

Abstract
Eisenmenger syndrome is the most severe form of pulmonary arterial hypertension and arises on the basis of congenital heart disease with a systemic-to-pulmonary shunt. Due to the chronic slow progressive hypoxemia with central cyanosis, adult patients with the Eisenmenger syndrome suffer from a complex and multisystemic disorder including coagulation disorders (bleeding complications and paradoxical embolisms), renal dysfunction, hypertrophic osteoarthropathy, heart failure, reduced quality of life and premature death.
For a long time, therapy has been limited to symptomatic options or lung or combined heart-lung transplantation. As new selective pulmonary vasodilators have become available and proven to be beneficial in various forms of pulmonary arterial hypertension, this targeted medical treatment has been expected to show promising effects with a delay of deterioration also in Eisenmenger patients. Unfortunately, data in Eisenmenger patients suffer from small patient numbers and a lack of randomized controlled studies.
To optimize the quality of life and the outcome, referral of Eisenmenger patients to spezialized centers is required. In such centers, specific interdisciplinary management strategies of physicians specialized on congenital heart diseases and PAH should be warranted.
This medical update emphasizes the current diagnostic and therapeutic options for Eisenmenger patients with particularly focussing on the medical treatment and corresponding study results.
Keywords: Cardiovascular diseases, adult congenital heart defects, pulmonary hypertension, Eisenmenger syndrome, follow-up studies, Competence Network for Congenital Heart Defects.
1. MEDICAL MANAGEMENT - STUDY RESULTS
Timing of treatment in Eisenmenger patients is a sensitive issue, particularly in stable patients. There exist no studies or guidelines on this subject. Especially for the stable patient, the “noli-me-tangere” ruling the treatment strategies for Eisenmenger patients still holds -at least to some extend-with respect to the delicate balance of the many variables, in spite of the available specific pulmonary vasodilators. On the other hand, treatment may indeed appear indicated because of reduced exercise tolerance, increasing cyanosis, or increasing signs of heart failure [1].
However, it has to be kept in mind, that reduction of right ventricular afterload by pulmonary vasodilators is impossible in univentricular function. In other subtypes of congenital heart defects with Eisenmenger reaction (ER), the effect of these drugs may be more pronounced on the systemic vasculature, leading to systemic vasodilatation and, consecutively, in increased cyanosis. If indeed the pulmonary vascular system preferentially vasodilates, the increased pulmonary blood flow may cause left heart failure and pulmonary edema.
1.1. Anticoagulation
Systemic anticoagulation in patients with Eisenmenger syndrome (ES), remains controversial as randomized controlled data are lacking [2]. Eisenmenger patients suffer from multivarious haemostatic abnormalities. On the one hand, ES is associated with a variety of procoagulant biochemical aberrations [3]. Thromboembolic events in the pulmonary circulation occur in approximately 20% of these patients [4]. Therefore, anticoagulation seems logical for the prevention and treatment of thrombosis [5].
Otherwise, Eisenmenger patients are at increased risk of fatal and life threatening bleeding complications, particularly significant hemoptysis [4]. Besides a deficiency of coagulation factors and abnormal fibrinolysis, there is thrombocytopenia with platelet malfunction.
Moreover, it is difficult to assess the optimal level of anticoagulation by routine laboratory tests (see below). So far, systemic anticoagulation and the use of platelet aggregation inhibitors is still not generally recommended in Eisenmenger patients.
1.2. Long-Term Oxygen-Therapy
The use of long-term oxygen supplementation in adult patients with ES is controversial. There are few data and only one published study with a prospective controlled design [6]. Although some patients (e.g. with intense hypoxemia, dyspnea at rest and loss of vital capacity) might subjectively benefit from oxygen supplementation, the risk and side-effects of this therapy (e.g. desiccation of nasal mucosa, epistaxis, sleep disturbance, etc.) should be taken into account. In addition, the above-mentioned trial [6] showed no impact of nocturnal oxygen therapy on exercise capacity, natural history and survival of the patients within a follow up period of 2 years.
According to the guidelines, supplemental oxygen is a general recommendation for PAH patients. The routine use of supplemental oxygen at home is not recommended for Eisenmenger patients, the use should be at the treating physician’s discretion.
1.3. Nitric Oxide (NO)
Nitric oxide (NO) is a potent and selective pulmonary arteriolar vasodilator produced in endothelial cells [7]. It has a crucial function in regulating basal vascular resistance and may also affect platelets and vascular endothelial remodeling. Studies have shown inhaled NO to reduce pulmonary vascular resistance with minimal systemic effects in patients with ES and acute pulmonary hypertensive states of other etiologies [8, 9]. Amongst these, beneficial hemodynamic effects have been anecdotally described in children with ES [10].
