 flasks of confluent Vero cell cultures. Flasks were incubated for 1 hour at 37°C, with gentle rocking every 15 minutes. After the incubation for 1 hour, 9 mL of maintenance media (1× Earle's minimal essential medium, 2% fetal bovine serum, 200 U/mL penicillin, 200 μg/mL streptomycin, 2.5 μg/mL amphotericin B) were added to each flask, Cells were monitored daily for a cytopathic effect. If a cytopathic effect (CPE) was observed, the culture was confirmed as containing EEEV by RT-PCR.
RNA extraction, RT-PCR, and sequencing.
RNA was isolated from cell culture or tissue samples using the QIAmp Viral RNA kit (Qiagen, Valencia, CA). Viral RNA was reverse transcribed by using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) following the manufacture's recommendations, and reaction conditions using the random oligo and oligo dT primers in the kit and 3 μL of extracted RNA template.
EEEV cDNA was then amplified by PCR in 14 reactions to generate nearly complete genomes; primer sequences used in these reactions are available upon request. To amplify the genomic segments, 2 μL from each cDNA reaction was added to 25 μL PCR master mixture containing 1× PCR buffer, 0.2 mM dNTPs, 0.5 μM of each primer, and 2.0 units Taq DNA polymerase. Amplification was performed as follows: 1 cycle at 95°C for 4 minutes; 40 cycles at 95°C for 30 seconds, 55°C for 30 seconds, and 72°C for 1 minute and 20 seconds; and 1 cycle 72°C for 7 minutes. Amplification products were analyzed by gel electrophoresis on a 1% agarose gel. DNA from bands of the appropriate size were cleaned with the QIAquick PCR purification kit (Qiagen), and sequences determined by using a commercial DNA sequencing service (Genewiz, South Plainfield, NJ).
To amplify smaller segments of viral genomes, the same protocol was used as for amplifying the 14 segments used to determine the sequence of the complete genomes, except for modifications in the cycling conditions. The amplification cycling conditions consisted of 1 cycle at 95°C for 4 minutes; 35 cycles at 95°C for 20 seconds, 55°C for 30 seconds, and 72°C for 1 minute; and 1 cycle at 72°C for 7 minutes. The primer sequences used to amplify the pieces from the nonstructural genes were EEEnsp1 373c 5′-CGCTGAGACACCCTCGTTAT-3′ with EEEnsp 11268nc 5′-GAGTTTTGAAAGCCCAGCAG-3′; EEEnsp2 2064c 5′- TAGTAGACCCGCCATTCCAC-3′ with EEEnsp2 3227nc 5′-TGGTGTAAGTCAGCGGAACA-3′; and EEEnsp3 4641c 5′-CTAACAAGCAAGAAGCAAACG-3′ with EEEnsp3 5646nc 5′-TCGTACCGTCAATTCGAGTG-3′. The sequences for the structural region were obtained by using primers developed for genomic sequencing.
Sequence analyses.
EEEV genomes were constructed from data derived from the 14 overlapping segments amplified as described above, by using the SeqMan module of Lasergene (DNAstar, Madison, WI). The final contigs had at least two-fold coverage in all positions. The six genomes were aligned by using CLUSTAL W in MacVector (MacVector Inc., Cary, NC) and analyzed manually for location of parsimony informative sites. The alignment was then analyzed for sequence diversity by using the software program DnaSP.15 Sequence data used in this study have been deposited in the GenBank database with the accession numbers HM196169–HM196276, HM196169–HM196276, and HM210093–to HM210098.
Phylogenetic analyses.
Parsimony analyses were conducted by using subroutines available in the PAUP program package.16 The exhaustive search algorithm was used when possible. When the number of taxa exceeded the capacity of the program to conduct an exhaustive analysis, the heuristic algorithm was used. Statistical support for all groupings was evaluated by reanalysis of 1,000 synthetic bootstrap datasets.
The jModelTest17,18 was used to predict the best parameters in reconstructing Bayesian trees. It was also used to set the five substitution schemes, with the other values set as default (use base frequencies, rate variation with four categories), and ML-optimized base tree for likelihood calculations. The jModelTest predicted that the general time reversible plus proportion of invariant sites plus discretized gamma distribution (GTR + I + G) evolutionary model would be the best for the set of sequences in the first two Bayesian phylogenies analyzing the relationships among all Florida isolates whose sequence was determined.
The MrBayes software package19,20 was then used to calculate the phylogenetic tree. The GTR + G + I evolutionary model was used and the program was set to run for 1,000,000 generations with sampling every 1,000 generations. The first 25% (250) of sampled trees were discarded as burn-in. The average standard deviation of split frequencies at the end of the run was 0.01. The potential scale reduction factor for all parameters at the end of the run was 1.0 ± 0.004. For the tree including isolates from the northeastern United States, the methods were exactly the same as used, except that jModelTest predicted the best fitting model as GTR + I in both cases. This model was subsequently used on both phylogenetic reconstructions. The potential scale reduction factor on this tree was 1.00 ± 0.002, and the final SD of split frequencies was 0.01.
