

Abstract
Cisplatin (cisPt), Pt(NH3)2Cl2, is a cancer drug believed to kill cells via DNA binding and damage. Recent work has implied that the cellular copper (Cu) transport machinery may be involved in cisPt cell export and drug resistance. Normally, the Cu chaperone Atox1 binds Cu(I) via two cysteines and delivers the metal to metal-binding domains of ATP7B; the ATP7B domains then transfer the metal to the Golgi lumen for loading on cuproenzymes. Here, we use spectroscopic methods to test if cisPt interacts with purified Atox1 in solution in vitro. We find that cisPt binds to Atox1’s metal-binding site regardless of the presence of Cu or not: When Cu is bound to Atox1, the near-UV circular dichroism signals indicate Cu-Pt interactions. From NMR data, it is evident that cisPt binds to the folded protein. CisPt-bound Atox1 is however not stable over time and the protein begins to unfold and aggregate. The reaction rates are limited by slow cisPt dechlorination. CisPt-induced unfolding of Atox1 is specific because this effect was not observed for two unrelated proteins that also bind cisPt. Our study demonstrates that Atox1 is a candidate for cisPt drug resistance: By binding to Atox1 in the cytoplasm, cisPt transport to DNA may be blocked. In agreement with this model, cell line studies demonstrate a correlation between Atox1 expression levels, and cisplatin resistance.
Keywords: copper chaperone, protein unfolding, spectroscopy
Cisplatin (cis-diammine dichloro platinum(II); cisPt) is one of the most potent anticancer drugs in clinical use effective against several common types of solid tumors. The cytotoxic effect of cisPt is thought to be due to attack on DNA bases and this DNA damage causes induction of apoptosis in cancer cells (1). The effectiveness of cisPt-based chemotherapy can be hampered by several resistance mechanisms, including reduced uptake by the cell, rapid detoxification inside the cell, increased drug efflux from the cell, DNA repair, and defective apoptosis (1). It appears that cisPt is able to bind to a number of extra- and intracellular proteins which may be linked to both side effects of cisPt treatment and drug resistance. For example, cisPt is thought to bind to serum albumin in human blood plasma (2). In vitro studies have shown cisPt to bind to cytochrome c (at Met65) (3), superoxide dismutase (at His19) (4), lysozyme (at His residue) (5), and CopC (at Met residues) (6). Most often, large doses of cisPt (high micromolar) need to be administrated in the blood to assure sufficient cell uptake. Although there are no data on the level of cisPt inside cells during treatment, one estimate (7) suggests it to be in the low micromolar range.
It is today widely accepted that cellular copper (Cu) transporting proteins are involved in both cisPt uptake and efflux, at least to some degree (8). Whereas cisplatin uptake appears mediated by the copper transporter Ctr1, the copper transporting P1B-type ATPases ATP7A and ATP7B (Menkes and Wilson disease proteins) are suggested to be involved in cisplatin efflux. The initial link between cisplatin resistance and Cu transporters was due to early observations of cisplatin resistance being associated with overexpression of the ATP7B and ATP7A proteins (9, 10). In humans, cellular Cu homeostasis is maintained by the Cu chaperone Atox1 that obtains Cu from Ctr1 and then delivers it to metal-binding domains (MBDs) of ATP7A and ATP7B in the secretory pathway (11–13). These latter proteins couple ATP hydrolysis to Cu transport into the Golgi lumen, for metallation of cuproenzymes, such as apoceruloplasmin (14). Several diseases are related to imbalance in Cu homeostasis—for example, Menkes and Wilson diseases (15, 16) and aceruloplasminemia (17, 18). Atox1 is a 68-residue protein that, like the MBDs in ATP7A and ATP7B, has a ferredoxin-like fold with a compact βαββαβ structure and a conserved metal-binding motif MX1CX2X3C, located in the solvent-exposed β1-α1 loop, which binds a single Cu(I) (19, 20). Previous work has shown that apo- and Cu-loaded forms of Atox1 have similar ferredoxin-like folds apart from increased conformational dynamics in the Cu-binding loop in the holoform (21). Last year, using bacterial and hepatocytes/hepatoma cell assays, two studies reported that cisPt could bind to the MBDs of ATP7B, although it was unclear where in the polypeptide the binding site(s) were located (8, 22).
One may argue that because the MBDs of ATP7B are similar in structure to Atox1, the chaperone will also be able to interact with cisPt. Indeed, studies of wild-type (Atox1+/+) and knockout (Atox1−/−) mice, suggested Atox1 to be a cisPt-binding protein (23). Recently, this hypothesis was confirmed when two crystal structures of human Atox1 in complex with Pt were reported (24). In one of the crystal structures, a one-to-one complex was found and Pt was stripped of all its ligands and coordinated Cys12 and Cys15 in Atox1 along with an external tris(2-carboxyethyl)phosphine (reducing agent) ligand. In the other reported structure, two Atox1 molecules were bridged by a cisPt in which both ammine ligands remained in place. Coordination to the protein was facilitated by one Cys15 from each Atox1 in the dimer. Nonetheless, it must be noted that the reported crystal structures are only two of many Atox1-Pt species observed when 1∶1 mixtures of Atox1:cisPt (1 mM) were analyzed by electrospray ionization (ESI)-MS (24). To understand the interplay between cisPt and Atox1 in solution, we have investigated cisPt interactions with both apo- and Cu-loaded forms of Atox1 by far- and near-UV CD and NMR spectroscopy. We find that at cisPt and Atox1 concentrations similar to in vivo and physiological conditions, cisPt binds to the protein in the absence as well as in the presence of Cu. CisPt binding destabilizes both apo- and holoforms of Atox1, causing polypeptide unfolding and aggregation within hours to days.
Results
Probing cisPt Binding to Atox1 in Solution.