Due to the need of continuous inhalation, NO does not play any role in the long-term therapy and therefore only rare patients have been treated chronically. However, NO has an important role in the acute post-operative therapy. In addition, NO is established for the assessment of pulmonary vascular reactivity to identify patients with advanced pulmonary hypertension and ES, who could benefit from sustained vasodilator treatment [11].
1.4. Calcium channel blockers (CCBs)
The use of high doses of oral calcium channel blockers in pulmonary arterial hypertension remains very limited. There is a lack of randomized controlled trials and the available data are restricted to patients with iPAH.
Even though favorable clinical and prognostic effects of high doses of CCB drugs have been suggested [12], these effects were only demonstrable in patients with iPAH with an acute response to vasodilator testing (“responders”) [13, 14]. Hence, merely a minority subgroup of patients appears to benefit from this therapy. By contrast, CCB use in associated forms of PAH has been discouraged, particularly in patients who do not fulfill the criteria of hemodynamic responders, e.g. Eisenmenger patients [15].
As the effects of CCB are not restricted to the pulmonary circulation, vasodilator therapy with CCBs could even cause complications. In Eisenmenger patients, systemic vascular resistance could be lowered more than pulmonary vascular resistance, thus increasing the right-to-left shunt with worsening cyanosis and hypotension. For this reason, empiric CCB therapy in adult Eisenmenger patients is not recommended [15].
1.5. Endothelin-1 (ET-1) Receptor Antagonists
ET-1 is a powerful vasoconstrictor with elevated concentrations in the plasma and lung tissue of patients with PAH. It plays a key role in the pathogenesis of PAH including in vitro effects on proliferation, fibrosis and inflammation. As increased ET-1 plasma levels have been correlated with the severity and prognosis of PAH [16], the ET-1 pathway represents an important treatment target.
Bosentan
Bosentan is a non-selective endothelin receptor antagonist with dual activity on both ETA and ETB receptors and thus completely blocking the activity of ET-1. It is the first oral drug of this medical category, which has been approved by the FDA and EMEA in 2002 as orphan-drug for the treatment of pulmonary hypertension, and currently also for mildly symptomatic patients [17]. Furthermore, since July 2009 Bosentan is the only approved drug for the treatment of PAH in children, as there is a paediatric formulation approved for children with an age of at least 2 years [18, 19].
Particularly for the treatment of Eisenmenger patients, several case series and uncontrolled studies have been published, consistently demonstrating an improvement in exercise capacity and hemodynamics with bosentan treatment [20-23].
BREATHE-5 was designed as the first randomized, placebo-controlled and double-blind trial exclusively enrolling Eisenmenger patients. After a treatment period of 16 weeks receiving bosentan, patients showed a significant improvement in hemodynamics and 6 minute walking distance (6 MWD), without adversely affecting systemic arterial oxygen saturation [24]. In the BREATHE-5 open-label extension study, improvement in exercise capacity was maintained up to 40 weeks [25]. So far, the results of this follow-up were confirmed in two prospective, uncontrolled and open-label studies, which demonstrated an initial persistent improvement of objective exercise capacity, but a decline after one year [26] with reduction to baseline levels after two years [27]. In children, deterioration seemed to be more progressive, whereas in adult patients with the ES, the improvement appeared to last longer. However, these data have to be evaluated carefully due to the limited long-term experience, small subject groups and uncontrolled trial designs. In addition, natural progression of the disease cannot be distinguished from a possible tachyphylaxis.
Overall, bosentan related side effects include dose-dependent elevation of hepatic transaminases, edema and systemic hypotension. Bosentan may also interfere with the action of hormonal contraceptives.
In summary, based on the BREATHE-5 study as well as clinical evidence, bosentan seems to be safe and effective in PAH related to CHD, showing improvement in hemodynamic parameters, exercise capacity and functional class. Further experiences with bosentan in another large cohort of Eisenmenger patients, conducted by the German Competence Network for Congenital Heart Defects, are expected in the near future. Bosentan is currently approved for the treatment of severe PAH related to the ES.
Sitaxsentan
Sitaxsentan is a potent and highly selective ETA receptor antagonist with a distinctive oral bioavailability and a half-life of up to 7 hours, allowing effective once daily oral dosing. Since October 2006, sitaxsentan is the first ETA receptor antagonist approved for the treatment of PAH. There are few randomized-controlled studies (Table 1), demonstrating improvements in exercise capacity, hemodynamic parameters, WHO functional class and clinical events in patients with PAH of different etiologies [28, 29]. Similar to other pharmaceutical agents, the above-mentioned trials of sitaxsentan were predominantly focused on iPAH, while only a minority of patients suffered from PAH associated with CHD. Available data show that sitaxsentan has a lower incidence of hepatic toxicity than bosentan, but affects the metabolism of Warfarin.
Table 1.