Results
On the basis of an analysis of the records of EEEV isolates maintained by the Florida Department of Health Virology Laboratory, four regions were selected from which viral isolates for genomic sequence analysis were identified (Figure 1). These regions included the Western Panhandle, north central, east central, and southeastern regions of the state. These regions were selected because they are geographically distinct and represent different ecological biotomes. With the exception of the southeastern region of Florida, multiple archived viral samples collected over a relatively long period were available. In addition to these four regions in Florida, three isolates of EEEV from pools of mosquitoes collected at a well-characterized study site in the Tuskegee National Forest5,21,22 were included in the study. Descriptions of these isolates are shown in Table 1.
Figure 1.
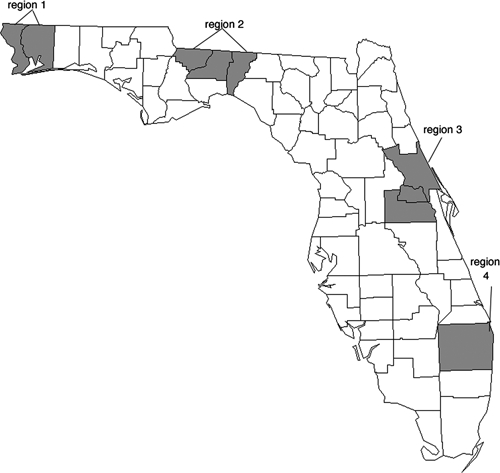
Regions in Florida from which viral isolates were chosen. Region 1 = Escambia and Santa Rosa Counties; Region 2 = Gadsden, Leon, and Jefferson Counties; Region 3 = Orange, Seminole, and Volusia Counties; Region 4 = Palm Beach County.
Initially, nucleic acid sequences of six isolates from Region 3 were determined. The sequence data covered almost the entire genome, encompassing all but the first 48 nucleotides from the 5′ untranslated region and all but the last 7 nucleotides from the poly-A tail, when compared with the complete NJ/60 genome sequence. These data were subjected to phylogenetic analysis by using maximum parsimony methods (Figure 2). This initial phylogeny supported the division of these isolates into two distinct clades separated by time, with all strains from the 1990s in one clade and remaining isolates from the 2000s in a separate clade. In contrast, no phylogenic grouping of isolates by host class (avian, equine, or mosquito) was found (Figure 2).
Figure 2.
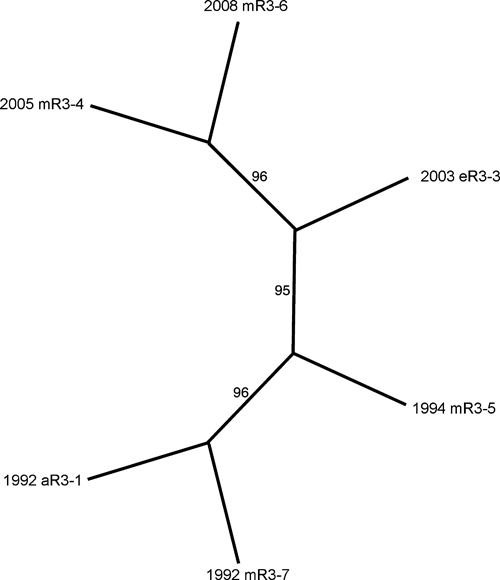
Phylogenetic analysis of eastern equine encephalitis virus genomic sequences of isolates from Volusia County, Florida (Region 3). The dataset was found to contain 17 informative sites distributed among the six taxa. An unrooted phylogeny was prepared by using the exhaustive search algorithm in the PAUP program package. This analysis returned one most parsimonious tree. The phylogeny has a consistency index of 0.98. Numbers on the figure indicate the percentage of times the grouping distal to the number were supported in a bootstrap re-analysis of 1,000 replicate datasets.
Sequence data from the six isolates were then aligned and the areas of greatest sequence diversity in the genomes were determined by using a sliding window with a window size of 300 nucleotides and a step size of 50 nucleotides (Figure 3). This information, along with the location of the parsimony informative sites, were used to select five segments of the genome with the greatest diversity and phylogenetically informative positions to target in the subsequent analysis of the additional isolates listed in Table 1. Overall, these segments covered 4,384 nucleotides, representing 37% of the total EEEV genome (Figure 3). In addition, to compare the Florida isolates with other EEEV sequences available in GenBank, the complete sequence of the structural polyprotein gene was also determined for each isolate shown in Table 1. This structural sequence covered 3,729 nucleotides, representing 32% of the EEEV genome (Figure 3).
Figure 3.
