

Abstract
Diabetic retinopathy (DR) has been classically considered to be a microcirculatory disease of the retina caused by the deleterious metabolic effects of hyperglycemia per se and the metabolic pathways triggered by hyperglycemia. However, retinal neurodegeneration is already present before any microcirculatory abnormalities can be detected in ophthalmoscopic examination. In other words, retinal neurodegeneration is an early event in the pathogenesis of DR which predates and participates in the microcirculatory abnormalities that occur in DR. Therefore, the study of the mechanisms that lead to neurodegeneration will be essential to identify new therapeutic targets in the early stages of DR. Elevated levels of glutamate and the overexpression of the renin- angiotensin-system play an essential role in the neurodegenerative process that occurs in diabetic retina. Among neuroprotective factors, pigment epithelial derived factor, somatostatin and erythropoietin seem to be the most relevant and these will be considered in this review. Nevertheless, it should be noted that the balance between neurotoxic and neuroprotective factors rather than levels of neurotoxic factors alone will determine the presence or absence of retinal neurodegeneration in the diabetic eye. New strategies, based on either the delivery of neuroprotective agents or the blockade of neurotoxic factors, are currently being tested in experimental models and in clinical pilot studies. Whether these novel therapies will eventually supplement or prevent the need for laser photocoagulation or vitrectomy awaits the results of additional clinical research.
Keywords: Diabetic retinopathy, Angiotensin II, Erythropoietin, Glutamate, Retinal neurodegeneration, Neuropeptides, Pigment epithelial derived factor, Somatostatin
INTRODUCTION
Diabetic retinopathy (DR) has been classically considered to be a microcirculatory disease of the retina due to the deleterious metabolic effects of hyperglycemia per se and the metabolic pathways triggered by hyperglycemia (polyol pathway, hexosamine pathway, DAG-PKC pathway, advanced glycation end-products and oxidative stress). However, retinal neurodegeneration is already present before any microcirculatory abnormalities can be detected in ophthalmoscopic examination. In other words, retinal neurodegeneration is an early event in the pathogenesis of DR which predates and participates in the microcirculatory abnormalities that occur in DR[1,2]. This concept was first introduced by Barber et al[3]. These authors observed that one month after inducing diabetes in rats by using streptozotocin there was a high rate of apoptosis (TUNEL positive cells) in the neuroretina without a significant apoptosis in endothelial cells. In the same paper, the authors found a higher rate of apoptosis in the neuroretina from diabetic donors compared to non-diabetic donors, even in the case of a diabetic donor without microvascular abnormalities. These findings have been further confirmed in experimental models. In addition, it has been demonstrated in experimental models that, apart from apoptosis, another feature of retinal neurodegeneration is glial activation[1-5]. Our research group has been able to demonstrate that both apoptosis and glial activation occur in the retina of diabetic patients and precede microvascular abnormalities[6,7] (Figure 1).
Figure 1.
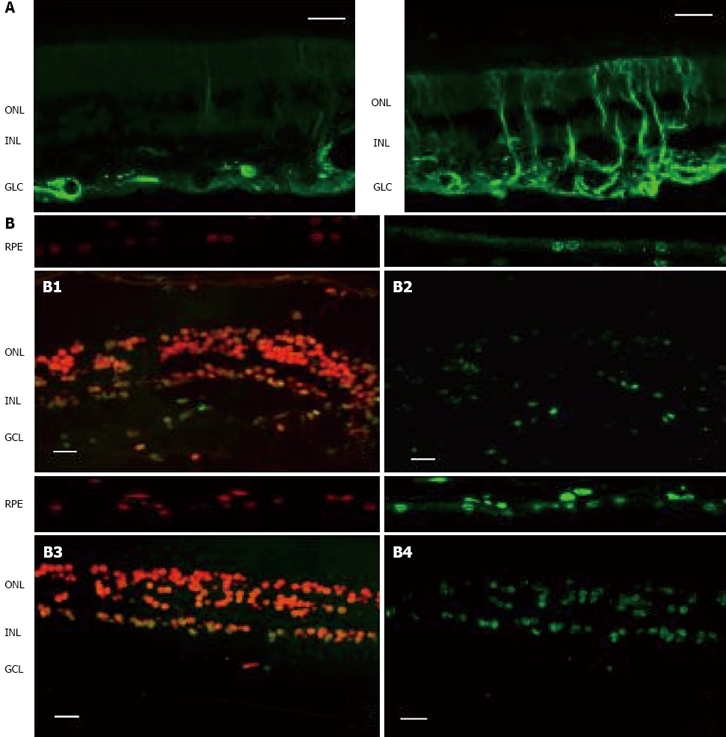
Comparison of the two key elements of neurodegeneration (glial activation and apoptosis) between a representative case of diabetic patient without DR and a non-diabetic subject. As can be seen, neurodegeneration is higher in the retina from the diabetic donor. A: Glial activation in the human retina. Glial fibrillar acidic protein (GFAP) immunofluorescence (green) from a non-diabetic donor (left panel) and a diabetic donor (right panel); B: Apoptosis in the human retina. Upper panel: Non-diabetic donor (B1: Propidium iodide, B2: TUNEL immunofluorescence). Low panel: Diabetic donor (B3: Propidium iodide, B4: TUNEL immunofluorescence). RPE: Retinal pigment epithelium; ONL: Outer nuclear layer; INL: Inner nuclear layer; GCL: Ganglion cell layer. The bar represents 20 μm.
Retinal ganglion cells are the earliest cells affected and have the highest rate of apoptosis[8]. However, an elevated rate of apoptosis has been also observed in the outer nuclear layer (photoreceptors)[9] and in the retinal pigment epithelium (RPE)[10].
