

Abstract
The active sites of enzymes are lined with side chains whose dynamic, geometric, and chemical properties have been finely tuned relative to the corresponding residues in water. For example, the carboxylates of glutamate and aspartate are weakly basic in water but become strongly basic when dehydrated in enzymatic sites. The dehydration of the carboxylate, although intrinsically thermodynamically unfavorable, is achieved by harnessing the free energy of folding and substrate binding to reach the required basicity. Allosterically regulated enzymes additionally rely on the free energy of ligand binding to stabilize the protein in a catalytically competent state. We demonstrate the interplay of protein folding energetics and functional group tuning to convert calmodulin (CaM), a regulatory binding protein, into AlleyCat, an allosterically controlled eliminase. Upon binding Ca(II), native CaM opens a hydrophobic pocket on each of its domains. We computationally identified a mutant that (i) accommodates carboxylate as a general base within these pockets, (ii) interacts productively in the Michaelis complex with the substrate, and (iii) stabilizes the transition state for the reaction. Remarkably, a single mutation of an apolar residue at the bottom of an otherwise hydrophobic cavity confers catalytic activity on calmodulin. AlleyCat showed the expected pH-rate profile, and it was inactivated by mutation of its active site Glu to Gln. A variety of control mutants demonstrated the specificity of the design. The activity of this minimal 75-residue allosterically regulated catalyst is similar to that obtained using more elaborate computational approaches to redesign complex enzymes to catalyze the Kemp elimination reaction.
Keywords: protein design, catalysis, pKa
The design of enzyme-like catalytic proteins is a challenging endeavour that has been approached using combinatorial (1) and computational strategies (2), as well as by following simple chemical principles (3, 4). The Kemp elimination (5) (Scheme 1) is an extensively studied benchmark for catalyst design (6–8) as a model for enzymatic C–H bond abstraction. Previously, computational methods were employed to alter the active sites of natural enzymes to catalyze this reaction (7). Mutation of a dozen or more side chains resulted in catalysts that showed rate enhancements of 104 to 105 relative to a given reference rate. Here, by combining a few physical principles with modern computational tools, we show that a single mutation can confer similar catalytic efficiency into a much simpler and allosterically switchable scaffold that previously had no catalytic activity. This approach circumvents often contentious questions concerning residual activities of the enzymatic framework or the appropriate reference background rate for expression of a “rate enhancement” while also suggesting a practical approach to the design of catalytically amplified sensors.
Scheme 1.

Kemp elimination reaction.
Here we applied a minimalist approach to protein design (9–11), which emphasizes the use of the simple scaffolds and the minimal number of mutations required to achieve a given activity. Dehydrated carboxylates are excellent catalysts of the Kemp elimination (12) due to their basicity toward carbon acids. Thus, an effective catalyst might be engineered by appropriately placing a single carboxylate at the bottom of a hydrophobic pocket, which would destabilize the anionic form and increase its basicity (13–16) and ability to deprotonate C–H bonds. The generation of such a site occurs at a thermodynamic cost, so the starting protein must have sufficient initial stability to withstand the substitution or, in switchable enzymes, receive additional stabilization by association with an allosteric ligand.
Several features were considered in choosing the particular starting scaffold protein for this application: (i) a lack of intrinsic catalytic activity, (ii) spatial separation between the substrate-binding and allosteric ligand-binding sites, (iii) the presence of a cavity of sufficient depth to accommodate the substrate, (iv) high thermodynamic stability, (v) small size to facilitate NMR as well as crystallographic studies and to allow facile preparation by either chemical synthesis or bacterial expression. These criteria were fulfilled by calmodulin (CaM), a prototypical member of the EF-hand class of regulatory proteins, and we chose its C-terminal domain (cCaM, M76-K148) for its stability and high affinity for Ca2+ (17, 18). Importantly, CaM is known to have no catalytic activity in the Kemp elimination reaction (19).
Results
Computational Design.