Diversity levels among the six Florida isolates of eastern equine encephalitis virus initially analyzed. Sequence data derived from the six isolates from Volusia County were analyzed for sequence polymorphisms as described in the text. Black bars indicate the polymorphic regions chosen for subsequent analysis in this study. Open bar indicates the entire structural region whose sequence was compared with the Genbank sequences from isolates listed in Table 2. Gray bars indicate partial regions of the structural polyprotein gene analyzed in previous studies of sequence diversity among northeastern isolates of eastern equine encephalitis virus.11
Sequence data derived from these selected regions were then used to construct two Bayesian phylogenetic trees by using the parameters described in the Materials and Methods. The first of these trees (Figure 4) used the concatenated segments from the variable regions of the nonstructural protein 1 (NSP1), NSP2, NSP3, capsid and envelope 1 genes shown in Figure 3, and the second phylogeny was prepared by using the data derived from the structural polyprotein gene (Figure 5). The two phylogenies generally agreed with one another, although there were some minor differences in the observed topologies. For example, the phylogeny prepared from the concatenated data grouped strains 2002 aR1-56 and 2003 eR3-40 together with a probability of 0.94 (Figure 4). In the tree derived from the polyprotein structural gene sequence, strains 2003 eR3-40 and 2003 eR3-3 are grouped together with a probability of 0.68, and isolate 2002 aR1-56 was grouped by itself as a polytomy (Figure 5). However, both analyses supported the existence of two major clades, with one clade containing three isolates obtained from the 1990s and an isolate from 2005, and the second clade contained all of the remaining 13 Florida isolates from 2001–2008, together with all of the Alabama isolates and three Florida isolates from the 1990s (Figures 4 and 5). The single Florida isolate obtained from the 1980s was distinct from any of the later isolates in both phylogenies (Figures 4 and 5).
Figure 4.
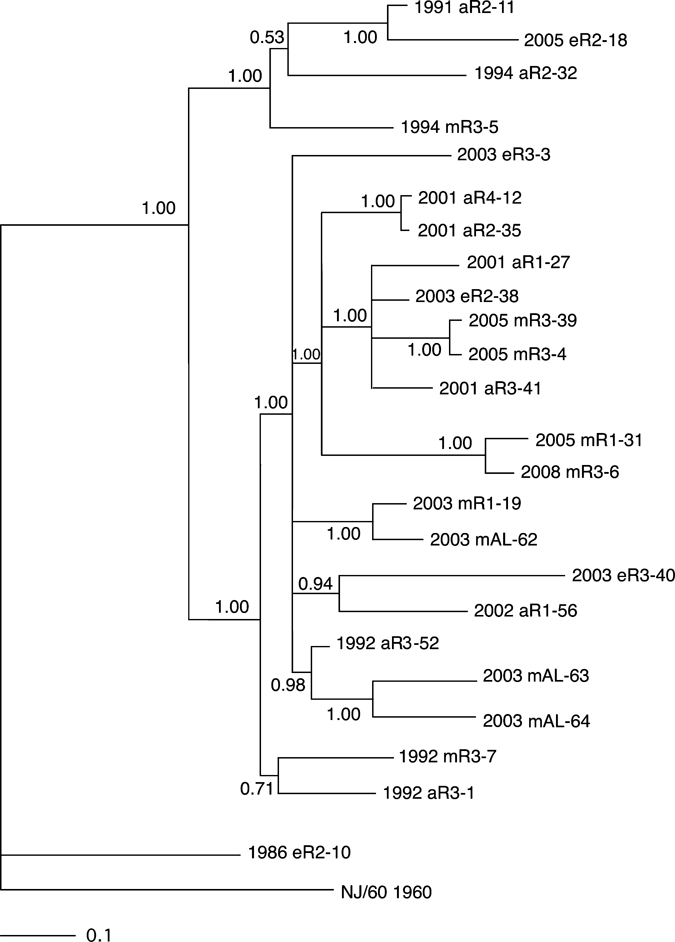
Bayesian analysis of eastern equine encephalitis virus isolates composed of concatenated regions of nonstructural protein 1 (NSP1), NSP2, NSP3, capsid, and envelope 1 genes from different regions of Florida. Bayesian phylogenies were prepared by using the MrBayes program package,19,20 as described in the Materials and Methods. Phylogenies were rooted using the NJ60 sequence. Branch lengths are proportional to distance (the number of nucleotide changes), and the distance scale at the bottom of the tree represents the number of expected substitutions per site. Values indicate the probability for each partition or clade in the tree.
Figure 5.