Neuroretinal damage produces functional abnormalities such as the loss of chromatic discrimination, contrast sensitivity and dark adaptation. These alterations can be detected by means of electrophysiological studies in diabetic patients with diabetes duration of less than two years, i.e. before microvacular lesions can be detected in ophthalmologic examination. In addition, neuroretinal degeneration will initiate and/or activate several metabolic and signalling pathways which will participate in the microangiopathic process as well as in the disruption of the blood-retinal barrier (a crucial element in the pathogenesis of DR)[11].
The underlying mechanisms that lead to neuronal deficits are likely to be broad. In addition, it is unknown which of the two primordial pathological elements (apoptotis or glial activation) is the first to appear and is consequently the primary event. Nevertheless, it seems that retinal glial cells play an essential role in maintaining the normal function of the retina. When Müller cells (the principal glial cells in the retina) become gliotic, display altered potassium siphoning, glutamate and GABA uptake are also known to express several modulators of angiogenesis[12].
IN VIVO EXPERIMENTAL MODELS TO STUDY RETINAL NEURODEGENERATION IN THE SETTING OF DR
Since neurodegeneration is an early event in the pathogenesis of DR, it is not necessary to use animal models with microangiopatic lesions in the eye such as Torii or Goto-Kakizaki rats. The experimental model currently used to study retinal neurodegeneration in DR is the rat with streptozotocin-induced diabetes (STZ-DM). In this model, electroretinographic abnormalities are present two weeks after inducing diabetes[13] and the presence of neural apoptosis and glial reaction can be clearly detected one month after starting diabetes[3,5]. However, it should be noted that the interpretation of the results of retinal neurodegeneration in STZ-DM rats might be hampered by the neurotoxic effect of STZ. Streptozotocin is a potent neurotoxic agent and is able to produce neural degeneration. Therefore, neurodegeneration (apoptosis + glial activation) observed in rats with STZ-DM could be due to STZ itself rather than the metabolic pathways related to diabetes. It is worthy of mention that pathological changes to the brain after an intraventricular injection of STZ are very similar to the neurodegeneration reported in DR[14]. Therefore, it may be advisable to use murine models with a spontaneous development of diabetes or at least experimental models in which diabetes has not been induced by a neurotoxic drug.
Mice have been used much less than rats as experimental models for the study of DR and retinal neurodegeneration. This is because they are more resistant to the STZ effect (mice need 3-5 doses of STZ to induce diabetes whereas in rats one dose is sufficient), have lower eye cups and present a lower degree of lesions compared to rats. This relative protection in developing pathological lesions related to diabetes can be partly attributed to a lower activity of aldose reductase (polyol pathway) compared to rats[5]. Nevertheless, because of its great potential for genetic manipulation, the mouse offers a unique opportunity to study the molecular pathways involved in disease development. Among mice, C57BL/KsJ-db/db is the model that best reproduces the neurodegenerative features observed in patients with DR. C57BL/KsJ-db/db mice carry a mutation in the leptin receptor gene and are a model for obesity-induced type 2 diabetes. They develop hyperglycemia starting at ~8 wk of age as a result of excessive food consumption. It is noteworthy that they present an abundant expression of aldose-reductase in the retina (an important differential trait from other mouse models)[15]. Therefore, C57BL/KsJ-db/db seems appropriate for investigating the underlying mechanisms of retinal neurodegeneration associated with diabetes and for testing new drugs.
NEUROTOXIC AND NEUROPROTECTIVE FACTORS
The main neurotoxic metabolite involved in diabetic retinal neurodegeneration is glutamate. In addition, there is emerging information about the neurotoxicity due to angiotensin II in the setting of the RAS overexpression that exists in DR. The role of other neurotoxic factors has yet to be elucidated.
Several types of insult cause the upregulation of neurotrophic factors and their receptors in the retina resulting in decreased photoreceptor cell death from subsequent injury. This phenomenon is more prominent in rats than in mice and neurotrophic factors are more efficacious in rats than mice[16]. Among the neuroprotective factors, pigment epithelial derived factor (PEDF), somatostatin (SST) and erythropoietin (Epo) seem to be the most relevant and will be reviewed in this paper. However, there are other neuroprotective factors such as neuroprotectin D1 (NPD1), brain-derived neurotrophic factor (BDNF), glial cell-line-derived neurotrophic factor (GDNF), ciliary neurotrophic factor (CNTF) and adrenomedullin (AM). It is worthy of mention that the balance between the neurotoxic and neuroprotective factors will determine the fate of the retinal neurons.
Glutamate
Glutamate is the major excitatory neurotransmitter in the retina and is involved in neurotransmission from photoreceptors to bipolar cells and from bipolar cells to ganglion cells. However, an elevated glutamate level (which results in excessive stimulation) is implicated in the so called “excitotoxicity” which leads to neurodegeneration. The excitotoxicity of glutamate is the result of overactivation of N-metil-D-aspartame receptors which have been overexpressed in DM-STZ rat receptors[17]. There are at least two mechanisms involved in glutamate-induced apoptosis: a caspase-3-dependent pathway and a caspase-independent pathway involving calpain and mitochondrial apoptosis inducing factor[18].
Elevated levels of glutamate in the retina have been found in experimental models of diabetes as well as in the vitreous fluid of diabetic patients with PDR[19,20]. However, there is no information about this issue in the earlier stages of DR.
The cause of the high levels of glutamate in DR has been related to a dysfunction of macroglia in metabolising glutamate[21]. The reason for this dysfunction seems to be related to impairment in the glutamate transporter of Müller cells due to diabetes-induced oxidative stress[22]. In addition, two enzymatic abnormalities in glutamate metabolism have been found in the diabetic retina: transamination to alpha-ketoglutarate and amination to glutamine. The reduced flux through these pathways may be associated with the accumulation of glutamate[23].
Angiotensin II
The blockade of the RAS with a converting enzyme inhibitor or by using angiotensin II type 1 (AT1) receptor blockers (ARBs) is one of the most used strategies for hypertension treatment in diabetic patients. Apart from the kidney, the RAS system is expressed in the eye. In the retina, RAS components are largely found and synthesized in two sites: neurons and glia cells in the inner retina and in blood vessels[24]. The finding of renin and angiotensin in glia and neurons suggests a role for these molecules in neuromodulation.