Controlled Clinical Trials with Entothelin-1 Receptor Antagonists in Patients with PAH. (Table Adapted from Galie [33])
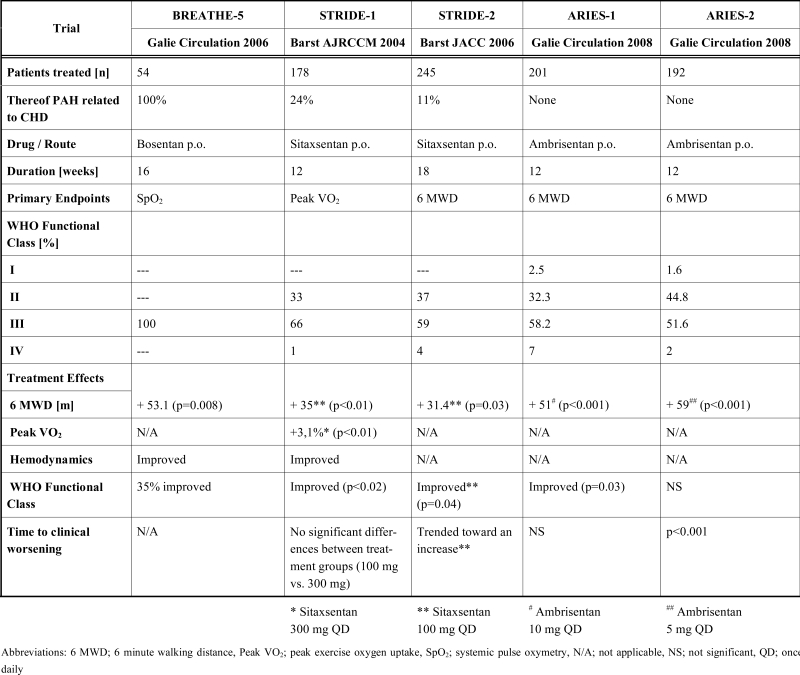 |
First long-term-results of a one year prospective, observational and open-label study have recently been published, suggesting sitaxsentan therapy to be safe and efficacious for patients with PAH. In an uncontrolled study arm, there was a suggestion that patients treated with sitaxsentan demonstrated a longer time to clinical worsening than those in the bosentan group [30]. Unfortunately, there was no subanalysis of the data for the various subgroups of PAH, such as PAH related to CHD. Therefore no conclusion can be drawn concerning Eisenmenger patients.
The experience with sitaxsentan specifically in Eisenmenger patients remains limited. Currently, there are few anecdotal reports and no randomized trials. Only Rosen-zweig [31] reported on a retrospective data analysis of sitaxsentan treatment in 14 Eisenmenger patients. After a follow-up period of up to 13 months, treatment appeared to be safe without a significant decrease in oxygen saturation. There was an improvement in hemodynamics and pulmonary to systemic vascular resistance ratio suggesting pulmonary selectivity. 6 MWD improved, but this was not statistically significant. Limited data underscore the need for further studies in patients with the ES.
Ambrisentan
Ambrisentan, another selective ETA receptor antagonist, has recently been approved for the treatment of PAH in WHO class II and III. ARIES-1 and ARIES-2 [32] (Table 1) demonstrated beneficial effects on exercise capacity (6 MWD), WHO class and time to clinical worsening. Regrettably, no patients with PAH related to CHD were included in the trials and currently there are no further data available for Ambrisentan treatment in such patients. Therefore, no conclusion can be drawn concerning the effect of Ambrisentan in patients with ES.
1.6. Phosphodiesterase-5 (PDE-5) inhibitors
Over the last years, PDE-5 inhibitors have been approved for the treatment of erectile dysfunction. As Type 5 PDE receptors are located predominantly in the penile and pulmonary vasculature, PDE-5 inhibitors are a potential group of medications for the treatment of pulmonary arterial hypertension. Currently, there are three different PDE-5 inhibitors, which have been studied in patients with PAH (Table 2).
Table 2.
PDE-5 Inhibitors in Patients with PAH [34]
| Sildenafil | Vardenafil | Tadalafil | |
|---|---|---|---|
| T max [min] | 60 | 40 - 45 | 75- 90 |
| T ½ [h] | ~ 3,5 | ~ 3,5 | 17,5 |
| PVR/SVR | Decrease | --- | Decrease |
| paO2 | significant improvement | --- | --- |
Abbreviations: T max; time to peak hemodynamic effects, T ½; mean half-life, PVR/SVR; pulmonary to systemic vascular resistance ratio, paO2; arterial oxygenation
Sildenafil and tadalafil1 have shown beneficial effects on pulmonary selectivity and arterial oxygenation.
SUPER-1, the pivotal study of Sildenafil [35], was the first large prospective multicenter blinded and controlled study demonstrating an improvement in exercise capacity, as assessed according to the six-minute walking test, functional class and hemodynamics in PAH patients. Therefore a treatment dose of 20 mg three times per day (TID) has been approved by the FDA and EMEA. Unfortunately, there is no sufficient experience for long-term-treatment. In those patients treated long-term, higher doses were used.
