

Abstract
The metastatic invasion of cancer cells from primary tumors to distant ecological niches, rather than the primary tumors, is the cause of much cancer mortality [Zhang QB, et al. (2010) Int J Cancer 126:2534–2541; Chambers AF, Goss PE (2008) Breast Cancer Res 10:114]. Metastasis is a three-dimensional invasion process where cells spread from their site of origin and colonize distant microenvironmental niches. It is critical to be able to assess quantitatively the metastatic potential of cancer cells [Harma V, et al. (2010) PLoS ONE 5:e10431]. We have constructed a microfabricated chip with a three-dimensional topology consisting of lowlands and isolated square highlands (Tepuis), which stand hundreds of microns above the lowlands, in order to assess cancer cell metastatic potential as they invade the highlands. As a test case, the invasive ascents of the Tepui by highly metastatic PC-3 and noninvasive LNCaP prostate cancer cells were used. The vertical ascent by prostate cancer cells from the lowlands to the tops of the Tepui was imaged using confocal microscopy and used as a measure of the relative invasiveness. The less-metastatic cells (LNCaP) never populated all available tops, leaving about 15% of them unoccupied, whereas the more metastatic PC-3 cells occupied all available Tepuis. We argue that this distinct difference in invasiveness is due to contact inhibition.
Keywords: biological physics, oncology, microfabrication
Sir Arthur Conan Doyle wrote a famous science fiction book called The Lost World, which posited that evolution would halt on the isolated mesas with very steep sides called Tepuis that exist in the Guiana Highlands of South America (1). Based on the view that cancer is a communal condition driven by stress (2), we asked if cancer cells, in particular metastatic cancer cells, would migrate away from a densely occupied region and proliferate where there is less competition for resources and space (3, 4). Thus, we designed experiments in the shape of microfabricated Tepuis, so that migration to less-populated regions occur along the steep sides of the microfabricated highlands.
There have been systematic studies of tumor migration and cancer metastasis (5–7), and relevant three-dimensional models have been generated (8). Metastasis is closely related to cell motility and proliferation, and it is known to be a highly controlled process, especially once a niche is established, due to the regulation of cell growth in secondary sites (9). However, the metastatic process is difficult to quantify because of the difficulties of real-time detection of cell behavior with high spatial and temporal resolution in vivo.
Cancer cell motility has already been systematically visualized and analyzed in microchannels and on two-dimensional substrates (10). However, the speed and character of cell motility is quite different from that normally seen in three-dimensional in vivo studies (11). Microfabricated three-dimensional structures have gained much interest in recent years. For example, high-density arrays of posts have been employed to sort and capture individual leukocytes (12), microwells are used for single-cell quantification and imaging (13), arrays of obstacles under flow can separate adherent cells (14), and techniques have been developed for patterning cells at specific locations to monitor cell–cell interactions (15).
Because different cancer cells have different metastatic potentials, a natural question to ask is: “How does the mobility of cancer cells and their proliferation affect the invasion potential in different steps during the metastasis?” To answer this, it is crucial to set up an in vitro microscale experiment where cancer cells can be released to invade vacant areas and where their spatial motility and proliferation can be analyzed to compare the invasion potentials quantitatively. Our experiments do not involve flow but rather use three-dimensional microstructures of very high aspect ratios that provide an approach for the study of the cancer cell invasion driven by cell motility and proliferation.
Fig. 1 shows the high-aspect-ratio Tepuis constructed to ascertain three-dimensional invasiveness. The Tepuis have an aspect ratio of about 3∶1, because they are 100 μm wide and 320 μm high. Two prostate cancer cell lines were used in this study (16): PC-3 cells derived from bone marrow, which are highly metastatic prostate cancer cells (17) and LNCaP cells derived from the left supraclavicular lymph node, which are phenotypically less metastatic (18).
Fig. 1.