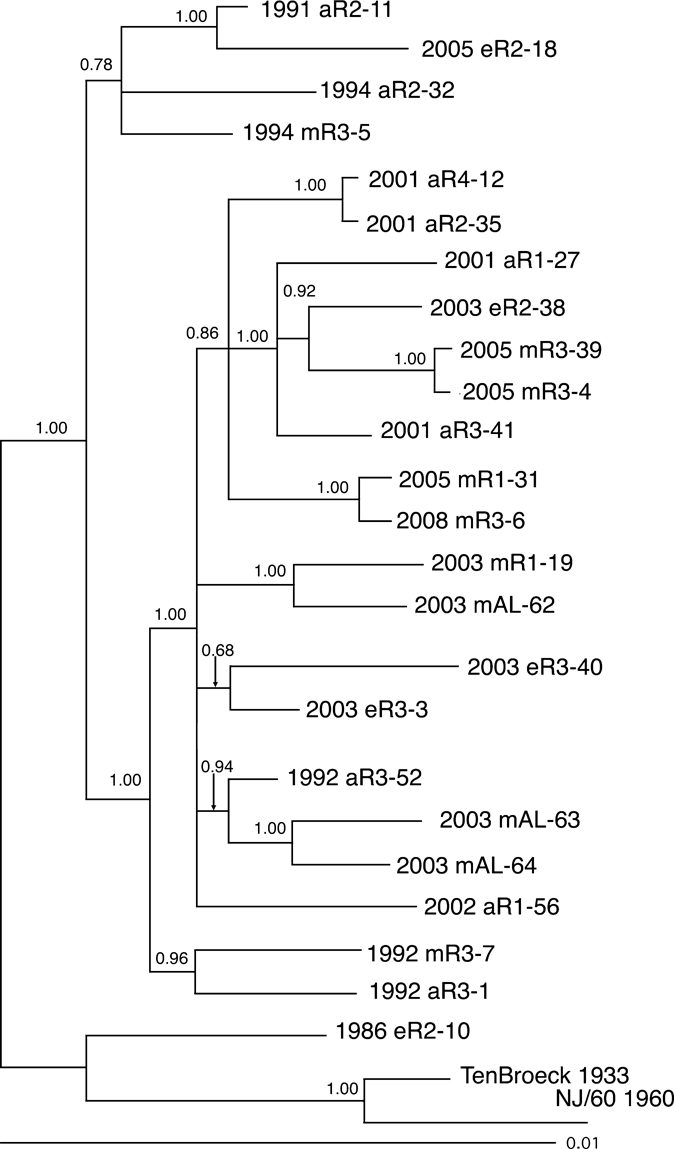
Bayesian analysis of eastern equine encephalitis virus isolates composed of the structural polyprotein from different regions of Florida. Bayesian phylogenies were prepared by using the MrBayes program package,19,20 as described in the Materials and Methods. Phylogenies were rooted by using the NJ60 and Ten Broeck sequences. Branch lengths are proportional to distance (the number of nucleotide changes), and the distance scale at the bottom of the tree represents the number of expected substitutions per site. Values indicate the probability for each partition or clade in the tree.
Both phylogenies, when considered together, generally grouped isolates from the same region and collection year together, although there were some exceptions to this grouping. There were two pairs of isolates examined that were derived from the same region and same year (pairs 2003 eR3-3 + 2003 eR3-40 and 2005 mR3-4 + 2005 mR3-39). The first of these was monophyletic in the structural phylogeny but not the concatentated gene phylogeny, and the second pair was monophyletic on both phylogenies. Similarly, there were two sets of isolates containing three isolates each that were derived from the same region and year (1992 aR3-1 + 1992 mR3-7 + 1992 aR3-52 and 2003 mAL-62 + 2003 mAL-63 + 2003 mAL-64). In both of these three isolate groups, both phylogenies identified pairs of isolates that were monophyetic, and classified the remaining isolate as distinct from the monophyletic pair (Figures 4 and 5). Neither phylogeny supported the grouping of isolates by either host type or geographic region.
Recently, published studies based upon analyses of the structural genes have proposed the hypothesis that EEEV foci in the northeastern United States arise from periodic importations of the virus from Florida.11,14 To test this hypothesis, published structural gene sequences from 18 EEEV isolates obtained from regions outside Florida were analyzed with structural gene data obtained from the Florida isolates. The sequences from the GenBank isolates included in this analysis are shown in Table 2. Of these isolates, 12 contained the full structural polyprotein gene sequence. These isolates varied greatly in when they were isolated and where they originated. To compare more sequences from a more tightly temporally and spatially distributed group, data from six additional isolates available on GenBank from the northeastern United States were also analyzed. However, these latter sequences included only portions of the structural polyprotein gene (Figure 3). Thus, this analysis was limited to the 1,559 nucleotides for which data were available from all isolates. The resulting phylogeny contained more polytomies among the Florida isolates than did the phylogeny prepared using the entire structural gene sequences, as would be expected considering the more limited dataset analyzed.
Table 2.
