

Abstract
The DNA-dependent activator of IFN-regulatory factors (DAI), also known as DLM-1/ZBP1, initiates an innate immune response by binding to foreign DNAs in the cytosol. For full activation of the immune response, three DNA binding domains at the N terminus are required: two Z-DNA binding domains (ZBDs), Zα and Zβ, and an adjacent putative B-DNA binding domain. The crystal structure of the Zβ domain of human DAI (hZβDAI) in complex with Z-DNA revealed structural features distinct from other known Z-DNA binding proteins, and it was classified as a group II ZBD. To gain structural insights into the DNA binding mechanism of hZβDAI, the solution structure of the free hZβDAI was solved, and its bindings to B- and Z-DNAs were analyzed by NMR spectroscopy. Compared to the Z-DNA–bound structure, the conformation of free hZβDAI has notable alterations in the α3 recognition helix, the “wing,” and Y145, which are critical in Z-DNA recognition. Unlike some other Zα domains, hZβDAI appears to have conformational flexibility, and structural adaptation is required for Z-DNA binding. Chemical-shift perturbation experiments revealed that hZβDAI also binds weakly to B-DNA via a different binding mode. The C-terminal domain of DAI is reported to undergo a conformational change on B-DNA binding; thus, it is possible that these changes are correlated. During the innate immune response, hZβDAI is likely to play an active role in binding to DNAs in both B and Z conformations in the recognition of foreign DNAs.
DNA can activate immune responses in the innate immune system. Nonmethylated CpG sequences are recognized by toll-like receptor 9, and this results in inducing type-I interferon (IFN) (1). Double-stranded DNA, when placed in the cytosol of a cell by invading microbes or left by incomplete clearance of DNA damage, also can evoke immune responses (2–4). A recent study demonstrated that, in addition, the DNA-dependent activator of IFN-regulatory factors (DAI), formerly known as DLM-1 or ZBP1, also detects cytosolic DNAs and activates the innate immune response (5). DAI mediates activation of the innate immune system by facilitating DNA-mediated induction of type-I IFN and the expression of other related genes. DAI contains three DNA binding domains in the N terminus: two Z-DNA binding domains (ZBDs), Zα and Zβ, and an adjacent D3 region, a putative B-DNA binding domain. The D3 region plays pivotal roles in DNA binding, but all three domains are indispensable for the full activation of DAI (6). An intriguing question in innate immunity is how diverse foreign DNAs in the B- or Z-DNA conformations can be efficiently recognized by the DNA binding domains of DAI.
For the last decade there have been extensive structural and biochemical studies on ZBDs. The editing enzyme ADAR1 (double-stranded RNA adenosine deaminase) was the first Zα-containing protein to be identified. Zα domains were also found in the vaccinia virus E3L protein (7) and in PKZ (protein kinase containing Z-DNA binding domain). Crystal structures of the Zα domains of ADAR1, DAI, and Yaba-like pox disease virus complexed with Z-DNAs have been reported (8–10). Solution structures of the DNA-free Zα domain from human ZαADAR1 (hZαADAR1) and from vaccinia virus ZαE3L (vvZαE3L) are also available (11, 12). A typical Zα domain consists of an α + β helix-turn-helix motif and a β sheet. Z-DNA binding is mediated by helix α3, called the recognition helix; the tyrosine residue in the recognition helix is the centerpiece in this process. The β-loop between strands β2 and β3 participates in Z-DNA binding in conjunction with the recognition helix and forms the “wing” structure. The wing contains conserved proline residues that make hydrophobic contacts with the phosphate backbone of Z-DNA.
Contrary to the wealth of structural information on Zα domains, few studies have reported the structures of Zβ domains, the second Z-DNA binding domains identified in ADAR1, DAI, and PKZ. To date, the crystal structure of Zβ domain from human ZβADAR1 is known (13), but it lacks the central tyrosine on the recognition helix and does not have Z-DNA binding activity. In contrast, the Zβ domain of human DAI (hZβDAI) is unusual because it has Z-DNA binding activity, despite the lack of some residues that are known to be essential for Z-DNA binding in Zα domains (7, 14). The recent crystal structure of hZβDAI/Z-DNA complex revealed a unique mode of Z-DNA recognition (15), and thus it is considered to be one of the group II ZBDs.
In order to gain further structural insights into the unusual Z-DNA binding mode of hZβDAI, and to elucidate its DNA binding mechanism, we determined the solution structure of the DNA-free hZβDAI and analyzed its binding to B- and Z-DNAs using NMR spectroscopy. Here, we show that hZβDAI in solution exhibits structural deviations from the crystal structure of the complex, suggesting that hZβDAI undergoes conformational alterations upon DNA binding. In addition, residues involved in the weaker B-DNA binding are revealed by chemical-shift perturbation (CSP) experiments, and its binding mode to B-DNA is proposed. These results provide clues for understanding how hZβDAI contributes to the recognition of foreign DNA in the innate immune response.