Similar to other vasodilators, sildenafil showed promising effects in patients with iPAH. Recently, favorable effects have been reported in patients with the ES. However, so far there are only a number of individual cases, several case series [36], observational studies and few randomized placebo-controlled trials [37] with increasing evidence for sildenafil in Eisenmenger patients.
In summary, preliminary results have demonstrated that sildenafil is safe and improved symptomatic status, functional class, exercise capacity (6 MWD and exercise duration) and pulmonary hemodynamic parameters in patients with severe pulmonary hypertension related to the ES [38-40]. As these data are based on relatively small subject groups, appropriate studies with larger cohorts are necessary, and are currently being conducted by the German Competence Network for Congenital Heart Defects.
Tadalafil is another PDE-5 inhibitor reported to affect PAH in patients with the ES in an observational study. In one study from India, oxygen saturation and the mean functional class improved after a 12-weeks treatment with tadalafil in selected symptomatic Eisenmenger patients [41]. At this time, the limited available data show tadalafil to be safe and effective in these patients, even though further investigations are required.
1.7. Prostacyclin and Prostacyclin Analogs
Prostanoids can be administered by continuously intravenous or subcutaneous infusion, by inhalation and orally. Due to their active profile with vasodilatory, antiproliferative, anti-inflammatory and anticoagulant effects, they are suitable drugs for the treatment of PAH (Table 3). In current treatment algorithms for patients with WHO Functional Class III and IV, prostacyclin analogs are indicated particularly in patients with right heart failure.
Table 3.
Controlled Clinical Trials with Prostacyclin Analogs in Patients with PAH. (Table Adapted from Galie [33])
| Trial | Treprostinil | ALPHABET | Beraprost-LT | AIR |
|---|---|---|---|---|
| Simonneau AJRCCM 2002 | Galie JACC 2002 | Barst JACC 2003 | Olschewski N Engl J Med 2002 | |
| Patients [n] | 469 | 130 | 116 | 203 |
| Thereof PAH related to CHD | 23% | 18% | 16% | None |
| Drug / Route | Treprostinil s.c. | Beraprost p.o. | Beraprost p.o. | Iloprost inh. |
| Duration [months] | 3 | 3 | 12 | 3 |
| Primary Endpoints | 6 MWD | 6 MWD | Disease progression | 6 MWD |
| Peak VO2 | WHO Functional Class | |||
| WHO Functional Class [%] | ||||
| I | --- | --- | --- | --- |
| II | 11 | 49 | 53 | 59 |
| III | 82 | 51 | 47 | 41 |
| IV | 7 | --- | --- | --- |
| Treatment Effects | ||||
| 6 MWD [m] | + 16 (p=0.006) | + 25 (p=0.036) | + 23 (p=0.18) | + 36 (p=0.06) |
| Peak VO2 | N/A | N/A | Trend to increase (NS) | N/A |
| Hemodynamics | Improved | No Change | No Change | Improved |
| WHO Functional Class | N/A | NS | 58% unchanged (p=0.155) |
65% unchanged |
| Time to clinical worsening | N/A | N/A | N/A | N/A |
Abbreviations: 6 MWD; 6 minute walking distance, Peak VO2; peak exercise oxygen uptake, N/A; not applicable, NS; not significant
Epoprostenol
Prostacyclin is a potent endogenous vasodilator produced in the vascular endothelium. Epoprostenol was the first synthetic prostacyclin analog, which became standard therapy of severe PAH in many countries. Due to a short half-life of a few minutes, continuous i.v.-infusion of epoprostenol is required, which exposes patients to significant side-effects and associated risks. Epoprostenol is well studied in patients with iPAH, and randomized controlled trials have shown improvements in exercise capacity, quality of life and hemodynamics [33, 42]. In patients with PAH caused by congenital cardiac lesions in whom conventional therapy has failed, long-term prostacyclin therapy has shown amelioration in hemodynamics and quality of life following one year of treatment [43, 44], even though there were serious adverse events reported, including cerebrovascular accidents. Another case series studied i.v.-epoprostenol in Eisenmenger patients, showing improved oxygenation and 6 MWD [45]. Unfortunately, the data are limited and there is a lack of randomized controlled trials in patients with ES. Although Eisenmenger patients might also benefit from this treatment option with intravenous epoprostenol, there exist limited data on the efficacy and safety [43].