The C-terminal domain of CaM has a cavity that binds aromatic side chains of peptides, suggesting that it could accommodate the similarly sized benzisoxazole substrate. We computationally scanned the entire cavity using a three-step procedure to identify a site where a Glu or Asp would be accommodated within the pocket projecting the carboxylate toward the target C–H bond of the substrate (Fig. 1). Although the replacement of a hydrophobic side chain with Glu or Asp should lead to some destabilization, it is important that they not entirely disrupt the structure of the protein. Thus, in the first step, individual Glu and Asp substitutions were first examined in all low-energy rotamers, allowing variation in the conformation of the remaining wild-type residues (Fig. S1). The substrate must also be bound productively in the Michaelis complex. It was therefore docked into the single-site mutants remaining after the first step of screening by using an automated procedure (see Materials and Methods) to determine whether the benzisoxazole C–H hydrogen would be appropriately positioned in the Michaelis complex. Finally, a “superrotamer” library (20–22), in which the Glu carboxylate was virtually fused to the substrate in the transition state geometry (23), was evaluated at each position (Fig. S2 and Table S1). Only one position, F92, could accommodate an Asp or Glu in the calcium bound state as well as productively interact with the substrate in the transition state. In particular, Glu gave the lower computed energy, it better positioned its sidechain in a low-energy rotameric configuration to interact with incoming substrate, and it is intrinsically more basic than Asp. Thus, cCaM F92E, which we named AlleyCat (for ALLostEricallY Controlled cATalyst), was strongly predicted to be optimal of the possible mutations.
Fig. 1.

Summary of the computational procedure. A) Identification of potential points to introduce a catalytic residue into cCaM (coordinates from X-ray structure 1EXR). (B) Examination of the Glu residues in the identified positions/docking of the substate. (C) Transition state docking based on a search of a superrotamer library for the transition state.
To test the specificity of the design, we also prepared a total of 13 mutants in which each of the hydrophobic residues in the binding pocket was individually mutated to Asp and Glu (Fig. S3). The active site Glu92 residue was also mutated to Gln, which eliminates the carboxylate, or His, which substitutes the carboxylate for an imidazole base.
Characterization of AlleyCat’s Activity.
AlleyCat exhibited Kemp elimination activity in the presence of 10 mM Ca2+, while F92D was 16-fold less active, F92H showed very low activity (at least 20-fold lower than that of F92E); the remaining 11 control mutants had activities that were not above the background and at least 25-fold lower than AlleyCat. AlleyCat is active for over 40 turnovers and shows no evidence of product inhibition. The initial rate is linearly dependent on substrate concentration at low substrate concentrations and shows the onset of apparent saturation near 1 mM (Fig. S4), as has been observed for other designed proteins (7). However, because the activity coefficient (γ) of this poorly soluble substrate is likely to deviate from unity at high concentrations, we restricted our analysis of the data to the linear region to obtain kcat/KM rather than the individual parameters.
The pH-rate profile and a mutational analysis support the expected mechanism, in which Glu92 with an elevated pKa acts as a general base. The pH-rate profile is typical of a single ionizable group; the value of kcat/Km for the deprotonated form is 5.8 ± 0.3 M-1 s-1, with an apparent pKa = 6.9 ± 0.1 (Fig. 2C). The protonated form showed no activity over background. Also, the wild-type protein and the mutant F92Q were at least 50-fold less active than AlleyCat, providing strong evidence that the measured pKa was associated with the Glu92 (Fig. 2A). Finally, to assure that the activity was not associated with a trace contaminant, we examined a variety of different constructs containing F92E in either the full-length protein or the isolated C-terminal domain, with or without a His-tag and purified using different procedures. Under the same buffer and substrate conditions (see Materials and Methods) the activity of these constructs was constant within 20%, indicating the activity was indeed associated with the C-terminal domain of CaM.
Fig. 2.

(A) Comparison of the benzisoxazole elimination activity of AlleyCat (circles) with cCaM-F92Q (diamonds), [Protein] = 16 μM, [Substrate] = 0.5 mM. (B) Dependence of the activity of AlleyCat on [Ca2+] at pH 6.9, solid line shows the simulated two-site binding isotherm with an average Kd of 10-5 M. (C) pH profile of activity of AlleyCat. (D) Chemical (guanidinium hydrochloride, GdmCl) denaturation profiles of AlleyCat (circles) and cCaM (triangles).
Allosteric Regulation and Ca2+-Dependent Conformational Change.