Results
Overall Structure of hZβDAI and Comparison to Other Z-DNA Binding Domains.
We determined the three-dimensional solution structure of hZβDAI using NMR spectroscopy. The backbone RMSD of 20 lowest energy structures superimposed is 0.13 ± 0.04 Å (Table S1 and Fig. S1). The overall fold of hZβDAI in solutions appears similar to other known ZBDs with the canonical α + β helix-turn-helix motif consisting of three helices and three β-strands (called “winged helices”) (Fig. 1A). The overall conformation of the DNA-free hZβDAI is similar with those of hZαADAR1 and vvZαE3L (Fig. 1B). Notable conformational differences are found in the recognition helix (α3) and the wing (β2 and β3), as previously observed for the crystal structure of hZβDAI complexed with Z-DNA, in comparison with other ZBD/DNA complexes (15). It appears that the recognition helix of hZβDAI is bent with a mixed conformation of 310- and α-helices, rather than a straight α-helix, and the tip of the wing is seen to pull out of the canonical position critical in Z-DNA binding (Fig. 1B). The residual dipolar coupling (RDC) experiments also confirmed that the DNA-free hZβDAI has a bent helix rather than a straight helix (Table S2 and Fig. S2). In this analysis, the RDC values calculated from a modified structure containing the recognition helix in a straight conformation showed larger deviations than those calculated from the NMR or X-ray structures (Fig. S2).
Fig. 1.

The solution structure of free hZβDAI and structural comparison with homologous proteins. (A) The solution structure of free hZβDAI is represented in the ribbon diagram. It contains an α + β helix-turn-helix fold typical of B-DNA binding proteins. (B) Superimposition of the solution structures defined by NMR spectroscopy, free hZβDAI, free hZαADAR1, and free vvZαE3L (blue, red, and yellow, respectively). Dashed black circles indicate the N-terminal region of α3 and β turns.
Conformational Changes of hZβDAI upon Z-DNA Binding.
From the crystal and solution structures of hZαADAR1, it is known that Zα does not show any significant conformational changes upon Z-DNA binding, which indicates that it is a prepositioned binding surface for Z-DNA (11). However, hZβDAI shows notable changes in the N-terminal region of the recognition helix (Fig. 2A and Fig. S3). This suggests that conformational rearrangements occur in the DNA-recognition regions upon DNA binding. Free and DNA-bound hZβDAI commonly have a 310-helix (T136–D139) at the N-terminal end of the α3-helix; however, they display distinct conformations. Whereas the helicity of the recognition helix is well maintained in the free hZβDAI via hydrogen bonds between the N-terminal 310-helix and the C-terminal α-helix, the helicity is broken in the DNA-bound hZβDAI because of the absence of such backbone hydrogen bonds (Fig. S3A) (15). In the free hZβDAI, the backbone carbonyl oxygens of A137 and K138 in the 310-helix form hydrogen bonds with the side chain and backbone NH groups of N141 in the α3-helix of 2.9 and 3.6 Å, respectively. But, in the case of the DNA-bound hZβDAI, the distance between the backbone carbonyl oxygen of K138 and the backbone NH group of N141 is 4.1 Å, and the side chain NH group of N141 no longer forms a hydrogen bond with A137 because of the interaction with DNA [Protein Data Bank (PDB) ID code 3EYI, chain A]. The conformational change of the 310-helix in the DNA-bound hZβDAI was also confirmed by measuring the distances between Cα atoms in the 310-helix and the α3-helix (Table S3). The solution conformation would not be favorable for Z-DNA interaction because K138 and R142, which play a pivotal role in Z-DNA binding, point away from the DNA backbone (Fig. S3B). However, Z-DNA binding causes the extension of the N-terminal 310-helix and the proper binding of K138 and R142 to Z-DNA (Fig. S3A). It is thought that these conformational changes could be occurring because the energy gained by the interactions of K138, N141, and R142 with the phosphate backbone of Z-DNA is more than the energy loss caused by the dissociation of the hydrogen bond between the side chain of N141 and the carbonyl group of A137 and the backbone hydrogen bond in K138/N141.
Fig. 2.

Comparison of the free and DNA-bound hZβDAI structures. (A) Superimposition of the free (blue) and DNA-bound (magenta) hZβDAI structures (15). Z-DNA is depicted by a stick diagram. The secondary structures are labeled. (B) Comparison of the tyrosine conformation on the α3-helix in Z-DNA binding proteins; free hZβDAI and hZαADAR1 (blue and red, respectively) and DNA-bound hZβDAI and hZαADAR1 (magenta and green, respectively), and free vvZαE3L (yellow). The tyrosine residue in Z-DNA bound structures (magenta and green) shows CH-π interactions (17) with the tryptophan residue in the β3-strand. A similar conformation is found with the free hZαADAR1 structure (red). (C) Superimposition of six residues contacting Z-DNA in the hZβDAI/Z-DNA complex (magenta) (15) with those in the ensemble of 20 lowest energy structure of free hZβDAI (blue). The Z-DNA backbone is in dark red; bases are in gray.