Iloprost
Intravenous iloprost is a very stable prostacyclin analog and therefore an alternative to i.v.-prostacyclin. Inhaled iloprost has been studied extensively and it has been assumed that it is pulmonary selective, thus minimizing systemic side effects [46]. With a serum half-life of up to 25 minutes after inhalations, inhaled iloprost has to be administered 6-8 times per day. One randomized controlled trial with a 12-weeks treatment period with inhaled iloprost showed beneficial effects in terms of hemodynamics, exercise capacity, symptoms and clinical events [47] in patients with iPAH. So far, the efficacy in adult Eisenmenger patients has not yet been studied.
Treprostinil
Treprostinil is a stable prostacyclin analog with a half-life of three hours, currently available for subcutaneous and intravenous application. The effects of its continuous subcutaneous administration were studied in a large randomized controlled trial with patients suffering from iPAH (58%), PAH related to connective tissue disease (19%) and PAH caused by CHD (24%). There were beneficial effects on exercise capacity, hemodynamics and clinical events [48], but notably a high frequency of site pain limiting subcutaneous administration. For intravenous application, only few data exist with minuscule evidence for PAH in relation to congenital cardiac lesions [49]. Long-term use of treprostinil has resulted in similar survival benefits as i.v. epoprostenol [50].
Beraprost
Beraprost is the first orally active prostacyclin analog that is approved for treatment of iPAH only in Japan. After oral administration, peak concentrations are reached after 30 minutes.
There are two randomized controlled trials with beraprost and a relatively large trial size of 130 and 116 patients, suffering from iPAH and PAH associated with connective tissue disease and CHD, respectively. In the first trial, patients were randomized to receive the maximal tolerated dose of beraprost or placebo for 12 weeks [51]. Subgroup data analysis demonstrated that beraprost improved exercise capacity particularly in patients with iPAH, while those with associated conditions showed no significant changes. Moreover, there were no relevant beneficial effects in cardiopulmonary hemodynamics and WHO functional class. The second trial studied the long-term effects of beraprost treatment up to one year [52]. During earlier phases of treatment, data suggest less disease progression persisting up to 6 months, but after one year, there was no longer any difference between the beraprost and the placebo groups. Therefore, beraprost does not play a crucial role in the treatment of PAH related to congenital cardiac lesions and ES.
1.8. Combination Therapy
Combination of various drugs may have synergistic effects through interaction of different pathobiological pathways [53-56]. Data are limited, but combination therapy may be considered for symptomatic patients who failed to improve with first-line, monodrug treatment. Several small trials with limited recruiting numbers are under way, but so far there are only anecdotal reports and different single-center experiences of Eisenmenger patients treated with a combination of specific anti-pulmonary hypertensive agents [57, 58]. It could be expected that a combination therapy as goal oriented treatment in Eisenmenger patients may be a more beneficial standard therapy in the future, but caution should be advised to unknown interactions of the agents with regard to potential toxicity. Currently, controlled data justifying the use of combination therapy in ES patients are lacking.
1.9. Outlook for the Future
Future studies need to define valid and practical endpoints that are customized to ES patients. While what we learn and achieve here may serve as a model for pulmonary vascular disease in CHD in general. It will be an important question, whether drugs affecting remodeling processes will be usable.
2. ACKNOWLEDGEMENT
This work was supported, in part, by the Competence Network for Congenital Heart Defects, funded by the Federal Ministry of Education and Research (BMBF), FKZ 01GI0601.
Footnotes
Galie N, Brundage H, Ghofrani A, Oudiz RJ, Simonneau G, Beardsworth A, Chan M, Barst RJ. Tadalafil therapy in pulmonary arterial hypertension: results of a randomized, double-blind, placebo-controlled, phase III study. European Heart Journal 2008; 29 (Abstract Supplement): 519
REFERENCES