AlleyCat is allosterically regulated by Ca2+. Its catalytic activity of the protein is strictly Ca2+-dependent, showing less than 25-fold lower activity in the absence of Ca2+. The activity versus [Ca2+] profile conforms well to a tight two-site binding isotherm with a Kd < 10 μM (Fig. 2B). The CD spectrum of CaM shows an increase in the magnitude of the ellipticity at 222 nm (θ222) upon binding of Ca2+. The binding of Ca2+ to AlleyCat triggered an even larger increase in the magnitude of θ222; the value of θ222 was similar for cCaM and AlleyCat in the presence of Ca2+, but in the absence of Ca2+ AlleyCat appeared significantly less structured. This finding is consistent with the expectation that the F92E mutation destabilizes the protein, resulting in a partially folded apo state. Although the calcium bound state was essentially fully folded we expected that its thermodynamic stability might be lower than that of the wild type, because studies of natural and designed proteins show that enzymes often sacrifice thermodynamic stability to place their functional residues in nonoptimal environments (15, 24); for example, the degree of perturbation of a side chain’s pKa can be related to the corresponding decrease in the overall free energy of folding (25, 26). The high apparent pKa of Glu92 is consistent with this hypothesis and predicts that AlleyCat should be thermodynamically less stable than the corresponding wild-type domain. Indeed, the wild-type protein is 3.1 ± 0.6 kcal/mol more stable than AlleyCat in the presence of saturating concentrations of Ca2+ at pH 6.9 (Fig. 2D).
Structural Characterization.
The structure of AlleyCat was determined by heteronuclear NMR, using 1,205 distance and 140 dihedral restraints (average 16.2 restraints/residue, Fig. 3). The computed ensemble of structures (backbone rmsd 1.05 Å) is as well defined as previous NMR structures of cCaM (computed from a similar number of restraints). The centroid is in good agreement with the starting model and the NMR structure of wild-type cCaM (27) (rmsd 1.35 Å). Thus, the mutation has not caused large rearrangements in the backbone. The Glu92 side chain is located at the bottom of the hydrophobic pocket surrounded by apolar residues, where it is well positioned to serve as a general base. Also, the Glu92 carboxylate is well separated from the closest calcium atom, confirming the spatial segregation and distinct roles of the allosteric versus the catalytic sites.
Fig. 3.

NMR structure of AlleyCat. (A) Representation of protein’s surface showing E92 pointing into the hydrophobic binding pocket; carbons and the backbone atoms are shown in green. (B) The 20 conformers with the lowest energy representing the solution structure shown after superposition of the backbone heavy atoms N, Cα, and C′ atoms of the secondary structures. (C) “Sausage” representation of backbone and best-defined side chains: A spline function was drawn through the mean positions of Cα atoms with the thickness proportional to the mean global displacement of Cα atoms in the 20 conformers after superposition as in B. Calcium atoms are shown in yellow.
Discussion
These results illustrate how our emerging understanding of the delicate interplay between protein folding energetics, allosteric ligand binding, and functional group tuning (25, 26) can be combined with computational methods to design an allosterically regulated switch-like protein catalyst. By linking binding at one site to catalysis at a second site it has been possible to create a highly amplified sensor. Thus by using different natural or engineered proteins that bind other ligands it might be possible to design a variety of catalytically amplified sensors.
The primary goal of this work was to link a ligand-driven conformational change to the creation of a novel catalytic activity, which could easily be measured in vitro. The catalytic activity of AlleyCat was sufficient for this purpose, but how does it compare to earlier catalysts of Kemp elimination? A frequently used comparison is the ratio of the value of kcat/KM for AlleyCat versus the second order rate constant for acetate in water, which amounts to 2.8 × 105 in this case. The value of kcat/KM for AlleyCat (5.8 ± 0.3 M-1 s-1) is within the same range as observed for catalytic antibodies (8, 28, 29) (kcat/KM = 1.4–5,500 M-1 s-1). This is a significant accomplishment given that AlleyCat is switchable, its molecular weight is less than one tenth that of IgG antibodies, and AlleyCat was designed by a very simple, reproducible automated procedure.