Using 195Pt NMR, the ratio of the dichloro to the monoaqua forms of cisPt in the buffer solution used for all experiments was found to be 3∶1 (SI Appendix, Fig. S1A). Incubations of both apo- and holoforms of Atox1 with cisPt in 1∶1 ratios results in the disappearance of first the monoaqua form of cisPt, followed by slow disappearance of the dichloro form (SI Appendix, Fig. S1 B and C). There are no new peaks arising for the protein-bound Pt due to extensive line broadening of the Pt-Atox1 adduct. These findings demonstrate that cisPt binds to the protein in solution and that the reactive cisPt species is the monoaqua form. We independently confirmed cisPt binding to apo-Atox1 by ESI-MS. All samples (with and without cisPt additions) contained peaks at masses of 7,270 and 7,401, corresponding to the apoprotein without the first Met, and the apoprotein with the first Met residue in place, respectively. Samples with one- and fivefold molar excess of cisplatin over apo-Atox1 exhibited additional (dominant) adduct peaks at masses of 7,534 and 7,665 that were not found in the control apoprotein sample (SI Appendix, Fig. S2). The 7,534 adduct mass corresponds exactly to that of the apoprotein (without Met) in combination with a cisPt molecule that has lost one Cl ion (i.e., Pt(NH3)2Cl). Similarly, the 7,665 adduct mass corresponds to the apoprotein (with first Met) in combination with one Pt(NH3)2Cl.
Metal Titrations to Atox1 Probed by Near-UV CD.
Based on the reported crystal structures (24), we expected cisPt to bind to the Cys residues in the Cu-binding loop in apo-Atox1 although the binding-site for cisPt in Cu-Atox1 is less clear. We previously used near-UV CD to detect Cu binding to apo-Atox1 (25, 26). As seen in Fig. 1A, there are changes in the 260-nm region upon addition of 1 eq of Cu to apo-Atox1 (pH 6, 20 °C, 0.25 mM DTT). Earlier work has shown that, at these conditions, Atox1 binds one Cu with submicromolar to femtomolar affinity (27–29). Titration of cisPt to preformed Cu-Atox1 (1∶1) results in spectral CD features in the 260–370-nm range (Fig. 1B). Specifically, the peak at 260 nm becomes more negative, a new negative shoulder at 300 nm arises, and a positive peak at 370 nm appears. These changes are reminiscent of CD signals of metallothioneins (MTs) (30, 31), small Cys-rich peptides that bind many metals (up to 18) in distinct clusters involving bridging sulfurs. Similar to our observations, Neurospora Cu-MT has negative CD at 290 nm and positive CD at 360 nm (32). Moreover, mixed Zn-Cu MTs have positive CD features at 360 nm and negative ones at 280 nm that depend on total metal and Cu/Zn ratio (30). In MTs, the signals are proposed to arise from d10 - d10 interactions of adjacent metal ions when the metal–metal distances are relatively short (i.e., < 2.8 Å) (33).
Fig. 1.

Near-UV CD-monitored titrations of metals to apo/metallated Atox1 (pH 6, 20 °C): (A) 50 μM apo-Atox1 and 50 μM Cu-Atox1 (1∶1); (B) CisPt (up to fivefold excess as indicated) added to 50 μM Cu-Atox1; (C) CisPt (up to fivefold excess as indicated) added to 50 μM apo-Atox1; (D) Cu (up to fivefold excess as indicated) added to 50 μM cisPt-Atox1 (1∶1).
This similarity of our near-UV CD data to that of the metalloclusters in MTs indicates that cisPt is close to the Cu ion when bound to Atox1, as would be the case if both Cu and cisPt occupies Atox1’s metal-binding site. This binding is possible if cisPt makes use of one of the two Cys residues normally acting as Cu ligands (Cu could then coordinate a DTT molecule as its second ligand). Metal–metal d8 - d10 (i.e., Pt-Cu) interactions have been observed in model systems (exhibiting absorption above 300 nm) and theoretical calculations indicate that these metal–metal are similar to d10 - d10 interactions (34, 35). The changes in the CD spectra saturate when approximately 4 eq of cisPt have been added to Cu-Atox1, which agrees with 1∶1 binding because, within the time frame of this experiment, only one-third of the cisPt molecules is in the monoaqua form (two-thirds are still in the dichloro form; see above and SI Appendix, Fig. S1). As expected, incubation of cisPt and Cu-Atox1 mixed in 1∶1 ratio for many hours results in an exponential increase in the near-UV CD bands, which occurs after the initial appearance of a signal change. These bands appear with a rate constant of 0.38 h-1 (pH 6, 20 °C). Previously, the development of the monoaqua form of cisPt was reported to be 0.37 h-1 (37 °C, pH 6.5) and rate limiting for the formation of cisPt–DNA complexes (36).
Addition of cisPt to apo-Atox1, on the other hand, results in no changes in the near-UV CD signal (Fig. 2C). Because our Pt NMR and ESI-MS data demonstrated that cisPt binds to apo-Atox1 at our conditions, it appears that the formed complex lacks an induced CD signal in the near-UV wavelength range. Subsequent additions of Cu to a premixed cisPt + apo-Atox1 sample results in near-UV CD changes similar to those in Fig. 1B—i.e., negative CD at 300 nm and positive CD at 370 nm (Fig. 1D). Because these CD signals do not match those for Cu addition to apo-Atox1 (Fig. 1A), this result demonstrates that cisPt is already bound to Atox1 (but the complex is CD silent) when the Cu becomes bound. The similarities of the signals in Fig. 1 B and D indicate that a similar ternary Atox1-Cu-cisPt complex forms regardless of order of metal addition. As suggested above, this complex likely involves Cu coordination to one and cisPt coordination to the other Cys sulfur in Atox1’s metal-binding loop.