(A) Photo of the whole chip consisting of 4 × 4 arrays. A US 10-cent coin is shown on the same scale on the right. (B) SEM image corresponding to a single array in A consisting of 100 Tepuis. (C) SEM image of individual Tepuis; they are 100 × 100 × 320 μm in size and 50 μm apart.
The experiments described below show that, compared to highly metastatic cells (PC-3), LNCaP cells not only have a lower invasion rate but also have a lower invasion “efficiency,” where the efficiency is defined as the percentage of occupied Tepuis at steady state. We propose that the inability of the LNCaP cells to occupy all available Tepuis is due to contact inhibition, which is much less pronouced for PC-3 cells.
Results and Discussion
Before loading the cells into the microchips, PC-3 cells and LNCaP cells were cultured and trypsinized for collection. The suspended cells were uniformly plated on the chip surface with approximately 105 cells/cm2. The chips were then incubated in an environment kept at 37 °C and 5.0% CO2. The cells were allowed to settle for 24 h over both the Tepui tops and at the bottoms of the devices. After 24 h of incubation, polydimethylsiloxane blocks were used to gently swipe the Tepui tops, leaving the cells inside the gaps mostly on the bottom (Fig. 2A) (19). The surface-cleaned Tepuis were then covered with fresh RPMI medium and incubated again at 37 °C and 80% humidity in covered petri dishes (Fig. 2B). The culture medium was refreshed every 96 h thereafter. The dishes were removed from the incubator every 24 h for imaging in an upright microscope (90i, Nikon) to take bright-field pictures of the cell occupancy on the tops, or on a confocal microscope (SP5, Leica) to generate three-dimensional images of cell motility and morphology inside the gaps. A 40× oil immersion objective lens was used in confocal imaging to ensure high resolution with 100-μm limited axial scanning distance.
Fig. 2.

(A) After sweeping off the cancer cells from the tops of the Tepuis, the cancer cells are initially only occupying the bottom of the cavities in the microfabricated device. (B) The chip is incubated, and confocal microscopy is used to observe the movement of the motile PC-3 and LNCaP cells up along the sides of the Tepuis toward the tops.
Experimental Observations.
The initial condition for the experiment is shown in Fig. 3 following sweeping of the tops of the Tepuis. Fig. 3A shows that at t = 0 all tops were empty. By adjusting the focal plane of the microscope, bright-field images of the cells covering the floor between the Tepuis can also be recorded (Fig. 3B). Initially, the cells form a roughly 50% confluent layer covering the floor. Fig. 4 A and B are representative images of PC-3 cells on the Tepui array at 24 and 144 h after the cells were swept off, respectively. The PC-3 cells invade the Tepui tops and proliferate until 100% confluency is reached on all the Tepui tops. Fig. 4 C and D show arrays with LNCaP cells at 24 and 312 h after initial cell clearing, respectively. Unlike the PC-3 cells, the LNCaP cells do not invade all of the Tepui tops, leaving roughly 15% of the tops free, which results in a “checkerboard” pattern (Fig. S1). Further incubation past 312 h did not result in any further occupation of available Tepui tops.
Fig. 3.

Bright-field images at t = 0 of (A) the tops of Tepui, and (B) where the focal plane of the 10× objective is adjusted to image the cells at the base of the Tepui. In the latter, diffraction and waveguide effects result in the bright interference spots at the corners of the Tepui tops when looking down the 320 μm to the floor. The image reveals that the cancer cells form an approximately 50% confluent layer at the bottom.
Fig. 4.

Bright-field image of cancer cells on the Tepuis of a 10 by 10 array at different times. PC-3 cells at (A) t = 24 h and (B) t = 144 h. LNCaP cells at (C) t = 24 h and (D) 312 h.