There is growing evidence that RAS activation in the eye plays an important role in the pathogenesis of DR[24]. Therefore, apart from lowering BP, the blockade of the RAS could also be beneficial “per se” in reducing the development and progression of DR. In fact, recent evidence supports the concept that RAS blockade in normotensive patients has beneficial effects in the incidence and progression of DR[25-27].
The major components of RAS have been identified in ocular tissues and are overexpressed in the diabetic retina. Angiotensin II binds and activates two primary receptors, AT1-R and AT2-R. In adult humans activation of the AT1-R dominates the pathological states. AT1-R activation by angiotensin II produced by the retina stimulates several pathways involved in the pathogenesis of DR such as inflammation, oxidative stress, cell proliferation, pericyte migration, remodelling of extracellular matrix by increasing matrix metalloproteinases, angiogenesis and fibrosis (Figure 2)[24]. In addition, AT1-R activation by angiotensin II promotes leukostasis (the inappropriate adherence of leukocytes to the retinal capillaries) and neurodegeneration[24,28]. Apart from reducing microvascular disease, there is growing evidence pointing to neuroprotection as a relevant mechanism involved in the beneficial effects of ARBs in DR. In this regard, it has recently been reported that candesartan (the ARB with the best diffusion across the blood-brain barrier) has a neuroprotective effect after brain focal ischemia[29]. In addition, telmisartan and valsartan inhibit the synaptophysin degradation that exists in the retina of a murine model of DR[30]. Moreover, valsartan is able to prolong the survival of astrocytes and reduce glial activation in the retina of rats with hypoxia-induced retinopathy[31]. Furthermore, mitochondrial oxidative stress asociated with retinal neurodegeneration has been improved by using losartan in a model of spontaneously hypertensive rats[32]. Taken together, it seems that neuroprotection is a relevant mechanism involved in the beneficial effects of ARBs in DR.
Figure 2.

AT1-R activation by angiotensin II produced by the retina stimulates several pathways involved in the pathogenesis of DR such as inflammation, oxidative stress, leukostasis and angiogenesis. AT1-R:Angiotensin II type 1 receptor; AT2-R: Angiotensin II type 2 receptor; AGEs: Advanced glycated end products; BM: Basal membrane; BRB: Blood retinal barrier; ECM: Extracellular matrix; VEGF: Vascular endothelial growth factor.
PEDF
PEDF is a 50 kDa protein encoded by a single gene that is preserved across phyla from fish to mammals. It shares homology with the serine proteinase inhibitor (Serpin) family but lacks proteinase activity. PEDF was first purified from human RPE cells and was described as a neurotrophic factor with neuroprotective properties[33]. In this regard, it should be noted that intraocular gene transfer of PEDF significantly increases neuroretinal cell survival after ischemia-reperfusion injury[34] and excessive light exposure[35]. In addition, PEDF protects neurons from glutamate-mediated neurodegeneration[36].
Apart from its neurotrophic factor and neuroprotective properties, there is growing evidence that PEDF is among the most important natural inhibitors of angiogenesis and that it is the main factor accounting for the antiangiogenic activity of vitreous fluid where it is found in abundant quantities[37]. PEDF is responsible for the avascularity of the cornea and vitreous fluid and under hypoxic conditions its secretion is decreased. In addition, elevated glucose down-regulates PEDF expression in RPE cells. The receptors for PEDF are not known but experimental studies suggest that there may be receptors in the retina[38-40]. In addition, it has been suggested that antiangiogenic and neurotrophic activities reside in separate regions of the molecule, thus suggesting that more than one receptor exists[40,41].
Therefore, there are enough arguments to propose PEDF as a serious new candidate for DR treatment. PEDF can successfully be delivered to the eye by viral vectors. As an alternative to viral-mediated gene transfer, transplantation of autologous cells transfected with plasmids encoding for PEDF delivers therapeutic doses of PEDF to the eye. Another mechanism for delivering PEDF to the eye is to exploit its endogenous availability or production. It seems likely that much of the endogenous PEDF in the eye is bound to extracellular matrix molecules and thus may not be active. Drugs that release PEDF from these matrix molecules could increase free PEDF to therapeutic levels. In addition, levels of PEDF mRNA and secreted protein could be increased by either dexamethasone or retinoic acid[42]. Therefore, new strategies for diabetic retinopathy treatment based on PEDF activation are warranted.
SST
SST is a peptide that was originally identified as the hypothalamic factor responsible for the inhibition of the release of the growth hormone from the anterior pituitary. Subsequent studies have shown that SST has a much broader spectrum of inhibitory actions and is much more widely distributed in the body, occurring not only in many regions of the central nervous system but also in many tissues of the digestive tract and in the retina[43]. SST mediates its multiple biological effects via specific plasma membrane receptors that belong to the family of G-protein coupled receptors with seven transmembrane domains. So far, five SST receptor subtypes (SSTRs) have been identified (SSTRs 1-5).
Neuroretina and, in particular, the amacrine cells have been classically described as the main source of SST in the retina. However, we have found that SST expression and content is higher in RPE than in the neuroretina from human eye donors[6] (Figure 3). Therefore, RPE rather than neuroretina is the main source of SST, at least in humans. The amount of SST produced by the human retina is significant as deduced by the strikingly high levels found in the vitreous fluid[44,45]. Apart from SST, SSTRs are also expressed in the retina, with SSTR1 and SSTR2 being the most widely expressed[43,46,47]. The production of both SST and its receptors simultaneously suggests an autocrine action in the human retina.
Figure 3.