Fig. 2.

(A) Far-UV CD spectra as a function of time (0 h to 14 d) for apo-Atox1 (50 μM) mixed with 5 eq cisPt and incubated at 20 °C. Time points are 0, 5, 8, 24, 29, 32, 48, 73, 144, 168, 240, and 336 h. (B) CD changes at 220 nm (at 20 °C) plotted as a function of time for apo-Atox1 alone, apo-Atox1 (50 μM) mixed with 1 or 5 eq cisPt, and for Cu-Atox1 (50 μM) mixed with 1 or 5 eq cisPt. Also shown are a few time points for 0.5 mM apo-Atox1 mixed with 5 eq cisPt.
Metal Titrations at High Atox1 Concentrations.
To be able to link our CD data with NMR data (see below), we performed a set of metal titration experiments at higher protein concentration (now 0.5 mM instead of 50 μM). Notably, we find the same near-UV CD signals to appear upon cisPt additions to Cu-Atox1, or Cu additions to cisPt-Atox1, as observed at lower protein concentrations (SI Appendix, Fig. S3 A and B), indicating that the same metal–Atox1 complex with both Cu and cisPt bound simultaneously forms also at higher concentrations of the components. However, when probed over time (hours, pH 6, 20 °C), we found that a sample of Atox1 (0.5 mM) mixed with 5 eq of cisPt slowly unfolds and in parallel the near-UV CD peaks disappear (Fig. 2B).
CisPt-Mediated Atox1 Unfolding.
The observation of cisPt-mediated unfolding at high protein concentration (above) prompted us to test for possible slow unfolding of the cisPt-Atox1 complexes at 20 °C also at micromolar Atox1 concentrations. In the near-UV CD titrations (Fig. 1) performed within an hour, no time dependence was observed (in accord with one-fourth cisPt being in the monoaqua form and reactive; the rest of the cisPt existing as dichloro and not reactive on this timescale). Nonetheless, incubation of cisPt-Atox1 (5 eq cisPt+apo-Atox1) at 20 °C (50 μM Atox1) for more than 1 wk resulted in slow unfolding with a half-life of approximately 30 h (Fig. 2 A and B). An identical mixture in terms of concentrations was also studied by NMR (see NMR section below) and the distribution of observed disappearance rates for peaks corresponding to the folded protein were similar to the CD-detected unfolding rate constant (SI Appendix, Fig. S4). Similarly to the apo-Atox1 case, incubation of cisPt-Cu-Atox1 (5 eq cisPt+Cu-Atox1) at 20 °C (50 μM protein) for several days resulted in protein unfolding as detected by far-UV CD (half-life of approximately 6 h; Fig. 2B). Importantly, at the same conditions, apo-Atox1 alone did not show any signs of unfolding for at least 2 wk. When only 1 eq of cisPt was added to apo-Atox1, there was little, if any, protein unfolding observed over the course of 2 wk. Addition of 1 eq cisPt to holo-Atox1, in contrast, resulted in some unfolding with a half-life of the reaction estimated to about 2 d (Fig. 2B). These observations imply that cisPt promotes slow Atox1 unfolding at low protein concentrations.
Protein-Metal Interactions Probed by NMR.
To characterize cisPt’s interactions with Atox1 further, we turned to NMR and 15N-labeled protein. Although NMR samples with 50 μM protein gave reasonable signals, we obtained better quality data at 500 μM protein; therefore, all NMR experiments reported in this section were performed with this protein concentration. The 1H-15N heteronuclear single quantum coherence (HSQC) spectra of apo- and Cu-loaded protein match published data (21) and most amino acid residues could be assigned. Distinct spectral changes occur within 60 min of addition of cisPt in twofold molar excess to Atox1 (Fig. 3). In addition to a subspectrum of the apoprotein, a set of well dispersed peaks with attenuated signal intensity appear, which we attribute to folded Atox1 in complex with cisPt. Cross-peaks corresponding to the cisPt-bound Atox1 complex could not be assigned to specific residues because of their short lifetime, which impeded acquisition of extended assignment experiments. A third set of narrowly distributed peaks are present in the random-coil region of the spectrum and are assigned to cisPt-induced unfolded Atox1 conformation(s). As in the case of the apoprotein, addition of cisPt to holo-Atox1 gives an NMR spectrum that is a superimposition of three distinct states: holo-Atox1, cisPt-induced unfolded protein, and a state with weak but well-dispersed resonances attributed to a folded cisPt-Cu-Atox1 complex. Notably, upon cisPt binding to Cu-Atox1, peaks indicative of the holo-form remains (e.g., glycine-14) implying that a ternary complex (i.e., Cu is retained upon cisPt binding) is formed (SI Appendix, Fig. S5). This binding is in agreement with the conclusions made based on the near-UV CD data. In addition, independent experiments using size exclusion chromatography linked to inductively coupled plasma MS support that both metals interact with the protein simultaneously (SI Appendix, Fig. S6).
Fig. 3.

Interaction between cisPt and apo-Atox1 probed with NMR spectroscopy. 1H-15N HSQC NMR spectra of apo-Atox1 (green contours) superimposed on that of a 2∶1 cisPt:Atox1 complex (orange contours). Selected peaks corresponding to the folded cisPt:Atox1 complex (*) and unfolded species (#) are indicated.