For both cell types, the numbers of cells on each Tepui as a function of time are highly nonuniform, which indicates that the cells do not simply move up the sides and proliferate uniformly once on top (Figs. S2 and S3). Furthermore, the experiments also show two characteristic features that distinguish the behavior of the two cell types (see Fig. 5): (i) At any given time, the PC-3 cells occupy a larger number of Tepui than the LNCaP cells, showing a higher invasion rate for PC-3 cells than for LNCaP cells, and (ii) although the PC-3 cells occupy all Tepui within a finite time (5 d), the LNCaP cells leave some of the Tepui unoccupied, no matter how long the cells were cultured (even longer than 312 h) (Figs. S4 and S5).
Fig. 5.

Fractional occupation of Tepui tops as a function of time from top clearing for LNCaP cells (red solid circle) and PC-3 (blue solid circle) cells. The logistic Eq. 1 is fit to the points to guide the eye. The fitting for LNCaP cells yields Kn = 0.88 and Rn = 0.028, and for PC-3 cells Kn = 0.99 and Rn = 0.085. The horizontal line shows limiting occupation for LNCaP cells.
These observations raise the questions of how do the cells reach the tops of the Tepui, and why do LNCaP cells not ultimately cover all the tops? To address these issues, confocal microscopy (SP5, Leica) was employed for 3D imaging of GFP-expressing PC-3 cells and LNCaP cells in the microfabricated arrays. The images were recorded after the cell densities on the Tepui reached a steady state: 120 h for PC-3 cells and 312 h for LNCaP cells (see Fig. S6).
Fig. 6 shows typical z-slices and three-dimensional reconstructions of GFP-tagged PC-3 cells or LNCaP cells on the chip. Both cell types were found climbing along the Tepui sides. At steady state, PC-3 cells occupied the tops of all the Tepuis and were found all along the sides. The LNCaP cells behave strikingly differently: Some of the Tepuis are fully occupied with cells confluent on the tops; on other Tepui, the cells barely reached the top and formed much higher density clusters surrounding the sides of the Tepui (see Movie S1).
Fig. 6.

Confocal images and the corresponding three-dimensional reconstructions showing the cell-chip system after reaching steady state. The green channel represents fluorescence of GFP expressed within the cells, and the red channel represents the direct reflection from the surface of the chip. (A) A confocal z-slice of PC-3 cells cultured on the microchip. The focal plane is 25 μm below the top of the Tepui. (B) The three-dimensional reconstruction shows PC-3 cells attached to the sides and densely packed on the top. (C) A confocal z-slice of LNCaP cells cultured on an identical microchip. The focal plane is 25 μm below the top of the Tepui. (D) The three-dimensional reconstruction of LNCaP cells attached to the surface, showing that they barely reached the top.
Data Analysis and Modeling.
The above observation confirmed the expected result that PC-3 cells are more invasive than LNCaP cells, which is known from extensive in vivo studies (20, 21). The most puzzling aspect of the data is the fairly large fraction of the Tepui tops that remain unoccupied during LNCaP invasion. In Fig. 5, we plot the number of occupied Tepui versus time for both of the cell lines. These data were obtained from 16 identical copies each consisting of 100 Tepuis as shown in Fig. 1. A Tepui was counted as occupied when cells appeared on the top, irrespective of the number of cells. Although we did observe cells both entering and exiting a Tepui top, basically a top surface once occupied remained occupied and did not clear.
At the most basic level, we can view the growth and progression of the cells up the Tepui walls and occupation of the tops as a logistic process where increasing cell densities result in a decrease of the proliferation rate due to decrease in the carrying capacity of the environment (22). The inability of the LNCaP cells to occupy the tops of the Tepuis can then be viewed as a result of presumed sensitivity of relatively noninvasive LNCaP cells to the local cell density, which we relate to the stronger contact inhibition in LNCaP cells as opposed to the highly invasive PC-3 cells (23). The logistic equation is the simplest macroscopic model with these features. The model contains the growth rate Rn of the occupied fraction n and the carrying capacity Kn of the microenvironment:
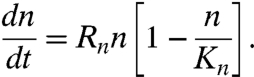 |
[1] |
This gives 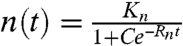 , where C is an integral constant. In this model, although we can not ascertain why there is a variance in the occupied tops without further information, fits of Eq. 1 yield a growth rate Rn of 1/36 h-1 for LNCaP cells, whereas the metastatic PC-3 cells occupy the Tepuis three times faster with Rn = 1/12 h-1. As a comparison, notice that the doubling time of LNCaP is only twice as long as that of PC-3 cells. The puzzle remains why the slowly growing LNCaP do not invade all the Tepui tops.