Isolates of eastern equine encephalitis virus from GenBank included in the analysis
| Isolate | Strain | Location of isolation* | Date isolated | GenBank accession no. |
|---|---|---|---|---|
| TenBroeck 1933 | Ten Broeck | VA | 1933 | U01558 |
| LA 1947 | Decuir | LA | 1947 | U01552 |
| NJ/60 1959 | NJ/60 | NJ | 1959 | U01554 |
| MA 1977 | ME77132 | MA | 1977 | U01555 |
| MS 1983 | MS-4789 | MS | 1983 | AF159552 |
| CT 1990 | Williams | CT | 1990 | U01557 |
| FL 1991-4697 | FL91-4697 | FL | 1991 | AY705241 |
| FL 1993-939 | FL93-939 | FL | 1993 | EF151502 |
| FL 1993-969 | FL93-969 | FL | 1993 | GU001911 |
| TX 1995 | PV5-2547 | TX | 1995 | AF159555 |
| GA 1997 | GA97 | GA | 1997 | AY705240 |
| TX 2003 | TX1634 | TX | 2003 | GU001914 |
| MA 2006 | MA06 | MA | 2006 | GU108612 |
| TN 2008 | TN08 | TN | 2008 | GU001921 |
| CT-1996† | 310-96 | CT | 1996 | EU573664 |
| CT-2006† | 8746-06 | CT | 2006 | EU573660 |
| CT-2001† | 10116-01 | CT | 2001 | EU573627 |
| CT-2003† | 13243-03 | CT | 2003 | EU573638 |
| CT-2003† | 14955-03 | CT | 2003 | EU573656 |
| NY-2005† | N155-05 | NY | 2005 | EU573680 |
VA = Virginia; LA = Louisiana; NJ = New Jersey; MA = Massachusetts; MS = Mississippi; CT = Connecticut; FL = Florida; TX = Texas; GA = Georgia; TN = Tennessee; NY = New York.
Partial structural polyprotein gene sequences.
Despite this finding, the resulting phylogeny supported a rough temporal association of the isolates from the northeastern United States and Florida (Figure 6). Most isolates obtained in the first decade of the 21st century from outside Florida were included in the large clade containing most of the 2001–2008 Florida isolates shown in Figures 4 and 5 (Figure 6). Similarly, isolate CT 1996 grouped with the Florida isolates 1986 eR2-10 and FL 1993-939 (Figure 3). Within the large clade containing most isolates from Florida from 2001–2008, some evidence of association between specific Florida isolates and those collected elsewhere was also evident. For example, CT 2003-13243 was contained within clade that included a number of Florida isolates collected from 2001–2005 (Figure 6).
Figure 6.
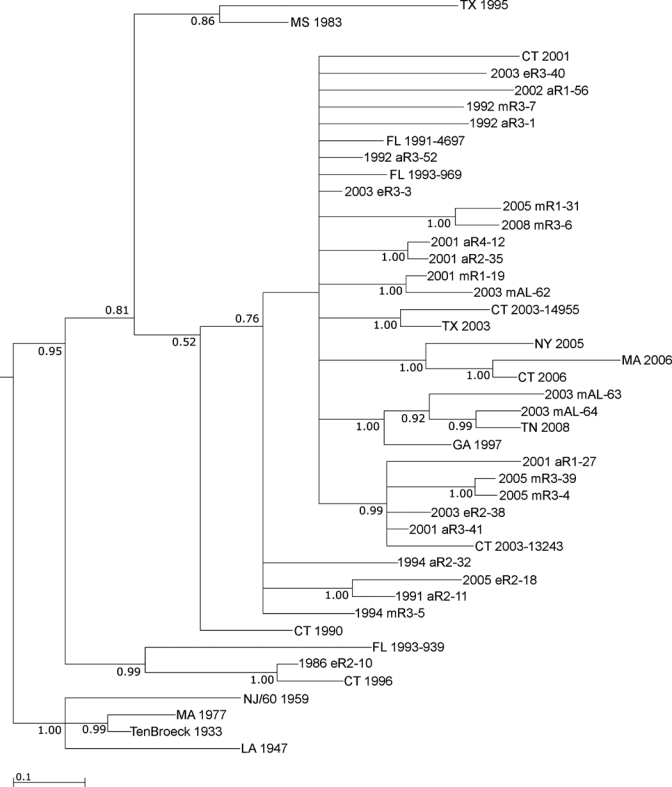
Bayesian analysis of eastern equine encephalitis virus isolates from Florida and other locations in the United States. Bayesian phylogenies were prepared by using the MrBayes program package,20 as described in the Materials and Methods. Phylogenies were rooted by using the NJ60 and Ten Broeck sequences. Branch lengths are proportional to distance (the number of nucleotide changes), and the distance scale at the bottom of the tree represents the number of expected substitutions per site. Values indicate the probability for each partition or clade in the tree.