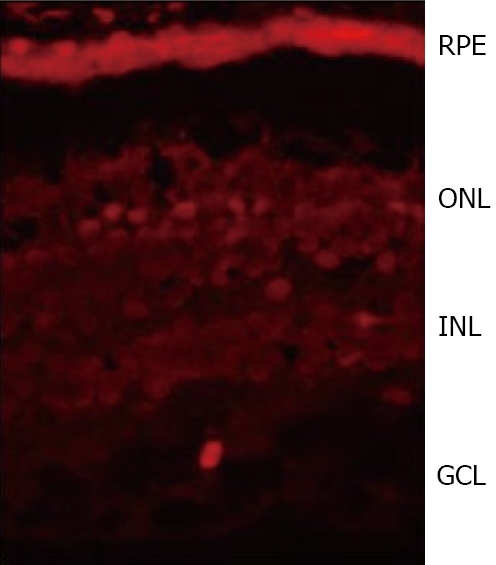
SST immunofluorescence (red) in the human retina showing a higher expression in RPE than in the neuroretina from a human eye donor. RPE:Retinal pigment epithelium; ONL: Outer nuclear layer; INL: Inner nuclear layer; GCL:Ganglion cell layer.
The main functions of SST for retinal homeostasis are the following: (1) SST acts as a neuromodulator through multiple pathways, including intracellular Ca2+ signaling, nitric oxide function and glutamate release from the photoreceptors. In addition, a loss of SST immunoreactivity occurs after degeneration of the ganglion cells. Therefore, the neuroretinal damage that occurs in DR might be the reason for the decreased SST levels detected in the vitreous fluid of these patients. In fact, we have recently found that low SST expression and production is an early event in DR and is associated with retinal neurodegeneration (apoptosis and glial activation)[6]; (2) SST is a potent angiostatic factor. SST may reduce endothelial cell proliferation and neovascularisation by multiple mechanisms including the inhibition of postreceptor signalling events of peptide growth factors such as IGF-I, VEGF, epidermal growth factor and PDGF[48]; and (3) SST has been involved in the transport of water and ions. Various ion/water transport systems are located on the apical side of the RPE adjacent to the subretinal space and a high expression of SST-2 has been shown in this apical membrane of the RPE[46].
In DR there is a downregulation of SST associated with retinal neurodegeneration[6]. The lower expression of SST in RPE and neuroretina is associated with a dramatic decrease of intravitreal SST levels in both PDR[39,40] and DME[49]. As a result, the physiological role of SST in preventing both neovascularisation and fluid accumulation within the retina could be reduced and consequently the development of PDR and DME is favoured. In addition, the loss of neuromodulator activity could also contribute to neuroretinal damage. For all these reasons, intravitreal injection of SST analogues or gene therapy has been proposed as a new therapeutic approach in DR[50].
Epo
Erythropoietin (Epo) was first described as a glycoprotein produced exclusively in fetal liver and adult kidney that acts as a major regulator of erythropoiesis. However, Epo expression has also been found in the human brain and in the human retina[51,52]. In recent years, we have demonstrated that both Epo and its receptor are expressed in the adult human retina[53]. Epo and EpoR mRNAs are significantly higher in RPE than in the neuroretina[53]. In addition, intravitreal levels of Epo are ~3.5 fold higher that those found in plasma[52]. The role of Epo in the retina remains to be elucidated but it seems that it has a potent neuroprotective effect[54,55].
Epo is upregulated in DR[52,53,56,57]. Epo overexpression has been found in both the RPE and neuroretina of diabetic eyes (Figure 4)[52,53]. This is in agreement with the elevated concentrations of Epo found in the vitreous fluid of diabetic patients (~30 fold higher than plasma and ~10 fold higher than in non-diabetic subjects)[52]. Hypoxia is a major stimulus for both systemic and intraocular Epo production. In fact, high intravitreous levels of Epo have recently been reported in ischemic retinal diseases such as PDR[52,56-58]. In addition, it has been reported that Epo has an angiogenic potential equivalent to VEGF[57,59]. Therefore, Epo could be an important factor involved in stimulating retinal angiogenesis in PDR. However, intravitreal levels of Epo have been found at a similar range in PDR to that in DME (a condition in which hypoxia is not a predominant event)[52]. In addition, intravitreal Epo levels are not elevated in non-diabetic patients with macular edema secondary to retinal vein occlusion[60]. Finally, a higher expression of Epo has been detected in the retinas of diabetic donors at early stages of DR compared to non-diabetic donors and this overexpression is unrelated to mRNA expression of hypoxic inducible factors (HIF-1α and HIF-1β)[53]. Therefore, stimulating agents other than hypoxia/ischemia are involved in the upregulation of Epo that exists in the diabetic eye.
Figure 4.
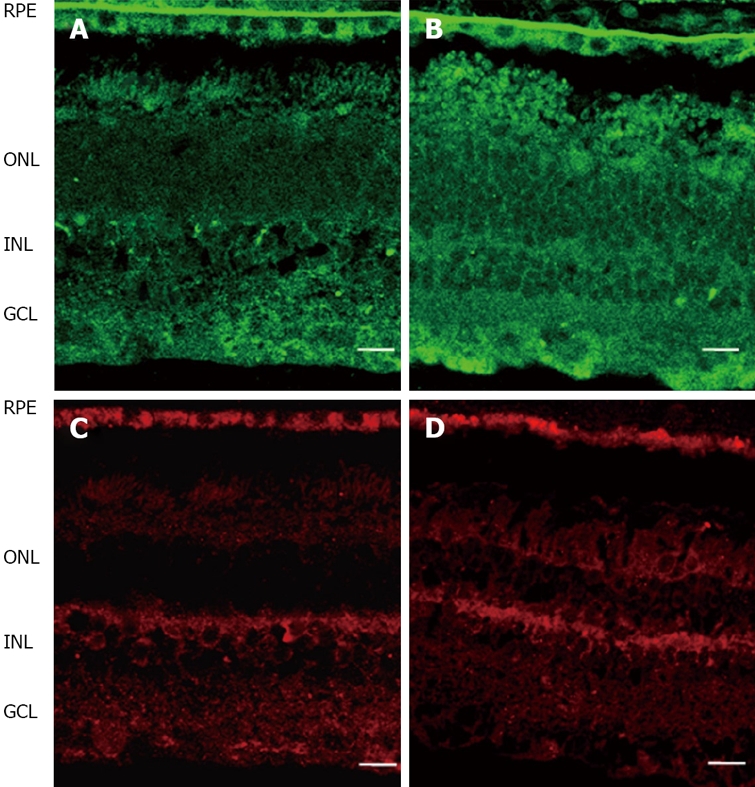
Comparison of Epo (upper pannel, green) and EpoR (lower pannel, red) immunofluorescence in the human retina between representative samples. A, C: Non-diabetic donor; B, D: Diabetic donor. RPE: Retinal pigment epithelium; ONL: Outer nuclear layer; INL: Inner nuclear layer; GCL: Ganglion cell layer. The bar represents 20 mm.