- 1.Dimopoulos K, Inuzuka R, Goletto S, et al. Improved survival among patients with Eisenmenger syndrome receiving advanced therapy for pulmonary arterial hypertension. Circulation. 2010;121(1):20–5. doi: 10.1161/CIRCULATIONAHA.109.883876. [DOI] [PubMed] [Google Scholar]
- 2.Benistry J, Landzberg M. Eisenmenger’s syndrome. Curr Treat Options Cardiovasc Med. 1999;1(4):355–62. doi: 10.1007/s11936-999-0031-7. [DOI] [PubMed] [Google Scholar]
- 3.De P S, Soares R, Maeda NY, Bydlowski SP, Lopes AA. Markers of endothelial dysfunction and severity of hypoxaemia in the Eisenmenger syndrome. Cardiol Young. 2005;15(5):504–13. doi: 10.1017/S1047951105001381. [DOI] [PubMed] [Google Scholar]
- 4.Silversides CK, Granton JT, Konen E, Hart MA, Webb GD, Therrien J. Pulmonary thrombosis in adults with Eisenmenger syndrome. J Am Coll Cardiol. 2003;42(11):1982–7. doi: 10.1016/j.jacc.2003.07.022. [DOI] [PubMed] [Google Scholar]
- 5.Broberg C, Ujita M, Babu-Narayan S, et al. Massive pulmonary artery thrombosis with haemoptysis in adults with Eisenmenger's syndrome: a clinical dilemma. Heart. 2004;90(11):e63. doi: 10.1136/hrt.2004.039198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sandoval J, Aguirre JS, Pulido T, et al. Nocturnal oxygen therapy in patients with the Eisenmenger syndrome. Am J Respir Crit Care Med. 2001;164(9):1682–7. doi: 10.1164/ajrccm.164.9.2106076. [DOI] [PubMed] [Google Scholar]
- 7.Wessel DL, Adatia I, Thompson JE, Hickey PR. Delivery and monitoring of inhaled nitric oxide in patients with pulmonary hypertension. Crit Care Med. 1994;22(6):930–8. doi: 10.1097/00003246-199406000-00009. [DOI] [PubMed] [Google Scholar]
- 8.Adatia I, Perry S, Landzberg M, Moore P, Thompson JE, Wessel DL. Inhaled nitric oxide and hemodynamic evaluation of patients with pulmonary hypertension before transplantation. J Am Coll Cardiol. 1995;25(7):1656–64. doi: 10.1016/0735-1097(95)00048-9. [DOI] [PubMed] [Google Scholar]
- 9.Roberts JD Jr, Lang P, Bigatello LM, Vlahakes GJ, Zapol WM. Inhaled nitric oxide in congenital heart disease. Circulation. 1993;87(2):447–53. doi: 10.1161/01.cir.87.2.447. [DOI] [PubMed] [Google Scholar]
- 10.Wimmer M, Schlemmer M. Long-term hemodynamic effects of nifedipine on congenital heart disease with Eisenmenger's mechanism in children. Cardiovasc Drugs Ther. 1992;6(2):183–6. doi: 10.1007/BF00054569. [DOI] [PubMed] [Google Scholar]
- 11.Post MC, Janssens S, Van de Werf F, Budts W. Responsiveness to inhaled nitric oxide is a predictor for mid-term survival in adult patients with congenital heart defects and pulmonary arterial hypertension. Eur Heart J. 2004;25(18):1651–6. doi: 10.1016/j.ehj.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 12.Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med. 1992;327(2):76–81. doi: 10.1056/NEJM199207093270203. [DOI] [PubMed] [Google Scholar]
- 13.Olschewski H. [Current recommendations for the diagnosis and treatment of pulmonary hypertension] Dtsch Med Wochenschr. 2006;131(49 Suppl 9):S334–7. doi: 10.1055/s-2006-957206. [DOI] [PubMed] [Google Scholar]
- 14.Sitbon O, Humbert M, Jais X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation. 2005;111(23):3105–11. doi: 10.1161/CIRCULATIONAHA.104.488486. [DOI] [PubMed] [Google Scholar]
- 15.Depta J, Krasuski R. Evidence-based Medical Management of Pulmonary Hypertension 2008: Review of Updated 2007 ACCP Guidelines. Adv Pulm Hyperten. 2008;7(1):222–7. [Google Scholar]
- 16.Galie N, Manes A, Branzi A. The endothelin system in pulmonary arterial hypertension. Cardiovasc Res. 2004;61(2):227–37. doi: 10.1016/j.cardiores.2003.11.026. [DOI] [PubMed] [Google Scholar]
- 17.Galie N, Rubin L, Hoeper M, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet. 2008;371(9630):2093–100. doi: 10.1016/S0140-6736(08)60919-8. [DOI] [PubMed] [Google Scholar]
- 18.Beghetti M, Hoeper MM, Kiely DG, et al. Safety experience with bosentan in 146 children 2-11 years old with pulmonary arterial hypertension: results from the European Postmarketing Surveillance program. Pediatr Res. 2008;64(2):200–4. doi: 10.1203/PDR.0b013e318179954c. [DOI] [PubMed] [Google Scholar]