Discussion
Our data support the conclusion that EEEV isolates from Florida generally cluster by year of isolation. For example, the oldest Florida isolate examined in the study (1986 eR2-10) was found to be distinct from all of the other Florida isolates from the 1990s and 2000s. Furthermore, analysis supported the existence of two major clades into which the other Florida isolates grouped. The smaller of these clades consisted primarily of Florida isolates from the 1990s, but also included a single isolate from 2005. The larger clade consisted primarily of isolates from the 2000s, although it also included three isolates from 1992. In contrast, the data failed to show any evidence for spatial clustering of EEEV of the Florida isolates. Such spatial clustering would have been expected if EEEV transmission were localized in isolated foci in the different regions of the state. Some of the most closely related virus isolates were from widely separated regions (e.g., 2001 aR2-35 and 2001 aR4-12). These data suggest that the virus is not geographically isolated in Florida and that it is therefore capable of disseminating across fairly large distances in the state. Similarly, the data also did not support any evolutionary grouping of viral isolates based upon the source from which the virus was isolated; viral isolates from mosquitoes, birds, or equine sources did not group together. These data therefore do not support the hypothesis of distinct virus isolates circulating in different host species in Florida, as has been recently reported in studies of EEEV in South America.23
Although phylogenies developed from the data tended to group isolates obtained from the same period together, they did not provide any evidence for a progressive temporal evolution of the virus, as is seen with influenza, One potential explanation for this finding is that the limited degree of diversity in the virus provided insufficient phylogenetically informative data to detect such an orderly temporal evolutionary pattern. However, the phylogenies reported appear relatively robust; both datasets produced nearly identical phylogenies in which major groupings received strong statistical support, suggesting the data were informative enough to perform an accurate phylogenic analysis. However, the relatively short branch lengths observed underscores the overall high degree of sequence conservation previously reported in North American EEEV.11–14 This lack of sequence diversity reflects the conserved evolutionary history of the virus. It has been suggested that one reason for the high degree of sequence conservation in EEEV may relate to its need to infect multiple hosts with different physiologies.24,25 Mutations in many different positions might affect the ability of the virus to efficiently infect one of these diverse hosts, which would limit the genetic variability seen in naturally circulating virus populations.
Alternative explanations for the lack of a clear temporal evolutionary pattern may relate to the biology of the virus in Florida. First, unlike the pattern seen in the northeastern United States, EEEV is stably endemic, with year round transmission in Florida. Such a pattern might lead to the production of a genetically diverse virus population, which would in turn lead to many strains co-circulating simultaneously as competing clusters of viruses.26 Stochastic processes driven by local conditions could then lead to the predominance of a particular viral type during a given year. Second, EEEV is generally a non-fatal viral infection of the passerine birds of North America, with infection of these species leading to long-term immunity.27 This type of infection would not favor a gradual temporal evolution of the virus through antigenic drift, such as is seen with human influenza A virus, where the host retains partial immunity to future influenza strains. Rather, a strong level of immunity in the avian host population would lead to a transmission pattern more similar to that seen with human measles, where stochastic events give rise to certain dominant strains that then tend to remain dominant for a given period.28 In human measles, this transmission pattern leads to a pattern similar to what is seen in our study, where little evidence of an orderly evolution of the virus over time can be detected.
The phylogenetic pattern of Florida viral isolates differs from that shown in recent studies of isolates from the northeastern United States. Those studies have demonstrated that in the northeastern United States, EEEV tends to occur in successive waves of genetically fairly uniform virus populations that circulate for a number of years and then disappear, only to be replaced by another population of nearly genetically identical viruses.11,14 It has been suggested that this pattern is the result of periodic introduction of EEEV into the northeastern United States, resulting in establishment of foci that remain active for a few years before dying out and being replaced by a subsequent viral introduction.11
It has been further hypothesized that Florida might be the source of these viral introductions to the northeastern United States.14 Our data provide some support for this hypothesis. For example, the Connecticut isolate CT-2003-13243 grouped with Florida isolates from 2001–2005, suggesting that the CT 2003–2004 clade previously identified may have arisen by an introduction from a Florida viral reservoir. Similarly, as reported, the CT 310-96 isolate (CT 1996 in Figure 6) grouped with the FL 1993-93911 isolate, and was even more closely related to the FL 1986 mR2-10 isolate reported, supporting the hypothesis of a Florida origin. Finally, the three isolates from Alabama grouped with two Florida isolates, suggesting that the Alabama virus might also have been introduced from Florida. Two of the AL isolates (2003 mAL-63 and 2003 mAL-64) also grouped with isolates from Tennessee and Georgia, indicating that EEEV may be also be introduced into the southeastern United States from a Florida reservoir. However, although the data in general support a relationship between the Florida isolates and those obtained from elsewhere, in many cases it is not possible to deduce a direct relationship between a particular Florida isolate and those collected outside Florida because of the presence of polytomies and poor statistical support for some of the direct pairings present in the phylogeny. However, it appears that the stable endemic transmission pattern of EEEV in Florida may have resulted in the development of a highly diverse virus population, and it is thus possible that these isolates arose from a Florida progenitor strain that has not yet been characterized. Additional studies comparing more isolates from Florida to those from the northeastern United States may be useful in resolving this issue.