The reason why Epo is increased in DR remains to be elucidated but the bulk of the available information points to a protective effect rather than a pathogenic effect at least in the early stages of DR. In addition, Epo is a potent physiological stimulus for the mobilization of endothelial progenitor cells (EPCs) and therefore could play a relevant role in regulating the traffic of circulating EPCs towards injured retinal sites[61]. In this regard, the increase of intraocular synthesis of Epo that occurs in DR can be contemplated as a compensatory mechanism to restore the damage induced by the diabetic milieu. In fact, exogenous Epo administration by intravitreal injection in early diabetes may prevent retinal cell death and protect the blood retinal barrier function in STZ-DM rats[62]. Nevertheless, in advanced stages, the elevated levels of Epo could potentiate the effects of VEGF thus contributing to neovascularisation and consequently worsening PDR[61,63].
The potential advantages of Epo or EpoR agonists in the treatment of DR include neuroprotection, vessel stability and enhanced recruitment of EPCs to the pathological area. However, as mentioned above, timing is critical since if Epo is given at later hypoxic stages the severity of DR could increase. However, in the case of the eye, disease progression is easy to follow without invasive investigation and allows timing of the administration of drugs to be carefully monitored hopefully resulting in better clinical outcomes.
CONCLUSION
Neurodegeneration is an early event in the pathogenesis of DR. Elevated levels of glutamate and the overexpression of the RAS system play an essential role in the neurodegenerative process that occurs in the diabetic retina. Among the neuroprotective factors, PEDF, SST and Epo seem to be the most important contributing factors but the role of NPD1, BDNF, GDNF, CNTF and AM should also be taken into account. In fact, the balance between neurotoxic and neuroprotective factors rather than the levels of neurotoxic factors alone will determine the presence or not of retinal neurodegeneration in the diabetic eye.
Finally, it should be stressed that the study of the mechanisms that lead to neurodegeneration will be essential for identifying new therapeutic targets in the early stages of DR. At present this is a growing and increasingly important field in medicine which involves several areas of knowledge such as diabetology, ophthalmology and the neurosciences and consequently clinical trials with a multidisciplinary approach are needed.
Acknowledgments
M Villarroel is a recipient of a grant from the Fundació Institut de Recerca Hospital Universitari Vall d’Hebron. CIBER for Diabetes and Associated Metabolic Diseases is an initiative of the Instituto de Salud Carlos III.
Footnotes
Supported by Grants from the Ministerio de Ciencia e Innovacion, No. SAF2009-07408, CIBER de Diabetes y Enfermedades Metabólicas Asociadas and Generaltitat de Catalunya, No. 2009SGR739
Peer reviewers: Renu A Kowluru, PhD, Professor of Ophthalmology, Anatomy/Cell Biology, and Endocrinology, K404, Kresge Eye Institute, Wayne State University, 4717 St Antoine, Detroit, MI 48201, United States; Gregory I Liou, PhD, Professor, Department of Ophthalmology, Medical College of Georgia, Augusta, GA 30912-3400, United States
S- Editor Zhang HN L- Editor Roemmele A E- Editor Liu N
References
- 1.Barber AJ. A new view of diabetic retinopathy: a neurodegenerative disease of the eye. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:283–2902. doi: 10.1016/S0278-5846(03)00023-X. [DOI] [PubMed] [Google Scholar]
- 2.Antonetti DA, Barber AJ, Bronson SK, Freeman WM, Gardner TW, Jefferson LS, Kester M, Kimball SR, Krady JK, LaNoue KF, et al. Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes. 2006;55:2401–2411. doi: 10.2337/db05-1635. [DOI] [PubMed] [Google Scholar]
- 3.Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998;102:783–7916. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lorenzi M, Gerhardinger C. Early cellular and molecular changes induced by diabetes in the retina. Diabetologia. 2001;44:791–804. doi: 10.1007/s001250100544. [DOI] [PubMed] [Google Scholar]
- 5.Asnaghi V, Gerhardinger C, Hoehn T, Adeboje A, Lorenzi M. A role for the polyol pathway in the early neuroretinal apoptosis and glial changes induced by diabetes in the rat. Diabetes. 2003;52:506–511. doi: 10.2337/diabetes.52.2.506. [DOI] [PubMed] [Google Scholar]
- 6.Carrasco E, Hernández C, Miralles A, Huguet P, Farrés J, Simó R. Lower somatostatin expression is an early event in diabetic retinopathy and is associated with retinal neurodegeneration. Diabetes Care. 2007;30:2902–2908. doi: 10.2337/dc07-0332. [DOI] [PubMed] [Google Scholar]
- 7.Carrasco E, Hernández C, de Torres I, Farrés J, Simó R. Lowered cortistatin expression is an early event in the human diabetic retina and is associated with apoptosis and glial activation. Mol Vis. 2008;14:1496–1502. [PMC free article] [PubMed] [Google Scholar]