To study the time dependence of the interactions between cisPt and Atox1, we mixed apo-Atox1 with 5 eq of cisPt and acquired a series of 1H-15N HSQC spectra over 16 h (Fig. 4). Initially cross-peaks corresponding to three states are observed: apo-Atox1, cisPt-bound folded Atox1 (the intermediate species), and the cisPt-induced unfolded form of Atox1. In subsequent spectra, the unfolded peaks grow in intensity paralleled by a decrease in the folded apoprotein and cisPt-bound Atox1 resonance intensities. After 16 h, the protein has unfolded completely because only the unfolded conformation is observable in the spectrum. Kinetics of the spectral changes were extracted by fitting peak volumes from the series of 1H-15N HSQC spectra to exponentially decaying functions (SI Appendix, Fig. S7). All aporesonances can be accurately fitted with a single exponential decay function (Eq. 1) yielding very similar rate constants with an average (global) value of 0.30 ± 0.08 h-1. The convergence between individual rates shows that apo-Atox1 is responding to one concerted process. Notably, the rate of depletion of the apoform closely resembles the previously measured rate-limiting development of the reactive monoaqua species (0.37 h-1 at 37 °C and pH 6.5) for the formation of cisPt–DNA complexes (36). Our observations suggest that dechlorination is rate limiting also for the interaction between cisPt and Atox1.
Fig. 4.

CisPt-induced unfolding of Atox1. 1H-15N HSQC NMR spectra acquired after 4, 8, and 16 h after mixing of a 5∶1 cisPt:apo-Atox1 sample.
Meanwhile, the time-dependent cross-peak volumes for resonances assigned to the Atox1–cisPt intermediate are not well fit to monoexponential decays. This observation is consistent with a consecutive reaction model in which primarily the monoaqua (and not the dichloro) form of cisPt forms a complex with folded Atox1. This intermediate then reacts further to generate an unfolded Atox1–cisPt complex. In accordance with this model, we fit the time-dependent changes in signal intensities of the cisPt-Atox1 intermediate with an initial buildup rate of 0.3 h-1 (from the rate of apodepletion) and a decay rate that was allowed to float (Eq. 2). The intermediate peaks decay with an average rate of 0.19 ± 0.09 h-1 which is significantly slower than the rate of apodepletion. Protein-bound cisPt can undergo a second dechlorination step to generate a reactive species. The rate constant for this second event is 0.35 h-1 in the already mentioned cisPt–DNA adduct (36). Similarly, we propose that the observed decay rate of the intermediate species (involving folded Atox1 bound to cisPt) is rate limited by the second dechlorination step of cisPt occurring with a rate of 0.19 h-1. The cisPt-triggered unfolding of Atox1 is likely occurring following the second dechlorination step and is an irreversible process.
Aggregation States of Species from NMR Spin Relaxation and Electrophoresis.
A fundamental question of mechanistic importance during the cisPt-induced unfolding of Atox1 is the aggregation state of the unfolded species. We addressed this question by measuring R2 relaxation rates for various Atox1 species, which offer a sensitive probe of motion on fast (picosecond to nanosecond) and slow (microsecond to millisecond) timescales. The assigned backbone 15N R2 relaxation rates of holo-Atox1 at 20 °C range between 4.6–8.6 s-1, with most values clustered around 6.5 s-1 (SI Appendix, Fig. S8). The rates are in good agreement with previously reported values (21). Moreover, the intrinsic R2 relaxation rate calculated with the Lipari and Szabo model free formalism (37) (assuming a compact spherical protein and τC = 3.8 ns, S2 = 0.90, and τe = 100 ps) is 6.5 s-1, which adequately matches the measured relaxation rates. The convergence between calculated and measured R2 rates shows that holo-Atox1 is monomeric at the millimolar concentrations employed in our study.
An additional relaxation experiment with a holo-Atox1 sample containing 5 eq of cisPt and incubated for 24 h provided values of R2 for the cisPt-induced denatured protein. Peaks attributed to the denatured species have markedly lower relaxation rates compared to folded holo-Atox1 (SI Appendix, Fig. S8; R2 range 2.8–8.2 s-1, with most values clustered around 4.7 s-1). Reference R2 relaxation rates for the fully unfolded protein were derived from a sample of Atox1 denatured with 8 M urea (SI Appendix, Fig. S8). The relaxation rates of Atox1 in the presence of 8 M urea are smaller compared to the holoprotein denatured with 5 eq of cisPt (range 1.3–4.8 s-1, with an average value of 2.7 s-1).
Employing the relaxation rates in 8 M urea as a benchmark for that of a fully unfolded (monomeric) protein of this size suggests that the denatured Atox1 species formed during incubation with cisPt in buffer undergoes comparatively restricted internal motions or is present as oligomers. Formation of higher order aggregates is feasible because cisPt contains two chloro groups that can be readily exchanged to form intermolecular cross-links. We propose that networks of unfolded polypeptides can form between Pt linkages from one protein to a Cys- or Met-sulfur in another protein, or to a DTT molecule that interacts with a Cu in another protein. In support, Pt-sulfur-Pt bridges between cisPt molecules have been reported (38). Presence of oligomeric species in protein-cisPt samples that appeared unfolded by far-UV CD was analyzed by gel electrophoresis. In addition to larger species not entering the gel, there were a range of smaller oligomers detected (SI Appendix, Fig. S9). The identification of oligomeric species suggests that the increase in R2 rates in the cisPt-unfolded, as compared to the urea-denatured, sample is due to an increase in the apparent molecular weight in the cisPt-Atox1 sample.
CisPt-mediated unfolding of Atox1 is specific because an unrelated protein (S16 from Aquifex aeolicus; ref. 39) was stable over weeks (at 20 °C) at a concentration of 1 mM protein after mixing with 5 eq of cisPt although NMR data showed binding. Also far-UV CD data on a sample of 50 μM S16 mixed with 5 eq of cisPt revealed no protein unfolding during 2 wk of incubation. Moreover, cytochrome c, known to bind cisPt via Met65 (3), did not unfold when incubated in presence of excess cisPt for 2 wk. Pt NMR experiments demonstrated that cisPt binds to cytochrome c at these conditions.
Biological Conditions in Vitro.