- 19.Beghetti M, Haworth SG, Bonnet D, et al. Pharmacokinetic and clinical profile of a novel formulation of bosentan in children with pulmonary arterial hypertension: the FUTURE-1 study. Br J Clin Pharmacol. 2009;68(6):948–55. doi: 10.1111/j.1365-2125.2009.03532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Apostolopoulou SC, Manginas A, Cokkinos DV, Rammos S. Effect of the oral endothelin antagonist bosentan on the clinical, exercise, and haemodynamic status of patients with pulmonary arterial hypertension related to congenital heart disease. Heart. 2005;91(11):1447–52. doi: 10.1136/hrt.2004.051961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christensen DD, McConnell ME, Book WM, Mahle WT, et al. Initial experience with bosentan therapy in patients with the Eisenmenger syndrome. Am J Cardiol. 2004;94(2):261–3. doi: 10.1016/j.amjcard.2004.03.081. [DOI] [PubMed] [Google Scholar]
- 22.Gatzoulis MA, Rogers P, Li W, et al. Safety and tolerability of bosentan in adults with Eisenmenger physiology. Int J Cardiol. 2005;98(1):147–51. doi: 10.1016/j.ijcard.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 23.Schulze-Neick I, Gilbert N, Ewert R, et al. Adult patients with congenital heart disease and pulmonary arterial hypertension: first open prospective multicenter study of bosentan therapy. Am Heart J. 2005;150(4):716. doi: 10.1016/j.ahj.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 24.Galie N, Beghetti M, Gatzoulis MA, et al. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study. Circulation. 2006;114(1):48–54. doi: 10.1161/CIRCULATIONAHA.106.630715. [DOI] [PubMed] [Google Scholar]
- 25.Gatzoulis MA, Beghetti M, Galie N, et al. Longer-term bosentan therapy improves functional capacity in Eisenmenger syndrome: results of the BREATHE-5 open-label extension study. Int J Cardiol. 2008;127(1):27–32. doi: 10.1016/j.ijcard.2007.04.078. [DOI] [PubMed] [Google Scholar]
- 26.van Loon RL, Hoendermis ES, Duffels MG, et al. Long-term effect of bosentan in adults versus children with pulmonary arterial hypertension associated with systemic-to-pulmonary shunt: does the beneficial effect persist? Am Heart J. 2007;154(4):776–82. doi: 10.1016/j.ahj.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 27.Apostolopoulou SC, Manginas A, Cokkinos DV, Rammos S. Long-term oral bosentan treatment in patients with pulmonary arterial hypertension related to congenital heart disease: a 2-year study. Heart. 2007;93(3):350–4. doi: 10.1136/hrt.2006.100388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barst RJ, Langleben D, Frost A, et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;169(4):441–7. doi: 10.1164/rccm.200307-957OC. [DOI] [PubMed] [Google Scholar]
- 29.Barst RJ, Langleben D, Badesch D, et al. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J Am Coll Cardiol. 2006;47(10):2049–56. doi: 10.1016/j.jacc.2006.01.057. [DOI] [PubMed] [Google Scholar]
- 30.Benza RL, Barst RJ, Galie N, et al. Sitaxsentan for the treatment of pulmonary arterial hypertension: a one year, prospective, open label, observation of outcome and survival. Chest. 2008;134(4):775–82. doi: 10.1378/chest.07-0767. [DOI] [PubMed] [Google Scholar]
- 31.Rosenzweig E, Rowan C, Barker L. Sitaxsentan Treatment In Patients With Eisenmenger Syndrome. Circulation. 2007;116:II_457. [Google Scholar]
- 32.Galie N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117(23):3010–9. doi: 10.1161/CIRCULATIONAHA.107.742510. [DOI] [PubMed] [Google Scholar]
- 33.Galie N, Seeger W, Naeije R, Simonneau G, Rubin LJ. Comparative analysis of clinical trials and evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):81S–8S. doi: 10.1016/j.jacc.2004.02.038. [DOI] [PubMed] [Google Scholar]
- 34.Ghofrani HA, Voswinckel R, Reichenberger F, et al. Differences in hemodynamic and oxygenation responses to three different phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension: a randomized prospective study. J Am Coll Cardiol. 2004;44(7):1488–96. doi: 10.1016/j.jacc.2004.06.060. [DOI] [PubMed] [Google Scholar]
- 35.Galie N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148–57. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 36.Lim ZS, Salmon AP, Vettukattil JJ, Veldtman GR. Sildenafil therapy for pulmonary arterial hypertension associated with atrial septal defects. Int J Cardiol. 2007;118(2):178–82. doi: 10.1016/j.ijcard.2006.06.045. [DOI] [PubMed] [Google Scholar]