, where C is an integral constant. In this model, although we can not ascertain why there is a variance in the occupied tops without further information, fits of Eq. 1 yield a growth rate Rn of 1/36 h-1 for LNCaP cells, whereas the metastatic PC-3 cells occupy the Tepuis three times faster with Rn = 1/12 h-1. As a comparison, notice that the doubling time of LNCaP is only twice as long as that of PC-3 cells. The puzzle remains why the slowly growing LNCaP do not invade all the Tepui tops.
A more sophisticated model is to view the experiment as essentially a wound-healing problem, where the Tepuis create cylindrical “wounds” in the otherwise flat two-dimensional surface. The cells attempt to close the wound by climbing up the sides with some characteristic speed c. Wound-healing dynamics have been a powerful tool in studying collective cell migration, and recent work connects the wound-healing front to the propagation of a wave of cells (24, 25). A simple equation that takes into account both contact inhibition and logistic growth is the Fisher’s equation with a density-dependent diffusion coefficient D(ρ), where ρ(x,t) is the cell number per unit volume (26):
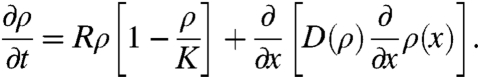 |
[2] |
Because contact inhibition affects both proliferation and migration (23), both R and D should in principle be density dependent. For simplicity, we will assume the carrying capacity K being renormalized while R remains a constant.
To complete the model, we need an assumption for the diffusion coefficient. Several forms have been proposed in the literature to describe evolution, biogeography, and wound healing by epithelium cells. Here, we consider a model where contact inhibition strength I is used to mildly modify the effective diffusion coefficient of the cells (26):
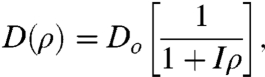 |
[3] |
where Do is the diffusion constant in the absence of contact inhibition. As I → ∞ (that is, as the contact inhibition becomes very strong), we simply have the logistic Eq. 1. On the other hand, when the contact inhibition is weak, Eq. 2 reduces to the normal Fisher’s equation. It is known that the normal Fisher’s equation (where the diffusion coefficient is a constant) admits traveling wave solutions of the form u(x - ct), where 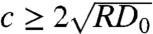 is the speed of the wave front. Moreover, numerical studies suggest that asymptotically (t → ∞) there is a unique wave speed
is the speed of the wave front. Moreover, numerical studies suggest that asymptotically (t → ∞) there is a unique wave speed 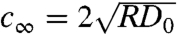 irrespective of the initial condition (27). For PC-3 cells, which we propose to have little contact inhibition, the asymptotic front velocity can be estimated from their diffusion coefficient (D0 = 10 μm2 h-1; see Movie S2) and growth rate (R = 1/60 h-1) to be 0.8 μm/h. However, in the experiment we observed most of the Tepuis were occupied in less than 150 h with PC-3 cells. This discrepancy implies that the early stage of Eq. 2, rather than the long time evolution, is relevant to our experiment. Interestingly, although the model of Eq. 3 has similar asymptotic behavior as the normal Fisher’s equation, contact inhibition strength I does slow down the front propagation at the early stage (26). The failure to describe our experiment by asymptotic dynamics of Eqs. 2 and 3 also points to the importance of boundary condition set by the Tepui top. It is possible that near the Tepui top, propagation slows down and cell density saturates on the side walls, which further enhances the effect of contact inhibition that may finally stop the wave.