Inside cells, the pH is approximately 7.3, the salt concentration is approximately 160 mM, the temperature is 37 °C, and there is glutathione instead of DTT (as we have used here). Therefore, to assess the relevance of our findings at physiological conditions, we repeated the cisPt-binding experiments, and cisPt-promoted protein unfolding experiments at pH 7.3, 160 mM NaCl, 0.25 mM glutathione (SI Appendix, Fig. S10). We find the same phenomena occurring at these conditions: cisPt binds to Cu-Atox1 as the same near-UV CD signals are induced (SI Appendix, Fig. S10A) as in Fig. 1. Furthermore, Atox1 unfolding is triggered with a speed very similar to that found at 20 °C and low salt (SI Appendix, Fig. S10 B and C). We speculate that the higher temperature increases the rate of the reaction, whereas the increased ionic strength slows it down; together, the net difference is small.
Atox1 Expression in Tumor Cells.
To relate our in vitro findings to in vivo conditions, we studied cellular Atox1 expression in different human tumor cell lines (melanoma T289 and ovarian cancer 2008 cells) before and after exposure to cisplatin (SI Appendix). We found that the cell line with fivefold higher resistance to cisplatin (melanoma T289 cells) (40) has considerably higher levels (3.3-fold) of Atox1 than the cell line with lower cisplatin resistance (ovarian cancer 2008 cells) (41) (SI Appendix, Fig. S11). In addition, there is an indication that 2-h exposure to 20 μM cisplatin increases cellular levels of Atox1 by 17–75% in these cell lines.
Discussion
Although many different systems are implicated in cisPt trafficking within the cell, there is evidence for a strong linkage between cisPt resistance and the human Cu transport system. The relationship between cisPt and Cu transporters is underscored by recent observations that cisPt therapy combined with siRNA-mediated ATP7B silencing significantly reduces tumor growth in a mouse ovarian cancer model (42). Recent in vitro data have suggested that there are direct interactions between cisPt and the metal-binding domains of ATP7B (8, 22) but molecular details are lacking. Two different crystal structures of apo-Atox1 mixed with cisPt were reported last year, implying that Pt binds to the Cu site cysteines (24). Because the metal-binding domains of ATP7B are similar to Atox1 in terms of structural fold and Cu site, cisPt interactions with Atox1 may mimic its interactions with ATP7B domains. To add to the crystal work on Atox1 and the biological assays on ATP7B, we probed interactions of cisPt with Atox1 (in both apo- and holoforms) in solution using spectroscopic methods. Our solvent conditions were chosen to mimic the in vivo scenario as much as possible. Atox1 and cisPt may both be in the micromolar range in the cytoplasm (7, 43); the cytoplasm has 0.5–10 mM of glutathione (44), which is mimicked here by DTT in most experiments (but not all, see SI Appendix, Fig. S10). DTT has higher affinity for Cu(I) than glutathione in solution (45).
We found that cisPt binds to apo-Atox1 in solution, as was expected from the crystal structure work. However, based on Pt NMR and MS data, it appears that the dominant initial complex is Atox1 linked to one cisPt molecule that has exchanged one Cl ligand. This result agrees with earlier work on how cisPt binds to DNA (46): Initial DNA binding is limited by Cl dissociation, subsequent Pt-promoted DNA cross-linking depends on dissociation of the second Cl ligand from the cisPt molecule. The same mechanism appears to hold true for cisPt interaction with Atox1: The kinetics of apo-Atox1 disappearance and the subsequent kinetics for disappearance of the intermediate species (folded Atox1 linked to cisPt via one ligand) resemble reported values for cisPt first and second dechlorination (46), although the second step is slower at low protein concentrations.
We also demonstrated that cisPt binds to the holoform, Cu-Atox1, without loss of Cu—instead, both metals appear bound in Atox1’s metal-binding site. This conclusion is based on the high-wavelength CD signals (around 350 nm) that match signals found in multi-metal-cluster MTs (30, 31). It is possible that the folded Cu-cisPt-Atox1 complex is not monomeric but may to some degree resemble a recently reported Mo-Cu-Atx1 complex (47), formed when the copper-depleting drug tetrathiomolbydate was added to the yeast Cu chaperone, Atx1. In that reported crystal structure, a sulfur-bridged Mo-Cu cluster with 4 Cu, 1 Mo, and 10 sulfurs, connected 3 Atx1 molecules.
CisPt binding to Atox1 in solution results in slow protein unfolding at 20 °C (Fig. 2) and 37 °C (SI Appendix, Fig. S10), as well as thermal destabilization of the protein (SI Appendix, Fig. S12), which is true both with and without Cu bound to Atox1, although the absolute rates of unfolding depended on the presence of Cu or not (faster with Cu). Furthermore, NMR and gel electrophoresis data revealed that cisPt-promoted Atox1 unfolding is coupled to unfolded-state protein aggregation. It was reported for human albumin that cisPt promoted protein cross-links in vitro during the later stages of reaction (2), although the structural state of the protein molecules was not assessed. The lifetime of cells range from days (skin, gut) to many years (nerves), implying that the cisPt-mediated unfolding of Atox1 may be a biologically relevant phenomenon. Nonetheless, cancer cells treated with cisPt live for not more than 1 d and thus may mostly experience effects of the folded cisPt-Atox1 complex.