It is of more interest to compare the present strategy, which incorporated elements of the minimalist design, fundamental physical principles, and fully automated computational design to previous work of Houk, Tawfik, Baker, and coworkers (7). These researchers employed computational algorithms to redesign enzymatic scaffolds, which are arguably more favorable platforms for catalysis than the binding protein used here. Of 59 designed proteins, which were created by substitution of 10–15 aminoacid residues in each design, eight were active for the Kemp elimination, showing kcat/KM ranging from 5.9 M-1 s-1 (for protein KE71) to 163 M-1 s-1 (for protein KE59). These investigators emphasized the importance of structural studies to guide future computational studies, mechanistic studies, and comparison with other designs. Of the 59 proteins, the X-ray structure of one, KE07, was determined, providing an interesting comparison with AlleyCat, which has been also structurally characterized. AlleyCat has an activity similar to that of KE07 on a molar basis and ca. 2.5-fold greater activity on a weight basis. However, mutating the catalytic acid and base of KE07 led to only modest twofold to threefold increases or decreases in activity, suggesting that a step other than proton transfer is rate-determining or, possibly, that the complexity of the design led to a protein that employed an unanticipated mechanism. Although some of the other designs such as KE59 showed the expected response to mutations, their structures were not experimentally determined. By contrast, the simplicity of the minimalist approach not only allowed the design of a switchable catalyst, but the small scaffold and minimal number of mutations facilitated mechanistic determination. AlleyCat uses E92 as the catalytic base as inferred from the rate/pH profile and the F92Q mutation.
Our computational design strategy differs significantly from previous studies (7), which focused exclusively on optimizing interactions to satisfy a potential function developed to predict thermodynamic stability of a protein. As a result the carboxylate would be stabilized by hydrogen-bonded interactions to minimize the overall energy. However, catalysis often requires destabilizing specific groups to enhance their chemical reactivity. In this case, strong desolvation of a carboxylate in an environment replete of H-bond donors enhances its basicity, as was also inferred based on activity-enhancing substitutions observed during the directed evolution of KE07 (7, 30). Secondly, it is important to consider the ground state Michaelis complex to assume that the preferred bound orientation is competent for catalysis. Thus, it is likely that simple considerations such as these could enhance future design work, allowing more sophisticated computations to reach their full potential.
It is likely that the binding site of AlleyCat could be improved to optimize geometric complementarity with the substrate, which might lead to an enhanced KM, better dehydration of the substrate, and increased basicity of the active site Glu. Random mutagenesis might also be used to improve activity without any preconceived notion of the requirements for activity. For example, directed evolution of KE07 led to a 200-fold increase in activity (30). Thus, it will be particularly interesting to determine the extent to which catalytic efficiency of AlleyCat can be increased, given that it is smaller than almost all enzymes and lacked catalytic activity in the starting protein.
Materials and Methods
5-nitrobenzisoxazole has been prepared according to the literature procedure (19).
Calmodulin Mutagenesis and Expression.
The C-terminal portion of chicken calmodulin gene (31) (M76-K148) was cloned into pEXP5-NT/TOPO vector (Invitrogen) giving the gene corresponding to: MSGSHHHHHHGSSGENLYFQSLMKDTDSEEEIREAFRVFDKDGNGYISAA ELRHVMTNLGEKLTDEEVDEMIREADIDGDGQVNYEEFVQMMTAK. Mutagenesis was done with PfuTurbo DNA polymerase (Stratagene) using standard protocols. The proteins were expressed in Escherichia coli BL21(DE3) pLysS cells (Novagen) at 37° (cells were harvested after 1.5 h after induction) and purified over a Ni-NTA column (Qiagen).
The active mutant was additionally purified over DEAE column (Amersham Biosystems) on an ÄKTA FPLC system (Amersham Biosystems). To assess the effect of the His-tag, it was removed using TEV protease (Promega), and the protein purified as described in SI Text. Under standard conditions (500 μM substrate, 20 mM HEPES (pH 6.9); 150 mM NaCl, 1 mM CaCl2) activity (kcat/KM) of the protein with His-tag removed was approximately 95% of the starting His-tagged protein. Full-length CaM F92E mutant was also expressed and purified on phenyl sepharose (31). The ratio of kcat/KM for this protein relative to AlleyCat was 1.2. Thus, the catalytic activities of a variety proteins bearing the F92E mutation were essentially the same irrespective of the nature of the N-terminal domain, purification method, or the presence of a His-tag sequence. Identity of the proteins was confirmed by mass spectrometry. Isotopically labeled samples for NMR studies were expressed on M9 minimal medium containing 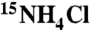 and 13C uniformly labeled glucose (Cambridge Isotopes).
and 13C uniformly labeled glucose (Cambridge Isotopes).