The phylogenetic relationships developed to date all support the hypothesis that Florida serves as the reservoir from which EEEV is periodically introduced into the northeastern United States. However, it is also possible for viruses that have undergone isolated evolution in the northeastern United States to migrate south and become established in Florida, further increasing viral diversity in this state. Arbovirus migration has already been documented to occur from the northeastern United States to Florida with the introduction of WNV to New York in 1999 and the subsequent appearance of the virus to Florida in 2001.
Our data suggest that a major switch in viral type occurred in the late 1990s or early 2000s. It is interesting to note that this finding corresponds to the period when WNV was first detected in Florida in 2001.29 Previous studies have suggested that introduction of WNV resulted in dramatic changes in the transmission of St. Louis encephalitis virus in Florida and elsewhere.30,31 It is therefore possible that the introduction of WNV might have also affected the ecology of EEEV transmission in Florida, resulting in a shift in the predominant circulating viral type. Such a change might have resulted from indirect effects of WNV on the enzootic passerine bird reservoir for EEEV, or other changes in the transmission dynamics of EEEV resulting from the introduction of another arbovirus into what has been a previously stable transmission system for EEEV. Laboratory and modeling studies examining the transmission dynamics of EEEV in the presence and absence of WNV would be useful in testing this hypothesis.
Footnotes
Financial support: This study was supported by a grant from the National Institute of Allergy and Infectious Diseases (Project # R01AI049724) to Thomas R. Unnasch.
Authors' addresses: Gregory S. White, Indio, CA, E-mail: gwhite@cvmvcd.org. Brett E. Pickett, Department of Pathology, University of Texas Southwestern Medical Center, Dallas, TX, E-mail: bpickett@uab.edu. Elliot J. Lefkowitz, Department of Microbiology, University of Alabama at Birmingham, Birmingham, AL, E-mail: elliotl@uab.edu. Amelia G. Johnson and Thomas R. Unnasch, Global Health Infectious Disease Research Program, Department of Global Health, University of South Florida, Tampa, FL, E-mails: ajohnso3@health.usf.edu and tunnasch@health.usf.edu. Christy Ottendorfer, Centers for Disease Control and Prevention, Atlanta, GA, E-mail: cottendorfer@gmail.com. Lillian M. Stark, Tampa, FL, E-mail: lillian_stark@doh.state.fl.us.
References
- 1.Sanmartin C, Trapido H, Barreto P, Lesmes CI. Isolations of Venezuelan and Eastern equine encephalomyelitis viruses from sentinel hamsters exposed in the Pacific lowlands of Colombia. Am J Trop Med Hyg. 1971;20:469–473. doi: 10.4269/ajtmh.1971.20.469. [DOI] [PubMed] [Google Scholar]
- 2.Walder R, Suarez OM, Calisher CH. Arbovirus studies in southwestern Venezuela during 1973–1981. II. Isolations and further studies of Venezuelan and eastern equine encephalitis, Una, Itaqui, and Moju viruses. Am J Trop Med Hyg. 1984;33:483–491. [PubMed] [Google Scholar]
- 3.Morris CD. In: The Arboviruses: Epidemiology and Ecology. Monath TP, editor. Boca Raton, FL: CRC Press; 1988. pp. 1–20. (Eastern equine encephalomyelitis). [Google Scholar]
- 4.Scott TW, Weaver SC. Eastern equine encephalomyelitis virus: epidemiology and evolution of mosquito transmission. Adv Virus Res. 1989;37:277–328. doi: 10.1016/s0065-3527(08)60838-6. [DOI] [PubMed] [Google Scholar]
- 5.Cupp EW, Klingler K, Hassan HK, Viguers LM, Unnasch TR. Transmission of eastern equine encephalomyelitis virus in central Alabama. Am J Trop Med Hyg. 2003;68:495–500. [PMC free article] [PubMed] [Google Scholar]
- 6.Cupp EW, Zhang D, Yue X, Cupp MS, Guyer C, Sprenger TR, Unnasch TR. Identification of reptilian and amphibian blood meals from mosquitoes in an eastern equine encephalomyelitis virus focus in central Alabama. Am J Trop Med Hyg. 2004;71:272–276. [PMC free article] [PubMed] [Google Scholar]
- 7.Elvinger F, Liggett AD, Tang KN, Harrison LR, Cole JR, Jr, Baldwin CA, Nessmith WB. Eastern equine encephalomyelitis virus infection in swine. J Am Vet Med Assoc. 1994;205:1014–1016. [PubMed] [Google Scholar]
- 8.Farrar MD, Miller DL, Baldwin CA, Stiver SL, Hall CL. Eastern equine encephalitis in dogs. J Vet Diagn Invest. 2005;17:614–617. doi: 10.1177/104063870501700619. [DOI] [PubMed] [Google Scholar]