Conclusions
The Cu chaperone Atox1 normally transports Cu(I) for delivery to ATP7A/B, an ATPase that makes Cu available for cuproenzymes. Here, we show in vitro that cisPt interacts specifically with folded apo-Atox1 in solution. In addition, we demonstrate that cisPt binds to Cu-loaded Atox1 without loss of Cu. Instead, based on the near-UV CD signals, cisPt appears to bind near the Cu ion. The cisPt-Atox1/cisPt-Cu-Atox1 complexes are not stable over time and the protein unfolds and aggregates within hours to days (at physiological conditions). Our study suggests a possible in-cell mechanism for cisPt drug resistance involving Atox1: By binding to Atox1 in the cytoplasm after cell uptake, cisPt transport to the cell nucleus is blocked. The folded Atox1–cisPt species may facilitate transfer of cisPt to ATP7B, for ATP7B-mediated cell export, or simply act as a “sink.” In agreement with this hypothesis, our cell line studies demonstrate that Atox1 expression levels are significantly higher in a cisPt resistance tumor cell line (SI Appendix, Fig. S11). Because the Cu-binding domains in ATP7B are structurally similar and have similar Cu sites, as in Atox1, we predict that cisPt may also bind to these domains in or near their Cu sites.
Materials and Methods
Protein Preparation.
Expression and purification of unlabeled and 15N-labeled Atox1 follow ref. 48 with some modifications (see SI Appendix). The plasmid was kindly provided by A. C. Rosenzweig (Northwestern University, Evanston, IL). Aquifex aeolicus S16 was purified following established procedures (39), and horse heart cytochrome c was purchased from Sigma.
CD Spectroscopy.
All CD spectra were recorded on a Chirascan (Applied Photphysics) at 20 °C. Copper was added to the samples in the form of CuCl2 (20 mM in H2O) and immediately reduced to CuI by the DTT in the buffer. Cisplatin (Sigma-Aldrich) was dissolved in H2O, microwaved for 20 s at 900 W and shaken until no insoluble particles were visible. The cisplatin solution was made fresh and used within 5 d. In titration experiments, samples were incubated for 10 min before measurement. Titrations were performed at 50 and 500 μM Atox1 with 0–5-fold excess of cisplatin added to preformed Cu-Atox1 or to apo-Atox1. Another set of titrations were made in which 0–5-fold excess of Cu was added to premixed 1∶1 cisPt-Atox1. Buffer was 50 or 200 mM NaCl, 20 mM MES, 0.25 mM DTT, pH 6. For samples with high-protein concentration, the DTT concentration was increased to 2 mM. The CD signals were corrected for volume changes and baselines were subtracted from each spectrum. Time dependences of cisPt-promoted Atox1 unfolding (20 °C), both apo- and holoforms mixed with different molar equivalents of cisPt (1 or 5), were measured at a protein concentration of 50 μM via far-UV CD during 2 wk.
Protein NMR.
NMR samples contained 0.5 mM 15N-labeled apo-Atox1 in a buffer of 50 mM NaCl, 20 mM MES, and 2 mM DTT at pH 6.0. For preparation of the holoprotein [i.e., Cu(I)-bound Atox1] equimolar amounts of Cu(II) (as CuCl2) was added to apo-Atox1 in the buffer with excess amounts of DTT. CisPt was added to apo- and holoforms of Atox1 in various amounts (up to fivefold molar excess) in different NMR experiments. In one set of experiments, Atox1 was first denatured by 8 M urea, followed by additions of Cu and cisPt in order to assess metal binding to the fully unfolded state of Atox1. The resulting NMR data however showed that there was no metal binding to the protein under these conditions. NMR experiments were carried out on a Bruker DRX 600 MHz spectrometer equipped with a 5-mm triple resonance z-gradient cryoprobe. All experiments were performed at 20 °C with temperature calibration using an external probe. 15N R2 relaxation experiments (49) were acquired in an interleaved manner to minimize the contribution from time-dependent changes in the spectra. All NMR spectra were processed with NMRPipe (50) and visualized in the program Ansig for Windows (51). Integration of cross-peaks volumes were performed with routines in NMRPipe. The evolution of the cross-peak volumes was fit in Matlab to Eq. 1 (apo-Atox1) and Eq. 2 (cisPt-bound Atox1 intermediate). All decays were normalized to the intensity of the first 1H-15N HSQC spectrum.
| [1] |
| [2] |
In both equations, k is the decay rate constant of apo-Atox1 in presence of cisPt, k1 is the decay of the cisPt-bound Atox1 species, and Ii are preexponential factors proportional to the starting concentration of the species.
195Pt NMR.
195Pt NMR experiments were conducted at 20 °C on a 400 and 500 MHz Bruker DRX instrument equipped with a Bruker broadband observe 10-mm probe. Spectra were acquired using a carrier frequency of 107.286 MHz, a spectral width of 100 kHz, 4,096 complex points, 20,000 transients, and a relaxation delay of 20 ms. Chemical shifts were referenced using an aqueous solution of PtCl6.
Supplementary Material
Acknowledgments.
This work was supported by the Kempe (E.B., M.W.-W., C.F.W. and P.W.-S.), Göran Gustafsson (P.W.-S.), and Wallenberg (P.W.-S.) Foundations, Umeå University Young Researcher Awards (M.W.-W. and P.W.-S.), and the Swedish Research Council (P.W.-S.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1012899108/-/DCSupplemental.