The tyrosine residue in the recognition helix is also critical in Z-DNA binding (8–12, 16). The conserved tyrosine confers CH-π interactions with both the edge of the tryptophan residue (17) in the β3-strand and the guanosine base in the syn-conformation in Z-DNA. Although the conserved tyrosine was observed to be in contact with Z-DNA in the Z-DNA complex structure of yatapox virus ZαE3L (10), it was proposed that the conformation of the vaccinia tyrosine residue in the free state vvZαE3L is strongly correlated with the lack of Z-DNA converting activity (12). Whereas hZαADAR1 has a prepositioned tyrosine to interact with Z-DNA even in the free state and shows high Z-DNA converting activity (11), vvZαE3L has the solvent-exposed tyrosine conformation with diverse rotamers, and its Z-DNA converting activity is reduced (12) (Fig. 2B). In the free hZβDAI, the corresponding Y145 does not overlap with that of the DNA-bound hZβDAI, but adopts a conformation close to that of vvZαE3L (Fig. 2B). Therefore, it is suggested that the nonprepositioned conformation of Y145 in the free hZβDAI may be one of the reasons for the lower Z-DNA converting activity of hZβDAI than that of hZαADAR1. Consistently, hZβDAI appears to have lower binding affinity to Z-DNA and slower kinetics in Z-DNA converting activity (18, 19) than hZαADAR1.
Overall, hZβDAI seems to have a more flexible binding surface than hZαADAR1. Among the nine residues of hZαADAR1 that are directly involved in DNA contact in the crystal structure of the complex, seven are already prepositioned in a free state (11). However, in the case of hZβDAI, four out of six Z-DNA contacting residues (R124, K138, R142, and Y145) rearrange upon Z-DNA binding, and only N141 and W162 are prepositioned (Fig. 2C).
Conformational Isomerization of the DNA-Free  .
.
The 1H-15N heteronuclear single quantum coherence (HSQC) spectrum analysis of the free hZβDAI showed that there are more than two sets of NMR signals, which indicates the existence of conformational isomers (Fig. S4A). It was found by analysis of the nuclear Overhauser enhancement spectroscopy (NOESY) spectra that this phenomenon is due to the cis-trans isomerization of proline residues. Analysis of chemical-shift differences between the major and minor signals along the residues revealed that the differences are larger near the proline residue, P122 (Fig. S4B). The large chemical-shift difference between major and minor signals from I164 could also be explained by the fact that I164 is close to P122 in three-dimensional space. To verify the presence of proline isomerization and test the functional role of this residue, the P122 residue was mutated to alanine and its NMR spectrum and Z-DNA converting activity were examined. Interestingly, the minor signals from the wild-type hZβDAI were not seen in the 1H-15N HSQC spectrum of the P122A mutant (Fig. S4A), thereby confirming that those signals were caused by the cis-trans isomerization of P122. However, P122A mutant did not show any functional difference in terms of Z-DNA conversion activity measured by CD spectroscopy, compared to the wild-type protein (Fig. S4C). Those findings suggest that the proline isomerization is not relevant to the Z-DNA converting activity of hZβDAI. Consistently, both cis- and trans-isomers were observed in the crystal structure of the hZβDAI/Z-DNA complex (15). Therefore, it is thought that both isomers are in equilibrium in solution and both contribute to Z-DNA binding. RDC data also support the presence of the two isomers in solution; that is, the experimental RDC value for E121 (4.0 Hz) is the middle of the calculated RDC values of the trans-conformer (-0.7 Hz) and the cis-conformer (8.1 Hz). The mechanism underlying the enhancement of Z-DNA conversion activity by proline isomerization and its functional role remains to be studied.
CSPs upon B- and Z-DNA Binding.