- 37.Singh TP, Rohit M, Grover A, Malhotra S, Vijayvergiya R. A randomized, placebo-controlled, double-blind, crossover study to evaluate the efficacy of oral sildenafil therapy in severe pulmonary artery hypertension. Am Heart J. 2006;151(4):851 e1–5. doi: 10.1016/j.ahj.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 38.Garg N, Sharma MK, Sinha N. Role of oral sildenafil in severe pulmonary arterial hypertension: clinical efficacy and dose response relationship. Int J Cardiol. 2007;120(3):306–13. doi: 10.1016/j.ijcard.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 39.Chau EM, Fan KY, Chow WH. Effects of chronic sildenafil in patients with Eisenmenger syndrome versus idiopathic pulmonary arterial hypertension. Int J Cardiol. 2007;120(3):301–5. doi: 10.1016/j.ijcard.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 40.Wort SJ. Sildenafil in Eisenmenger syndrome: safety first. Int J Cardiol. 2007;120(3):314–6. doi: 10.1016/j.ijcard.2007.03.136. [DOI] [PubMed] [Google Scholar]
- 41.Mukhopadhyay S, Sharma M, Ramakrishnan S, et al. Phosphodiesterase-5 inhibitor in Eisenmenger syndrome: a preliminary observational study. Circulation. 2006;114(47):1807–10. doi: 10.1161/CIRCULATIONAHA.105.603001. [DOI] [PubMed] [Google Scholar]
- 42.Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996;334(5):296–302. doi: 10.1056/NEJM199602013340504. [DOI] [PubMed] [Google Scholar]
- 43.Rosenzweig EB, Kerstein D, Barst RJ. Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation. 1999;99(14):1858–65. doi: 10.1161/01.cir.99.14.1858. [DOI] [PubMed] [Google Scholar]
- 44.McLaughlin VV, Genthner DE, Panella MM, Hess DM, Rich S. Compassionate use of continuous prostacyclin in the management of secondary pulmonary hypertension: a case series. Ann Intern Med. 1999;130(9):740–3. doi: 10.7326/0003-4819-130-9-199905040-00014. [DOI] [PubMed] [Google Scholar]
- 45.Fernandes SM, Newburger JW, Lang P, et al. Usefulness of epoprostenol therapy in the severely ill adolescent/adult with Eisenmenger physiology. Am J Cardiol. 2003;91(5):632–5. doi: 10.1016/s0002-9149(02)03328-3. [DOI] [PubMed] [Google Scholar]
- 46.Badesch DB, McLaughlin VV, Delcroix M, et al. Prostanoid therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):56S–61S. doi: 10.1016/j.jacc.2004.02.036. [DOI] [PubMed] [Google Scholar]
- 47.Olschewski H, Simonneau G, Galie N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347(5):322–9. doi: 10.1056/NEJMoa020204. [DOI] [PubMed] [Google Scholar]
- 48.Simonneau G, Barst RJ, Galie Net, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(6):800–4. doi: 10.1164/ajrccm.165.6.2106079. [DOI] [PubMed] [Google Scholar]
- 49.Tapson VF, Gomberg-Maitland M, McLaughlin VV, et al. Safety and efficacy of IV treprostinil for pulmonary arterial hypertension: a prospective, multicenter, open-label, 12-week trial. Chest. 2006;129(3):683–8. doi: 10.1378/chest.129.3.683. [DOI] [PubMed] [Google Scholar]
- 50.Lang I, Gomez-Sanchez M, Kneussl M, et al. Efficacy of long-term subcutaneous treprostinil sodium therapy in pulmonary hypertension. Chest. 2006;129(6):1636–43. doi: 10.1378/chest.129.6.1636. [DOI] [PubMed] [Google Scholar]
- 51.Galie N, Humbert M, Vachiery JL, et al. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2002;39(9):1496–502. doi: 10.1016/s0735-1097(02)01786-2. [DOI] [PubMed] [Google Scholar]
- 52.Barst RJ, McGoon M, McLaughlin V, et al. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2003;41(12):2119–25. doi: 10.1016/s0735-1097(03)00463-7. [DOI] [PubMed] [Google Scholar]
- 53.Hoeper MM, Markevych I, Spiekerkoetter E, Welte T, Niedermeyer J. Goal-oriented treatment and combination therapy for pulmonary arterial hypertension. Eur Respir J. 2005;26(5):858–63. doi: 10.1183/09031936.05.00075305. [DOI] [PubMed] [Google Scholar]
- 54.van Albada ME, Berger RM. Pulmonary arterial hypertension in congenital cardiac disease--the need for refinement of the Evian-Venice classification. Cardiol Young. 2008;18(1):10–7. doi: 10.1017/S1047951107001849. [DOI] [PubMed] [Google Scholar]
- 55.Ghofrani HA, Hoeper MM. Drug combination treatment for pulmonary arterial hypertension. Dtsch Med Wochenschr. 2006;131(49 Suppl 9):S330–3. doi: 10.1055/s-2006-957205. [DOI] [PubMed] [Google Scholar]
- 56.Barst RJ, Gibbs JS, Ghofrani HA, et al. Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S78–84. doi: 10.1016/j.jacc.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okyay K, Cemri M, Boyac B, Yalcn R, Cengel A. Use of long-term combined therapy with inhaled iloprost and oral sildenafil in an adult patient with eisenmenger syndrome. Cardiol Rev. 2005;13(6):312–4. doi: 10.1097/01.crd.0000181618.29506.1e. [DOI] [PubMed] [Google Scholar]
- 58.Lunze K, Gilbert N, Mebus S, et al. First experience with an oral combination therapy using bosentan and sildenafil for pulmonary arterial hypertension. Eur J Clin Invest. 2006;36(Suppl 3):32–8. doi: 10.1111/j.1365-2362.2006.01692.x. [DOI] [PubMed] [Google Scholar]