References
- 1.Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007;33:9–23. doi: 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ivanov AI, et al. Cisplatin binding sites on human albumin. J Biol Chem. 1998;273:14721–14730. doi: 10.1074/jbc.273.24.14721. [DOI] [PubMed] [Google Scholar]
- 3.Zhao T, King FL. Direct determination of the primary binding site of cisplatin on cytochrome C by mass spectrometry. J Am Soc Mass Spectrom. 2009;20:1141–1147. doi: 10.1016/j.jasms.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Calderone V, Casini A, Mangani S, Messori L, Orioli PL. Structural investigation of cisplatin-protein interactions: Selective platination of His19 in a cuprozinc superoxide dismutase. Angew Chem Int Ed Engl. 2006;45:1267–1269. doi: 10.1002/anie.200502599. [DOI] [PubMed] [Google Scholar]
- 5.Casini A, et al. ESI mass spectrometry and X-ray diffraction studies of adducts between anticancer platinum drugs and hen egg white lysozyme. Chem Commun (Camb) 2007;14:156–158. doi: 10.1039/b611122j. [DOI] [PubMed] [Google Scholar]
- 6.Sze CM, et al. Interaction of cisplatin and analogues with a Met-rich protein site. J Biol Inorg Chem. 2009;14:163–165. doi: 10.1007/s00775-008-0452-x. [DOI] [PubMed] [Google Scholar]
- 7.Tran MQ, Nygren Y, Lundin C, Naredi P, Bjorn E. Evaluation of cell lysis methods for platinum metallomic studies of human malignant cells. Anal Biochem. 396:76–82. doi: 10.1016/j.ab.2009.08.044. [DOI] [PubMed] [Google Scholar]
- 8.Dolgova NV, Olson D, Lutsenko S, Dmitriev OY. The soluble metal-binding domain of the copper transporter ATP7B binds and detoxifiescisplatin. Biochem J. 2009;419:51–56. doi: 10.1042/BJ20081359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katano K, et al. Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res. 2002;62:6559–6565. [PubMed] [Google Scholar]
- 10.Komatsu M, et al. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000;60:1312–1316. [PubMed] [Google Scholar]
- 11.Harris ED. Basic and clinical aspects of copper. Crit Rev Clin Lab Sci. 2003;40:547–586. [PubMed] [Google Scholar]
- 12.Hung IH, et al. Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae. J Biol Chem. 1997;272:21461–21466. doi: 10.1074/jbc.272.34.21461. [DOI] [PubMed] [Google Scholar]
- 13.Hamza I, Schaefer M, Klomp LW, Gitlin JD. Interaction of the copper chaperone HAH1 with the Wilson disease protein is essential for copper homeostasis. Proc Natl Acad Sci USA. 1999;96:13363–13368. doi: 10.1073/pnas.96.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hellman NE, et al. Mechanisms of copper incorporation into human ceruloplasmin. J Biol Chem. 2002;277:46632–46638. doi: 10.1074/jbc.M206246200. [DOI] [PubMed] [Google Scholar]
- 15.Gitlin JD. Wilson disease. Gastroenterology. 2003;125:1868–1877. doi: 10.1053/j.gastro.2003.05.010. [DOI] [PubMed] [Google Scholar]
- 16.Tao TY, Gitlin JD. Hepatic copper metabolism: Insights from genetic disease. Hepatology. 2003;37:1241–1247. doi: 10.1053/jhep.2003.50281. [DOI] [PubMed] [Google Scholar]
- 17.Kulkarni PP, She YM, Smith SD, Roberts EA, Sarkar B. Proteomics of metal transport and metal-associated diseases. Chemistry. 2006;12:2410–2422. doi: 10.1002/chem.200500664. [DOI] [PubMed] [Google Scholar]
- 18.Miyajima H, et al. Use of desferrioxamine in the treatment of aceruloplasminemia. Ann Neurol. 1997;41:404–407. doi: 10.1002/ana.410410318. [DOI] [PubMed] [Google Scholar]
- 19.Huffman DL, O’Halloran TV. Function, structure, and mechanism of intracellular copper trafficking proteins. Annu Rev Biochem. 2001;70:677–701. doi: 10.1146/annurev.biochem.70.1.677. [DOI] [PubMed] [Google Scholar]
- 20.Arnesano F, et al. Metallochaperones and metal-transporting ATPases: A comparative analysis of sequences and structures. Genome Res. 2002;12:255–271. doi: 10.1101/gr.196802. [DOI] [PubMed] [Google Scholar]
- 21.Anastassopoulou I, et al. Solution structure of the apo and copper(I)-loaded human metallochaperone HAH1. Biochemistry. 2004;43:13046–13053. doi: 10.1021/bi0487591. [DOI] [PubMed] [Google Scholar]
- 22.Leonhardt K, Gebhardt R, Mossner J, Lutsenko S, Huster D. Functional interactions of Cu-ATPase ATP7B with cisplatin and the role of ATP7B in the resistance of cells to the drug. J Biol Chem. 2009;284:7793–7802. doi: 10.1074/jbc.M805145200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Safaei R, Maktabi MH, Blair BG, Larson CA, Howell SB. Effects of the loss of Atox1 on the cellular pharmacology of cisplatin. J Inorg Biochem. 2009;103:333–341. doi: 10.1016/j.jinorgbio.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boal AK, Rosenzweig AC. Crystal structures of cisplatin bound to a human copper chaperone. J Am Chem Soc. 2009;131:14196–14197. doi: 10.1021/ja906363t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hussain F, Rodriguez-Granillo A, Wittung-Stafshede P. Lysine-60 in copper chaperone atox1 plays an essential role in adduct formation with a target Wilson disease domain. J Am Chem Soc. 2009;131:16371–16373. doi: 10.1021/ja9058266. [DOI] [PubMed] [Google Scholar]
- 26.Hussain F, Wittung-Stafshede P. Impact of cofactor on stability of bacterial (CopZ) and human (Atox1) copper chaperones. Biochim Biophys Acta. 2007;1774:1316–1322. doi: 10.1016/j.bbapap.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 27.Wernimont AK, Yatsunyk LA, Rosenzweig AC. Binding of copper(I) by the Wilson disease protein and its copper chaperone. J Biol Chem. 2004;279:12269–12276. doi: 10.1074/jbc.M311213200. [DOI] [PubMed] [Google Scholar]