- 8.Kern TS, Barber AJ. Retinal ganglion cells in diabetes. J Physiol. 2008;586:4401–4408. doi: 10.1113/jphysiol.2008.156695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park SH, Park JW, Park SJ, Kim KY, Chung JW, Chun MH, Oh SJ. Apoptotic death of photoreceptors in the streptozotocin-induced diabetic rat retina. Diabetologia. 2003;46:1260–1268. doi: 10.1007/s00125-003-1177-6. [DOI] [PubMed] [Google Scholar]
- 10.Aizu Y, Oyanagi K, Hu J, Nakagawa H. Degeneration of retinal neuronal processes and pigment epithelium in the early stage of the streptozotocin-diabetic rats. Neuropathology. 2002;22:161–170. doi: 10.1046/j.1440-1789.2002.00439.x. [DOI] [PubMed] [Google Scholar]
- 11.Tretiach M, Madigan MC, Wen L, Gillies MC. Effect of Müller cell co-culture on in vitro permeability of bovine retinal vascular endothelium in normoxic and hypoxic conditions. Neurosci Lett. 2005;378:160–165. doi: 10.1016/j.neulet.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 12.Fletcher EL, Phipps JA, Ward MM, Puthussery T, Wilkinson-Berka JL. Neuronal and glial cell abnormality as predictors of progression of diabetic retinopathy. Curr Pharm Des. 2007;13:2699–2712. doi: 10.2174/138161207781662920. [DOI] [PubMed] [Google Scholar]
- 13.Li Q, Zemel E, Miller B, Perlman I. Early retinal damage in experimental diabetes: electroretinographical and morphological observations. Exp Eye Res. 2002;74:615–625. doi: 10.1006/exer.2002.1170. [DOI] [PubMed] [Google Scholar]
- 14.Shoham S, Bejar C, Kovalev E, Schorer-Apelbaum D, Weinstock M. Ladostigil prevents gliosis, oxidative-nitrative stress and memory deficits induced by intracerebroventricular injection of streptozotocin in rats. Neuropharmacology. 2007;52:836–843. doi: 10.1016/j.neuropharm.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 15.Cheung AK, Fung MK, Lo AC, Lam TT, So KF, Chung SS, Chung SK. Aldose reductase deficiency prevents diabetes-induced blood-retinal barrier breakdown, apoptosis, and glial reactivation in the retina of db/db mice. Diabetes. 2005;54:3119–3125. doi: 10.2337/diabetes.54.11.3119. [DOI] [PubMed] [Google Scholar]
- 16.Wahlin KJ, Adler R, Zack DJ, Campochiaro PA. Neurotrophic signaling in normal and degenerating rodent retinas. Exp Eye Res. 2001;73:693–701. doi: 10.1006/exer.2001.1078. [DOI] [PubMed] [Google Scholar]
- 17.Ng YK, Zeng XX, Ling EA. Expression of glutamate receptors and calcium-binding proteins in the retina of streptozotocin-induced diabetic rats. Brain Res. 2004;1018:66–72. doi: 10.1016/j.brainres.2004.05.055. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Bhavnani BR. Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (AIF) and this process is inhibited by equine estrogens. BMC Neurosci. 2006;7:49. doi: 10.1186/1471-2202-7-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ambati J, Chalam KV, Chawla DK, D’Angio CT, Guillet EG, Rose SJ, Vanderlinde RE, Ambati BK. Elevated gamma-aminobutyric acid, glutamate, and vascular endothelial growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch Ophthalmol. 1997;115:1161–1166. doi: 10.1001/archopht.1997.01100160331011. [DOI] [PubMed] [Google Scholar]
- 20.Pulido JE, Pulido JS, Erie JC, Arroyo J, Bertram K, Lu MJ, Shippy SA. A role for excitatory amino acids in diabetic eye disease. Exp Diabetes Res. 2007;2007:36150. doi: 10.1155/2007/36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lieth E, Barber AJ, Xu B, Dice C, Ratz MJ, Tanase D, Strother JM. Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy. Penn State Retina Research Group. Diabetes. 1998;47:815–820. doi: 10.2337/diabetes.47.5.815. [DOI] [PubMed] [Google Scholar]
- 22.Li Q, Puro DG. Diabetes-induced dysfunction of the glutamate transporter in retinal Müller cells. Invest Ophthalmol Vis Sci. 2002;43:3109–3116. [PubMed] [Google Scholar]
- 23.Lieth E, LaNoue KF, Antonetti DA, Ratz M. Diabetes reduces glutamate oxidation and glutamine synthesis in the retina. The Penn State Retina Research Group. Exp Eye Res. 2000;70:723–730. doi: 10.1006/exer.2000.0840. [DOI] [PubMed] [Google Scholar]
- 24.Wilkinson-Berka JL. Angiotensin and diabetic retinopathy. Int J Biochem Cell Biol. 2006;38:752–765. doi: 10.1016/j.biocel.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 25.Chaturvedi N, Porta M, Klein R, Orchard T, Fuller J, Parving HH, Bilous R, Sjølie AK. Effect of candesartan on prevention (DIRECT-Prevent 1) and progression (DIRECT-Protect 1) of retinopathy in type 1 diabetes: randomised, placebo-controlled trials. Lancet. 2008;372:1394–1402. doi: 10.1016/S0140-6736(08)61412-9. [DOI] [PubMed] [Google Scholar]
- 26.Sjølie AK, Klein R, Porta M, Orchard T, Fuller J, Parving HH, Bilous R, Chaturvedi N. Effect of candesartan on progression and regression of retinopathy in type 2 diabetes (DIRECT-Protect 2): a randomised placebo-controlled trial. Lancet. 2008;372:1385–1393. doi: 10.1016/S0140-6736(08)61411-7. [DOI] [PubMed] [Google Scholar]