- 9.Centers for Disease Control and Prevention . Eastern Equine Encephalitis: Epidemiology and Geographic Distribution. 2010. http://www.cdc.gov/easternequineencephalitis/tech/epi.html Available at. Accessed May 10. [Google Scholar]
- 10.Bigler WJ, Lassing EB, Buff EE, Prather EC, Beck EC, Hoff GL. Endemic eastern equine encephalomyelitis in Florida: a twenty-year analysis, 1955–1974. Am J Trop Med Hyg. 1976;25:884–890. doi: 10.4269/ajtmh.1976.25.884. [DOI] [PubMed] [Google Scholar]
- 11.Armstrong PM, Andreadis TG, Anderson JF, Stull JW, Mores CN. Tracking eastern equine encephalitis virus perpetuation in the northeastern United States by phylogenetic analysis. Am J Trop Med Hyg. 2008;79:291–296. [PubMed] [Google Scholar]
- 12.Weaver SC, Hagenbaugh A, Bellew LA, Gousset L, Mallampalli V, Holland JJ, Scott TW. Evolution of alphaviruses in the eastern equine encephalomyelitis complex. J Virol. 1994;68:158–169. doi: 10.1128/jvi.68.1.158-169.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weaver SC, Scott TW, Rico-Hesse R. Molecular evolution of eastern equine encephalomyelitis virus in North America. Virology. 1991;182:774–784. doi: 10.1016/0042-6822(91)90618-l. [DOI] [PubMed] [Google Scholar]
- 14.Young DS, Kramer LD, Maffei JG, Dusek RJ, Backenson PB, Mores CN, Bernard KA, Ebel GD. Molecular epidemiology of eastern equine encephalitis virus, New York. Emerg Infect Dis. 2008;14:454–460. doi: 10.3201/eid1403.070816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 16.Swofford DL. PAUP: Phylogenetic Analysis Using Parsimony (v 4.0) Sunderland, MA: Sinauer Associates; 1998. [Google Scholar]
- 17.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 18.Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- 19.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 20.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 21.Graham SP, Hassan HK, Burkett-Cadena ND, Guyer C, Unnasch TR. Nestedness of ectoparasite-vertebrate host networks. PLoS ONE. 2009;4:e7873. doi: 10.1371/journal.pone.0007873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacob BG, Burkett-Cadena ND, Luvall JC, Parcak SH, McClure CJ, Estep LK, Hill GE, Cupp EW, Novak RJ, Unnasch TR. Developing GIS-based eastern equine encephalitis vector-host models in Tuskegee, Alabama. Int J Health Geogr. 2010;9:12. doi: 10.1186/1476-072X-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arrigo NC, Adams AP, Weaver SC. Evolutionary patterns of eastern equine encephalitis virus in North versus South America suggest ecological differences and taxonomic revision. J Virol. 2010;84:1014–1025. doi: 10.1128/JVI.01586-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weaver SC, Brault AC, Kang W, Holland JJ. Genetic and fitness changes accompanying adaptation of an arbovirus to vertebrate and invertebrate cells. J Virol. 1999;73:4316–4326. doi: 10.1128/jvi.73.5.4316-4326.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weaver SC. Evolutionary influences in arboviral disease. Curr Top Microbiol Immunol. 2006;299:285–314. doi: 10.1007/3-540-26397-7_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miralles R, Gerrish PJ, Moya A, Elena SF. Clonal interference and the evolution of RNA viruses. Science. 1999;285:1745–1747. doi: 10.1126/science.285.5434.1745. [DOI] [PubMed] [Google Scholar]
- 27.Hayes RO, Daniels JB, Maxfield HK, Wheeler RE. Field and laboratory studies on eastern encephalitis in warm- and cold-blooded vertebrates. Am J Trop Med Hyg. 1964;13:595–606. doi: 10.4269/ajtmh.1964.13.595. [DOI] [PubMed] [Google Scholar]
- 28.Grenfell BT, Pybus OG, Gog JR, Wood JL, Daly JM, Mumford JA, Holmes EC. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303:327–332. doi: 10.1126/science.1090727. [DOI] [PubMed] [Google Scholar]
- 29.Blackmore CG, Stark LM, Jeter WC, Oliveri RL, Brooks RG, Conti LA, Wiersma ST. Surveillance results from the first West Nile virus transmission season in Florida, 2001. Am J Trop Med Hyg. 2003;69:141–150. [PubMed] [Google Scholar]
- 30.Ottendorfer CL, Ambrose JH, White GS, Unnasch TR, Stark LM. Isolation of genotype V St. Louis encephalitis virus in Florida. Emerg Infect Dis. 2009;15:604–606. doi: 10.3201/eid1504.081094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reisen WK, Lothrop HD, Wheeler SS, Kennsington M, Gutierrez A, Fang Y, Garcia S, Lothrop B. Persistent West Nile virus transmission and the apparent displacement St. Louis encephalitis virus in southeastern California, 2003–2006. J Med Entomol. 2008;45:494–508. doi: 10.1603/0022-2585(2008)45[494:pwnvta]2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]