irrespective of the initial condition (27). For PC-3 cells, which we propose to have little contact inhibition, the asymptotic front velocity can be estimated from their diffusion coefficient (D0 = 10 μm2 h-1; see Movie S2) and growth rate (R = 1/60 h-1) to be 0.8 μm/h. However, in the experiment we observed most of the Tepuis were occupied in less than 150 h with PC-3 cells. This discrepancy implies that the early stage of Eq. 2, rather than the long time evolution, is relevant to our experiment. Interestingly, although the model of Eq. 3 has similar asymptotic behavior as the normal Fisher’s equation, contact inhibition strength I does slow down the front propagation at the early stage (26). The failure to describe our experiment by asymptotic dynamics of Eqs. 2 and 3 also points to the importance of boundary condition set by the Tepui top. It is possible that near the Tepui top, propagation slows down and cell density saturates on the side walls, which further enhances the effect of contact inhibition that may finally stop the wave.
The fact that LNCaP cells fail to fully occupy all the Tepui tops, coupled with the three-dimensional dense cell structures crowding on the walls of the Tepuis, whereas the PC-3 cells swiftly ascend the walls, would support our assumption that contact inhibition is crucial.
Conclusion
The metastatic prostate cancer cells (LNCaP and PC-3) under study exhibit strikingly different motility and proliferation in Tepui structures. The experiments reveal that highly metastatic PC-3 cells have a faster invasion rate compared to LNCaP cells. Interestingly, LNCaP cells fail to populate all Tepui tops and form dense three-dimensional structures that are not observed in two-dimensional cell culture. We argue that this phenomenon is due to cell contact inhibition and propose a model based on Fisher’s equation. Combined with confocal microcopy, the device shows promise to be a real-time platform for in vitro quantitative studies of cell invasion for a broad arrange of cancer cells. Future experiments will focus on introducing collagen into the Tepui arrays to study cell metastasis in extra cellular matrix structures. Stress gradients will also be applied to target cancer evolution in time and space.
Materials and Methods
The deep structures on our chip are created using a reactive ion etcher (Samco 800) using the Bosch deep-etching process. Fig. 1A is an image of the silicon chip with Tepui arrays inside. The scanning electron microscope (SEM) image in Fig. 1B shows an enlarged image of a single array. Each Tepui is separated by deep-etched channels.
PC-3 cells and LNCaP cells were grown in flasks to approximately 80% confluency and trypsinized with 0.25% Trypsin-EDTA (Mediatech Inc.) solution for collection. Cell suspensions were then centrifuged at 1,500 rpm (Centra CL3R, Thermo IEC) and 4 °C for 5 min. Cells were pelleted and resuspended in Roswell Park Memorial Institute Medium 1640 (RPMI 1640) (Invitrogen 11875-093) supplemented with 10% fetal bovine serum (Gemini 900108) and 1% penicillin/streptomycin (Sigma P4333). A gelatin-coated Tepui chip was placed with its structures facing up and fixed at the bottom of a polystyrene petri dish. Four milliliters of RPMI medium was pelleted into the petri dish to immerse the entire chip. The petri dish was then placed in a vacuum chamber for 20 min to eliminate bubbles inside the gaps. After that, the suspended cells were seeded onto the petri dish. The petri dish was well shaken to make sure that the cells were uniformly distributed. Each chip was plated with approximately 105 cells/cm2.