- 27.Mauer M, Zinman B, Gardiner R, Suissa S, Sinaiko A, Strand T, Drummond K, Donnelly S, Goodyer P, Gubler MC, et al. Renal and retinal effects of enalapril and losartan in type 1 diabetes. N Engl J Med. 2009;361:40–51. doi: 10.1056/NEJMoa0808400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen P, Scicli GM, Guo M, Fenstermacher JD, Dahl D, Edwards PA, Scicli AG. Role of angiotensin II in retinal leukostasis in the diabetic rat. Exp Eye Res. 2006;83:1041–1051. doi: 10.1016/j.exer.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 29.Krikov M, Thone-Reineke C, Müller S, Villringer A, Unger T. Candesartan but not ramipril pretreatment improves outcome after stroke and stimulates neurotrophin BNDF/TrkB system in rats. J Hypertens. 2008;26:544–552. doi: 10.1097/HJH.0b013e3282f2dac9. [DOI] [PubMed] [Google Scholar]
- 30.Kurihara T, Ozawa Y, Nagai N, Shinoda K, Noda K, Imamura Y, Tsubota K, Okano H, Oike Y, Ishida S. Angiotensin II type 1 receptor signaling contributes to synaptophysin degradation and neuronal dysfunction in the diabetic retina. Diabetes. 2008;57:2191–2198. doi: 10.2337/db07-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Downie LE, Pianta MJ, Vingrys AJ, Wilkinson-Berka JL, Fletcher EL. AT1 receptor inhibition prevents astrocyte degeneration and restores vascular growth in oxygen-induced retinopathy. Glia. 2008;56:1076–1090. doi: 10.1002/glia.20680. [DOI] [PubMed] [Google Scholar]
- 32.Silva KC, Rosales MA, Biswas SK, Lopes de Faria JB, Lopes de Faria JM. Diabetic retinal neurodegeneration is associated with mitochondrial oxidative stress and is improved by an angiotensin receptor blocker in a model combining hypertension and diabetes. Diabetes. 2009;58:1382–1390. doi: 10.2337/db09-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barnstable CJ, Tombran-Tink J. Neuroprotective and antiangiogenic actions of PEDF in the eye: molecular targets and therapeutic potential. Prog Retin Eye Res. 2004;23:561–577. doi: 10.1016/j.preteyeres.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 34.Takita H, Yoneya S, Gehlbach PL, Duh EJ, Wei LL, Mori K. Retinal neuroprotection against ischemic injury mediated by intraocular gene transfer of pigment epithelium-derived factor. Invest Ophthalmol Vis Sci. 2003;44:4497–4504. doi: 10.1167/iovs.03-0052. [DOI] [PubMed] [Google Scholar]
- 35.Imai D, Yoneya S, Gehlbach PL, Wei LL, Mori K. Intraocular gene transfer of pigment epithelium-derived factor rescues photoreceptors from light-induced cell death. J Cell Physiol. 2005;202:570–578. doi: 10.1002/jcp.20155. [DOI] [PubMed] [Google Scholar]
- 36.Bilak MM, Corse AM, Bilak SR, Lehar M, Tombran-Tink J, Kuncl RW. Pigment epithelium-derived factor (PEDF) protects motor neurons from chronic glutamate-mediated neurodegeneration. J Neuropathol Exp Neurol. 1999;58:719–728. doi: 10.1097/00005072-199907000-00006. [DOI] [PubMed] [Google Scholar]
- 37.Dawson DW, Volpert OV, Gillis P, Crawford SE, Xu H, Benedict W, Bouck NP. Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science. 1999;285:245–248. doi: 10.1126/science.285.5425.245. [DOI] [PubMed] [Google Scholar]
- 38.Aymerich MS, Alberdi EM, Martínez A, Becerra SP. Evidence for pigment epithelium-derived factor receptors in the neural retina. Invest Ophthalmol Vis Sci. 2001;42:3287–3293. [PubMed] [Google Scholar]
- 39.Notari L, Baladron V, Aroca-Aguilar JD, Balko N, Heredia R, Meyer C, Notario PM, Saravanamuthu S, Nueda ML, Sanchez-Sanchez F, et al. Identification of a lipase-linked cell membrane receptor for pigment epithelium-derived factor. J Biol Chem. 2006;281:38022–38037. doi: 10.1074/jbc.M600353200. [DOI] [PubMed] [Google Scholar]
- 40.Filleur S, Nelius T, de Riese W, Kennedy RC. Characterization of PEDF: a multi-functional serpin family protein. J Cell Biochem. 2009;106:769–775. doi: 10.1002/jcb.22072. [DOI] [PubMed] [Google Scholar]
- 41.Chader GJ. PEDF: Raising both hopes and questions in controlling angiogenesis. Proc Natl Acad Sci USA. 2001;98:2122–2124. doi: 10.1073/pnas.061024098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tombran-Tink J, Lara N, Apricio SE, Potluri P, Gee S, Ma JX, Chader G, Barnstable CJ. Retinoic acid and dexamethasone regulate the expression of PEDF in retinal and endothelial cells. Exp Eye Res. 2004;78:945–955. doi: 10.1016/j.exer.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 43.Cervia D, Casini G, Bagnoli P. Physiology and pathology of somatostatin in the mammalian retina: a current view. Mol Cell Endocrinol. 2008;286:112–122. doi: 10.1016/j.mce.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 44.Simó R, Lecube A, Sararols L, García-Arumí J, Segura RM, Casamitjana R, Hernández C. Deficit of somatostatin-like immunoreactivity in the vitreous fluid of diabetic patients: possible role in the development of proliferative diabetic retinopathy. Diabetes Care. 2002;25:2282–2286. doi: 10.2337/diacare.25.12.2282. [DOI] [PubMed] [Google Scholar]