- 28.Yatsunyk LA, Rosenzweig AC. Cu(I) binding and transfer by the N terminus of the Wilson disease protein. J Biol Chem. 2007;282:8622–8631. doi: 10.1074/jbc.M609533200. [DOI] [PubMed] [Google Scholar]
- 29.Xiao Z, Wedd AG. The challenges of determining metal-protein affinities. Nat Prod Rep. 2010;27:768–789. doi: 10.1039/b906690j. [DOI] [PubMed] [Google Scholar]
- 30.Presta A, Green AR, Zelazowski A, Stillman MJ. Copper binding to rabbit liver metallothionein. Formation of a continuum of copper(I)-thiolate stoichiometric species. Eur J Biochem. 1995;227:226–240. doi: 10.1111/j.1432-1033.1995.tb20380.x. [DOI] [PubMed] [Google Scholar]
- 31.Stillman MJ, Presta A, Gui Z, Jiang DT. Spectroscopic studies of copper, silver and gold-metallothioneins. Met Based Drugs. 1994;1:375–394. doi: 10.1155/MBD.1994.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beltramini M, Lerch K. Primary structure and spectroscopic studies of Neurospora copper metallothionein. Environ Health Perspect. 1986;65:21–27. doi: 10.1289/ehp.866521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pountney DL, Schauwecker I, Zarn J, Vasak M. Formation of mammalian Cu8-metallothionein in vitro: Evidence for the existence of two Cu(I)4-thiolate clusters. Biochemistry. 1994;33:9699–9705. doi: 10.1021/bi00198a040. [DOI] [PubMed] [Google Scholar]
- 34.Xia BH, et al. Metal-metal interactions in heterobimetallic d8-d10 complexes. Structures and spectroscopic investigation of [M′M′ '(mu-dcpm)2(CN)2]+ (M′ = Pt, Pd; M′′ = Cu, Ag, Au) and related complexes by UV-vis absorption and resonance Raman spectroscopy and ab initio calculations. J Am Chem Soc. 2003;125:10362–10374. doi: 10.1021/ja0355325. [DOI] [PubMed] [Google Scholar]
- 35.Xia BH, et al. Metallophilic attractions between d8-d10 heterometallic compounds trans-[Pt(PH3)2(CN)2] and M(PH3)2+ (M = Ag or Cu): Ab initio study. J Chem Phys. 2004;120:11487–11492. doi: 10.1063/1.1753563. [DOI] [PubMed] [Google Scholar]
- 36.Bancroft DP, Lepre CA, Lippard SJ. Pt-195 NMR kinetic and mechanistic studies of cis-diamminedichloroplatinum and trans-diamminedichloroplatinum(Ii) binding to DNA. J Am Chem Soc. 1990;112:6860–6871. [Google Scholar]
- 37.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules.1. Theory and range of validity. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
-
38.Hadi S. Reactions of the first cisplatin hydrolytes cis-
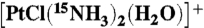 with N-acwetylcysteine. J Math Sci. 2006;11:146–152. [Google Scholar]
with N-acwetylcysteine. J Math Sci. 2006;11:146–152. [Google Scholar] - 39.Wallgren M, et al. Extreme temperature tolerance of a hyperthermophilic protein coupled to residual structure in the unfolded state. J Mol Biol. 2008;379:845–858. doi: 10.1016/j.jmb.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 40.Hemmingsson O, Zhang Y, Still M, Naredi P. ASNA1, an ATPase targeting tail-anchored proteins, regulates melanoma cell growth and sensitivity to cisplatin and arsenite. Cancer Chemother Pharmacol. 2009;63:491–499. doi: 10.1007/s00280-008-0762-2. [DOI] [PubMed] [Google Scholar]
- 41.Hemmingsson O, Nojd M, Kao G, Naredi P. Increased sensitivity to platinating agents and arsenite in human ovarian cancer by downregulation of ASNA1. Oncol Rep. 2009;22:869–875. doi: 10.3892/or_00000511. [DOI] [PubMed] [Google Scholar]
- 42.Mangala LS, et al. Therapeutic targeting of ATP7B in ovarian carcinoma. Clin Cancer Res. 2009;15:3770–3780. doi: 10.1158/1078-0432.CCR-08-2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banci L, et al. Affinity gradients drive copper to cellular destinations. Nature. 2010;465:645–648. doi: 10.1038/nature09018. [DOI] [PubMed] [Google Scholar]
- 44.Ostergaard H, Tachibana C, Winther JR. Monitoring disulfide bond formation in the eukaryotic cytosol. J Cell Biol. 2004;166:337–345. doi: 10.1083/jcb.200402120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao Z, et al. Unification of the copper(I) binding affinities of the metallo-chaperones Atx1, Atox1 and related proteins: Detection probes and affinity standards. J Biol Chem. 2011;286:11047–11055. doi: 10.1074/jbc.M110.213074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davies MS, Hall MD, Berners-Price SJ, Hambley TW. [1H, 15N] Heteronuclear single quantum coherence NMR study of the mechanism of aquation of platinum(IV) ammine complexes. Inorg Chem. 2008;47:7673–7680. doi: 10.1021/ic8006734. [DOI] [PubMed] [Google Scholar]
- 47.Alvarez HM, et al. Tetrathiomolybdate inhibits copper trafficking proteins through metal cluster formation. Science. 2010;327:331–334. doi: 10.1126/science.1179907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kihlken MA, Leech AP, Le Brun NE. Copper-mediated dimerization of CopZ, a predicted copper chaperone from Bacillussubtilis. Biochem J. 2002;368:729–739. doi: 10.1042/BJ20021036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Farrow NA, et al. Backbone dynamics of a free and a phosphopeptide-complexed Src homology-2 domain studied by N-15 NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 50.Delaglio F, et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 51.Helgstrand M, Kraulis P, Allard P, Hard T. Ansig for Windows: An interactive computer program for semiautomatic assignment of protein NMR spectra. J Biomol NMR. 2000;18:329–336. doi: 10.1023/a:1026729404698. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.