Cancer cells need a properly treated surface for adhesion, motility, and proliferation. In three dimensions, this is the extracellular matrix, but in this experiment we left the volume between the empty Tepuis and treated the silicon surfaces with a gelatin (Stemcell Tech). The process of the gelatin coating requires four steps: (i) First, the gelatin-buffer is pipetted onto the etched region of the chip. (ii) Then, the chip is put into a vacuum chamber for 20 s to remove any bubbles, making sure the buffer completely covers the structures. (iii) In the next step, the chip is blown dry before being placed in a sterile laminar flow hood at room temperature for 1 h to ensure uniform wetting of the device. (iv) Finally, the remaining gelatin-buffer solution is aspirated by a pipette, and the device is left in the sterile laminar flow hood for an additional hour to allow the silicon structure to dry completely. The PC-3 and LNCaP cell lines were obtained from the American Type Culture Collection (ATCCCRL 1435, ATCCCRL 1740). The doubling times of PC-3 cells and LNCaP cells are 38.5 and 60 h, respectively. The cells were cultured in RPMI medium with 80% humidity atmosphere at 37 °C with 5% CO2. The cells grew in a monolayer to approximately 80% confluency before being passaged.
To view the cells using confocal microscopy, GFP-tagged PC-3 cells and LNCaP cells were created to produce 3D images of the cell behavior inside the deep gaps. To prepare GFP-tagged cells, they were transfected using FuGene HD transfection reagent (Roche 04709691001) and Plasmid pAcGFP1-actin containing GFP-actin fusion protein (Clontech 632453). First, the cells were seeded in 6-well plates at a cell density of 4 × 104 cells per well and grown until cells reached 80% confluency. For transfection, 2 μg of plasmid DNA was first mixed in 100 μL of OPTI-MEM I Reduced-serum medium (Invitrogen 31985070). Eight microliters of FuGene HD transfection reagent was then added to the diluted plasmid DNA and mixed by vortexing for 2 s, and the mixture was then added to cells. Then, the cells were incubated for 48 h before 400 μg/mL Geneticin (Invitrogen 10131027) was added for selection. After 2 weeks, cells resistant to Geneticin were isolated and expanded into 48-well plates. Resistant clones were tested for expression of GFP-actin fusion by looking under a Nikon TE-2000 fluorescence microscope. Cells expressing GFP were further expanded. Because of the high heterogeneity and frequent chromosomal rearrangements, PC-3 cells are not likely to have uniform fluorescent expressions through transfection. Hence, some PC-3 cells with dim fluorescent light would not be visible under confocal microscope when they formed monolayer on the Tepui walls.
Supplementary Material
Acknowledgments.
This work was supported by National Cancer Institute (Grant 1U54CA143803-01), Carlsbergfondet, and The Danish Council for Independent Research—Natural Sciences. The project described was supported by Award U54CA143803 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Footnotes
The authors declare no conflict of interest.
See Commentary on page 6693.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1102808108/-/DCSupplemental.
References
- 1.Doyle AC. The Lost World. New York: Tom Doherty Associates; 1993. [Google Scholar]
- 2.Anand P, et al. Cancer is a preventable disease that requires major lifestyle changes. Pharm Res. 2008;25:2097–2116. doi: 10.1007/s11095-008-9661-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuan F. Stress is good and bad for tumors. Nat Biotechnol. 1997;15:722–723. doi: 10.1038/nbt0897-722. [DOI] [PubMed] [Google Scholar]
- 4.Sahai E. Illuminating the metastatic process. Nat Rev Cancer. 2007;7:737–749. doi: 10.1038/nrc2229. [DOI] [PubMed] [Google Scholar]