Kinetic Measurements.
Kinetic measurements were done on a SpectraMax M5 plate reader (Molecular Devices) monitoring absorbance at 380 nm at 22 °C using at least three independent measurements. In a typical experiment 2 μL of freshly prepared 5-nitrobenzisoxazole stock solution in acetonitrile was added to 200 mL of buffered (10–25 mM) protein (1–25 μM) solution so the final concentration of substrate ranges from 50 μM to 1 mM. Product’s extinction coefficient (15,800 cm-1 M-1) is taken from ref. 5. Kinetic parameters were obtained by fitting the data to the Michaelis–Menten equation {v0 = kcat[E]0[S]0/(KM + [S0])}. kcat/KM values were obtained by fitting the linear portion of the plot to v0 = (kcat/KM)[E]0[S]0. The following buffers (at 25 mM) were used for the pH profile studies: citrate (pH 4.0–5.5), MES (pH 5.5–6.5), HEPES (pH 6.5–8.0), bicine (8.0–9.0). kcat/KM values were obtained from fitting to kcat/KM = (kcat/KM)protonated + (kcat/KM)deprotonated × 10-pKa/(10-pH + 10-pKa), where pKa is the apparent pKa value of the active residue.
Chemical Denaturation Fits.
The chemical denaturation data were fit to (32):
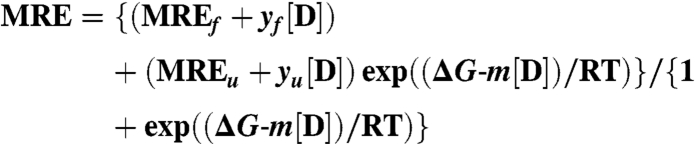 |
Where MRE is observed mean residue ellipticity; MREf and MREu are mean residue ellipticities in the folded and unfolded states, respectively; [D] concentration of denaturant; ΔG is free energy of unfolding, yf and yu are slopes for the folded and unfolded states, respectively. Experimental conditions: 4 mM HEPES (pH 6.9), 30 mM NaCl, 2 mM CaCl2. The fit yields the following thermodynamic parameters for unfolding: ΔG = 15 ± 2 kJ/mol and m = 7.0 ± 0.8 kJ/mol ∗ M for AlleyCat; ΔG = 23 ± 1 kJ/mol and m = 5.3 ± 0.3 kJ/mol ∗ M for cCaM. The ΔΔG value determined from these parameters is 3.1 ± 0.6 kcal/mol (the value was determined at the midpoint between the respective Cm to avoid a long extrapolation to [GdmCl] = 0).
Calmodulin Starting Structure Computational Modeling.
The sequence SLMKDTDSEEEIREAFRVFDKDGNGYISAAELRHVMTNLGEKLTDEEVDE MIREADIDGDGQVNYEEFVQMMTAK was modeled onto the structure of C-terminal domain of CaM (PDB ID code 1QS7) by maintaining the backbone and side chains of the common amino acids and building the amino acids that varied by finding the lowest energy (CHARMM force field) (33) conformation.
Docking.
AutoDock version 4.0 (34) was used to dock 5-nitrobenzisoxazole into CaM (maximum rotational/translational step sizes 50°, 2 Å; population size 150; 50,000 generations). The docking parameters were systematically varied in independent runs all achieving approximately the same docked pose.
Modelling the Structure of Mutants.
First, 100 side chain conformations (rotamers) for the asparate/glutamate position and 50 rotamers for any residue within 8 Å of the Asp/Glu were built using the Molecular Software Libraries (MSL, manuscript in preparation). Then, the pairwise energy table (CHARMM force field) was computed. The global minimum rotamer configuration was found using a cyclic simulated annealing Monte Carlo protocol, assuming the reference state energy to be similar. The final models were processed through a 100-step constrained CHARMM minimization using the Adopted Basis Newton–Raphson algorithm.
Superrotamers.
A superrotamer of glutamate-5-nitrobenzisoxazole was created based on QM calculations (7, 23). The superrotamer models were generated using the same protocol as specified above with the number of rotamers doubled. Partial charges were derived for 5-nitrobenzisoxazole using AM1-BCC (35). The bonded energy parameters for 5-nitrobenzisoxazole not present in CHARMM, were replaced with the most chemically similar terms.