- 45.Hernández C, Carrasco E, Casamitjana R, Deulofeu R, García-Arumí J, Simó R. Somatostatin molecular variants in the vitreous fluid: a comparative study between diabetic patients with proliferative diabetic retinopathy and nondiabetic control subjects. Diabetes Care. 2005;28:1941–1947. doi: 10.2337/diacare.28.8.1941. [DOI] [PubMed] [Google Scholar]
- 46.Lambooij AC, Kuijpers RW, van Lichtenauer-Kaligis EG, Kliffen M, Baarsma GS, van Hagen PM, Mooy CM. Somatostatin receptor 2A expression in choroidal neovascularization secondary to age-related macular degeneration. Invest Ophthalmol Vis Sci. 2000;41:2329–2335. [PubMed] [Google Scholar]
- 47.Klisovic DD, O’Dorisio MS, Katz SE, Sall JW, Balster D, O’Dorisio TM, Craig E, Lubow M. Somatostatin receptor gene expression in human ocular tissues: RT-PCR and immunohistochemical study. Invest Ophthalmol Vis Sci. 2001;42:2193–2201. [PubMed] [Google Scholar]
- 48.Davis MI, Wilson SH, Grant MB. The therapeutic problem of proliferative diabetic retinopathy: targeting somatostatin receptors. Horm Metab Res. 2001;33:295–299. doi: 10.1055/s-2001-15286. [DOI] [PubMed] [Google Scholar]
- 49.Simó R, Carrasco E, Fonollosa A, García-Arumí J, Casamitjana R, Hernández C. Deficit of somatostatin in the vitreous fluid of patients with diabetic macular edema. Diabetes Care. 2007;30:725–727. doi: 10.2337/dc06-1345. [DOI] [PubMed] [Google Scholar]
- 50.Hernández C, Simó R. Strategies for blocking angiogenesis in diabetic retinopathy by intravitreal therapy. From basic science to clinical practice. Exp Opin Investig Drug. 2007;16:1209–1226. doi: 10.1517/13543784.16.8.1209. [DOI] [PubMed] [Google Scholar]
- 51.Marti HH. Erythropoietin and the hypoxic brain. J Exp Biol. 2004;207:3233–3242. doi: 10.1242/jeb.01049. [DOI] [PubMed] [Google Scholar]
- 52.Hernández C, Fonollosa A, García-Ramírez M, Higuera M, Catalán R, Miralles A, García-Arumí J, Simó R. Erythropoietin is expressed in the human retina and it is highly elevated in the vitreous fluid of patients with diabetic macular edema. Diabetes Care. 2006;29:2028–2033. doi: 10.2337/dc06-0556. [DOI] [PubMed] [Google Scholar]
- 53.García-Ramírez M, Hernández C, Simó R. Expression of erythropoietin and its receptor in the human retina: a comparative study of diabetic and nondiabetic subjects. Diabetes Care. 2008;31:1189–1194. doi: 10.2337/dc07-2075. [DOI] [PubMed] [Google Scholar]
- 54.Jelkmann W. Effects of erythropoietin on brain function. Curr Pharm Biotechnol. 2005;6:65–79. doi: 10.2174/1389201053167257. [DOI] [PubMed] [Google Scholar]
- 55.Becerra SP, Amaral J. Erythropoietin--an endogenous retinal survival factor. N Engl J Med. 2002;347:1968–1970. doi: 10.1056/NEJMcibr022629. [DOI] [PubMed] [Google Scholar]
- 56.Katsura Y, Okano T, Matsuno K, Osako M, Kure M, Watanabe T, Iwaki Y, Noritake M, Kosano H, Nishigori H, et al. Erythropoietin is highly elevated in vitreous fluid of patients with proliferative diabetic retinopathy. Diabetes Care. 2005;28:2252–2254. doi: 10.2337/diacare.28.9.2252. [DOI] [PubMed] [Google Scholar]
- 57.Watanabe D, Suzuma K, Matsui S, Kurimoto M, Kiryu J, Kita M, Suzuma I, Ohashi H, Ojima T, Murakami T, et al. Erythropoietin as a retinal angiogenic factor in proliferative diabetic retinopathy. N Engl J Med. 2005;353:782–792. doi: 10.1056/NEJMoa041773. [DOI] [PubMed] [Google Scholar]
- 58.Inomata Y, Hirata A, Takahashi E, Kawaji T, Fukushima M, Tanihara H. Elevated erythropoietin in vitreous with ischemic retinal diseases. Neuroreport. 2004;15:877–879. doi: 10.1097/00001756-200404090-00029. [DOI] [PubMed] [Google Scholar]
- 59.Jaquet K, Krause K, Tawakol-Khodai M, Geidel S, Kuck KH. Erythropoietin and VEGF exhibit equal angiogenic potential. Microvasc Res. 2002;64:326–333. doi: 10.1006/mvre.2002.2426. [DOI] [PubMed] [Google Scholar]
- 60.Garcí-Arumí J, Fonollosa A, Macià C, Hernandez C, Martinez-Castillo V, Boixadera A, Zapata MA, Simo R. Vitreous levels of erythropoietin in patients with macular oedema secondary to retinal vein occlusions: a comparative study with diabetic macular oedema. Eye (Lond) 2009;23:1066–1071. doi: 10.1038/eye.2008.230. [DOI] [PubMed] [Google Scholar]
- 61.Chen J, Connor KM, Aderman CM, Smith LE. Erythropoietin deficiency decreases vascular stability in mice. J Clin Invest. 2008;118:526–533. doi: 10.1172/JCI33813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang J, Wu Y, Jin Y, Ji F, Sinclair SH, Luo Y, Xu G, Lu L, Dai W, Yanoff M, et al. Intravitreal injection of erythropoietin protects both retinal vascular and neuronal cells in early diabetes. Invest Ophthalmol Vis Sci. 2008;49:732–742. doi: 10.1167/iovs.07-0721. [DOI] [PubMed] [Google Scholar]
- 63.Grant MB, Boulton ME, Ljubimov AV. Erythropoietin: when liability becomes asset in neurovascular repair. J Clin Invest. 2008;118:467–470. doi: 10.1172/JCI34643. [DOI] [PMC free article] [PubMed] [Google Scholar]