- 5.Zaman MH, et al. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc Natl Acad Sci USA. 2006;103:10889–10894. doi: 10.1073/pnas.0604460103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minn AJ, et al. Lung metastasis genes couple breast tumor size and metastatic spread. Proc Natl Acad Sci USA. 2007;104:6740–6745. doi: 10.1073/pnas.0701138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lujambio A, et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci USA. 2008;105:13556–13561. doi: 10.1073/pnas.0803055105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamada KM, Cukierman E. Modeling tissue morphogenesis and cancer in 3D. Cell. 2007;130:601–610. doi: 10.1016/j.cell.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 9.Chambers A, Groom A, MacDonald I. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 10.Rolli CG, Seufferlein T, Kemkemer R, Spatz JP. Impact of tumor cell cytoskeleton organization on invasiveness and migration: A microchannel-based approach. PloS ONE. 2010;5:e8726. doi: 10.1371/journal.pone.0008726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Condeelis J, Segall J. Intravital imaging of cell movement in tumours. Nat Rev Cancer. 2003;3:921–930. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- 12.Irimia D, Toner M. Spontaneous migration of cancer cells under conditions of mechanical confinement. Integr Biol. 2009;1:506–512. doi: 10.1039/b908595e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Revzin A, Sekine K, Sin A, Tompkins R, Toner M. Development of a microfabricated cytometry platform for characterization and sorting of individual leukocytes. Lab Chip. 2005;5:30–37. doi: 10.1039/b405557h. [DOI] [PubMed] [Google Scholar]
- 14.Park MC, Hur JY, Kwon KW, Park SH, Suh KY. Pumpless, selective docking of yeast cells inside a microfluidic channel induced by receding meniscus. Lab Chip. 2006;6:988–994. doi: 10.1039/b602961b. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, et al. Micromolded arrays for separation of adherent cells. Lab Chip. 2010;10:2917–2924. doi: 10.1039/c0lc00186d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pickard MR, Darling D, Farzaneh F, Williams GT. Preparation and characterization of prostate cell lines for functional cloning studies to identify regulators of apoptosis. J Androl. 2009;30:248–258. doi: 10.2164/jandrol.108.005686. [DOI] [PubMed] [Google Scholar]
- 17.Voss MJ, Niggemann B, Zanker KS, Entschladen F. Tumour reactions to hypoxia. Curr Mol Med. 2010;10:381–386. doi: 10.2174/156652410791317020. [DOI] [PubMed] [Google Scholar]
- 18.Chen Q, et al. Gene expression in the LNCaP human prostate cancer progression model: Progression associated expression in vitro corresponds to expression changes associated with prostate cancer progression in vivo. Cancer Lett. 2006;244:274–288. doi: 10.1016/j.canlet.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 19.Liu LY, et al. A microfluidic device for continuous cancer cell culture and passage with hydrodynamic forces. Lab Chip. 2010;10:1807–1813. doi: 10.1039/c003509b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pulukuri SM, et al. RNA interference-directed knockdown of urokinase plasminogen activator and urokinase plasminogen activator receptor inhibits prostate cancer cell invasion, survival, and tumorigenicity in vivo. J Biol Chem. 2005;280:36529–36540. doi: 10.1074/jbc.M503111200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Laniado ME, et al. Expression and functional analysis of voltage-activated Na+ channels in human prostate cancer cell lines and their contribution to invasion in vitro. Am J Pathol. 1997;150:1213–1221. [PMC free article] [PubMed] [Google Scholar]
- 22.Spratt JA, Vonfournier D, Spratt JS, Weber EE. Decelerating growth and human breast-cancer. Cancer. 1993;71:2013–2019. doi: 10.1002/1097-0142(19930315)71:6<2013::aid-cncr2820710615>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 23.Chiao JW, et al. Endothelin-1 from prostate cancer cells is enhanced by bone contact which blocks osteoclastic bone resorption. Brit J Cancer. 2000;83:360–365. doi: 10.1054/bjoc.2000.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poujade M, et al. Collective migration of an epithelial monolayer in response to a model wound. Proc Natl Acad Sci USA. 2007;104:15988–15993. doi: 10.1073/pnas.0705062104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mark S, et al. Physical model of the dynamic instability in an expanding cell culture. Biophys J. 2010;98:361–370. doi: 10.1016/j.bpj.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai AQ, Landman KA, Hughes BD. Multi-scale modeling of a wound-healing cell migration assay. J Theor Biol. 2007;245:576–594. doi: 10.1016/j.jtbi.2006.10.024. [DOI] [PubMed] [Google Scholar]
- 27.Tang S, Weber RO. Numerical study of fishers equation by a petrov-galerkin finite-element method. J Aust Math Soc B. 1991;33:27–38. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.