Supplementary Material
Acknowledgments.
We thank A. Joshua Wand for providing a plasmid containing the gene of chicken calmodulin. We thank David Baker and Dan Tawfik for providing genes of KE07. We thank Guy Montelione for providing NMR time and Kathleen Molnar for assistance with mass spectrometry.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The NMR structure of AlleyCat has been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2kz2). Chemical shifts were deposited in the BioMagResBank http://www.bmrb.wisc.edu/ (accession code 16994).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1018191108/-/DCSupplemental.
References
- 1.Patel SC, Bradley LH, Jinadasa SP, Hecht MH. Cofactor binding and enzymatic activity in an unevolved superfamily of de novo designed 4-helix bundle proteins. Protein Sci. 2009;18:1388–1400. doi: 10.1002/pro.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lasilla JK, Privett HK, Allen BD, Mayo SL. Combinatorial methods for small-molecule placement in computational enzyme design. Proc Natl Acad Sci USA. 2006;103:16710–16715. doi: 10.1073/pnas.0607691103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broo KS, Brive L, Ahlberg P, Baltzer L. Catalysis of hydrolysis and transesterification reactions of p-Nitrophenyl esters by a designed helix—loop—helix dimer. J Am Chem Soc. 1997;119:11362–11372. [Google Scholar]
- 4.Johnsson K, Allemann RK, Widmer H, Benner SA. Synthesis, structure and activity of artificial, rationally designed catalytic polypeptides. Nature. 1993;365:530–532. doi: 10.1038/365530a0. [DOI] [PubMed] [Google Scholar]
- 5.Casey M, Kemp D, Paul K, Cox D. Physical organic chemistry of benzisoxazoles. I. Mechanism of the base-catalyzed decomposition of benzisoxazoles. J Org Chem. 1973;38:2294–2301. [Google Scholar]
- 6.Hollfelder F, Kirby AJ, Tawfik DS. Efficient catalysis of proton transfer by synzymes. J Am Chem Soc. 1997;119:9578–9579. [Google Scholar]
- 7.Rothlisberger D, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 8.Thorn SN, Daniels RG, Auditor M-TN, Hilvert D. Large rate acceleration in antibody catalysis by strategic use of haptenic charge. Nature. 1995;373:228–230. doi: 10.1038/373228a0. [DOI] [PubMed] [Google Scholar]
- 9.DeGrado WF, Wasserman ZR, Lear JD. Protein design, a minimalist approach. Science. 1989;243:622–628. doi: 10.1126/science.2464850. [DOI] [PubMed] [Google Scholar]
- 10.Kaplan J, DeGrado WF. De novo design of catalytic proteins. Proc Natl Acad Sci USA. 2004;101:11566–11570. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korendovych IV, et al. De novo design and molecular assembly of a transmembrane diporphyrin-binding protein complex. J Am Chem Soc. 2010;132:15516–15518. doi: 10.1021/ja107487b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kemp D, Casey M. Physical organic chemistry of benzisoxazoles. IV. Origins and catalytic nature of the solvent rate acceleration for the decarboxylation of 3-carboxybenzisoxazoles. J Am Chem Soc. 1975;97:7312–7318. [Google Scholar]
- 13.Isom DG, Cannon BR, Castaneda CA, Robinson A, Garcia-Moreno BE. High tolerance for ionizable residues in the hydrophobic interior of proteins. Proc Natl Acad Sci USA. 2008;105:17784–17788. doi: 10.1073/pnas.0805113105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pey AL, Rodriguez-Larrea D, Gavira JA, Garcia-Moreno BE, Sanchez-Ruiz JM. Modulation of buried ionizable groups in proteins with engineered surface charge. J Am Chem Soc. 2010;132:1218–1219. doi: 10.1021/ja909298v. [DOI] [PubMed] [Google Scholar]
- 15.Shoichet BK, Baase WA, Kuroki R, Matthews BW. A relationship between protein stability and protein function. Proc Natl Acad Sci USA. 1995;92:452–456. doi: 10.1073/pnas.92.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitten ST, Garcia-Moreno BE, Hilser VJ. Ligand effects on the protein ensemble: Unifying the descriptions of ligand binding, local conformational fluctuations, and protein stability. Method Cell Biol. 2008;84:871–891. doi: 10.1016/S0091-679X(07)84027-1. [DOI] [PubMed] [Google Scholar]
- 17.Falke JJ, Drake SK, Hazard AL, Peersen OB. Molecular tuning of ion binding to calcium signaling proteins. Q Rev Biophys. 1994;27:219–290. doi: 10.1017/s0033583500003012. [DOI] [PubMed] [Google Scholar]
- 18.Shaw GS, Hodges RS, Sykes BD. Calcium-induced peptide association to form an intact protein domain: 1H NMR structural evidence. Science. 1990;249:280–283. doi: 10.1126/science.2374927. [DOI] [PubMed] [Google Scholar]
- 19.Hollfelder F, Kirby AJ, Tawfik DS, Kikichi K, Hilvert D. Characterization of proton-transfer catalysis by serum albumins. J Am Chem Soc. 2000;122:1022–1029. [Google Scholar]
- 20.Bolton DN, Mayo SL. Enzyme-like proteins by computational design. Proc Natl Acad Sci USA. 2001;98:14274–14279. doi: 10.1073/pnas.251555398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clarke ND, Yuan SM. Metal search: A computer program that helps design tetrahedral metal-binding sites. Proteins. 1995;23:256–263. doi: 10.1002/prot.340230214. [DOI] [PubMed] [Google Scholar]
- 22.Regan L, Clarke ND. A tetrahedral zinc(II)-binding site introduced into a designed protein. Biochemistry. 1990;29:10878–10883. doi: 10.1021/bi00501a003. [DOI] [PubMed] [Google Scholar]
- 23.Hu Y, Houk KN, Kikuchi K, Hotta K, Hilvert D. Nonspecific medium effects versus specific group positioning in the antibody and albumin catalysis of the base-promoted ring-opening reactions of benzisoxazoles. J Am Chem Soc. 2004;126:8197–8205. doi: 10.1021/ja0490727. [DOI] [PubMed] [Google Scholar]
- 24.Faiella M, et al. An artificial di-iron oxo-protein with phenol oxidase activity. Nat Chem Biol. 2009;5:882–884. doi: 10.1038/nchembio.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baase WA, Liu G, Tronrud DE, Matthews BW. Lessons from the lysozyme of phage T4. Protein Sci. 2010;19:631–641. doi: 10.1002/pro.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isom DG, Castaneda CA, Cannon BR, Velu PD, Garcia-Moreno BE. Charges in the hydrophobic interior of proteins. Proc Natl Acad Sci USA. 2010;107:16096–16100. doi: 10.1073/pnas.1004213107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finn BE, et al. Calcium-induced structural changes and domain autonomy in calmodulin. Nat Struct Biol. 1995;2:777–783. doi: 10.1038/nsb0995-777. [DOI] [PubMed] [Google Scholar]
- 28.Debler EW, et al. Structural origins of efficient proton abstraction from carbon by a catalytic antibody. Proc Natl Acad Sci USA. 2005;102:4984–4989. doi: 10.1073/pnas.0409207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Genre-Grandpierre A, et al. Catalysis of the Kemp elimination by antibodies elicitied against a cationic hapten. Bioorg Med Chem Lett. 1997;7:2497–2502. [Google Scholar]
- 30.Khersonsky O, et al. Evolutionary optimization of computationally designed enzymes: kemp eliminases of the KE07 series. J Mol Biol. 2010;396:1025–1042. doi: 10.1016/j.jmb.2009.12.031. [DOI] [PubMed] [Google Scholar]
- 31.Urbauer JL, Short JH, Dow LK, Wand AJ. Structural analysis of a novel interaction by calmodulin: High-affinity binding of a peptide in the absence of calcium. Biochemistry. 1995;34:8099–8109. doi: 10.1021/bi00025a016. [DOI] [PubMed] [Google Scholar]
- 32.Masino L, Martin SR, Bayley PM. Ligand Binding and thermodynamic stability of a multidomain protein, calmodulin. Protein Sci. 2000;9:1519–1529. doi: 10.1110/ps.9.8.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brooks BR, et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morris GM, Huey R, Olson AJ. Using AutoDock for ligand-receptor docking. Curr Protoc Bioinformatics. 2008 doi: 10.1002/0471250953.bi0814s24. Chapter 8 Unit 8.14. [DOI] [PubMed] [Google Scholar]
- 35.Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.