An intriguing question to ask is how the ZBD converts B-DNA to Z-DNA. As for the role of the ZBD in Z-DNA conversion, there have been different explanations. The ZBD might take an active part in Z-DNA conversion by binding to B-DNA first and initiating the conversion process. Alternatively, the ZBD could be more passively involved; that is, it may bind to the preformed Z-DNA that might be transiently formed by Brownian motion and shift the B–Z equilibrium toward Z-DNA. In either case, the positively charged surface of the ZBD is expected to be involved in DNA binding. Regarding the role of DAI in the recognition of foreign DNA, it was anticipated that the ZBD would bind to various conformations of DNA (5). To obtain structural insight into the B-DNA binding mode of hZβDAI and possibly into the B-to-Z converting mechanism, CSP experiments with hZβDAI were performed in the presence of double-stranded (dCdG)6 (referred to as CG6), a Z-DNA forming sequence, and double-stranded (dCdCdG)(dAdT)6(dCdGdG) (referred to as AT6), a B-DNA forming one (Fig. 3 and Fig. S5). One-dimensional imino proton NMR and CD spectroscopy confirmed that CG6 formed Z-DNA upon the addition of hZβDAI (Fig. S5A) in the same way as observed for hZαADAR1 (20). Whereas resonances of the imino protons of CG6 changed during the titration of hZβDAI, AT6 showed no changes except the intensity reduction by the line broadening effect of hZβDAI binding. The B-to-Z transition of CG6 induced by hZβDAI binding was also monitored by CD spectra. The molar ellipticity exhibited significant alterations at 255 and 295 nm, which is characteristic of Z-DNA. However, no spectral change was observed in the case of AT6.
Fig. 3.

DNA binding experiments with CG6 and AT6. (A) Ribbon diagrams indicating the residues that show chemical-shift perturbations (Δδ) of the backbone; hZβDAI with CG6 (Left) and hZβDAI with AT6 (Right) (α, dark red, disappeared; β, red, ≥0.14 ppm; γ, orange, ≥0.10 ppm; δ, yellow, ≥0.06 ppm). (B) Surface mapping of the residues that show chemical-shift perturbations of the aliphatic side chains; 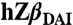 with CG6 (Left) and hZβDAI with AT6 (Right) (α, purple, disappeared; β, blue, ≥0.10 ppm; γ, light blue, ≥0.075 ppm; δ, cyan, ≥0.05 ppm).
with CG6 (Left) and hZβDAI with AT6 (Right) (α, purple, disappeared; β, blue, ≥0.10 ppm; γ, light blue, ≥0.075 ppm; δ, cyan, ≥0.05 ppm).
The CSPs for backbone amide signals in 1H-15N HSQC and the aliphatic side chain signals in 1H-13C HSQC were quantified and displayed on the ribbon models and surface diagrams of the free hZβDAI, respectively (Fig. 3 A and B). When the protein is bound to CG6 as a Z-DNA, the backbone amides of the residues from T136 to R146 in the α3 helix showed significant perturbations. These results can be anticipated from the fact they are located in the N-terminal region of the α3 recognition helix and have critical roles in Z-DNA binding (Fig. 3A). Surprisingly, A137–D143 were also strongly perturbed by AT6 as a B-DNA, which suggests that they may be involved in binding to B-DNA as well as to Z-DNA. Whereas the residues located in the N-terminal region of the α3 helix are important in the binding both CG6 and AT6, the backbone amide groups in the middle part of α3 helix (Y145, R146, and M147) and two β-strands (D154, D156, K160, W162, T163, and Y165) region seem to have preference to CG6 because they showed large CSP values only upon CG6 binding. In addition, residues 107–112 and 107–110 of α1 helix showed perturbation in CG6 and AT6 binding, respectively. Together with residue 126 of the α2 helix, the residues in the N-terminal part of the α1 helix interact with both Z- and B-DNA nonspecifically. Overall, Z-DNA binding showed CSP in a larger area than on B-DNA binding including the β-sheet, whereas B-DNA binding occurs mainly in the region near to the N-terminal of α3 helix.
The crystal structure of the hZβDAI/Z-DNA complex reveals a noncanonical DNA binding mode in that the wing is pulled away from the DNA (15). However, the hZβDAI mutant (E157A/ΔQ158/ΔS159/K160A) showed dramatic decreases in both Z-DNA binding and conversion activity (19). This observation prompted us to hypothesize that the wing structure may be involved in Z-DNA recognition to some extent via a mechanism yet unknown. CSP experiments upon addition of CG6 revealed significant changes in signals of both backbone atoms for the wing residues, H151, D154, D156, K160, W162, and T163, and side chain atoms from H151, M155, D156, E157, and W162, although they are not in direct contact with Z-DNA in the complex structure (15) (Fig. 3 A and B and Fig. S6). These findings may be explained by the fact that DNA binding could affect the magnetic environment of the wing directly or indirectly. H151, which is located near the hinge between α3 and the wing, showed altered conformations in the free and the DNA-bound hZβDAI. Thus, DNA-induced conformational changes in the wing might affect the conformation of H151. The CSP of the side chain of M155 seems to be due to the CH-π or hydrophobic interaction of M155 with W162, a key residue in Z-DNA binding. Additionally, on CG6 binding, R146 and D143 show large CSPs on both backbone and side chain signals, which is consistent with the fact that these two residues form a salt bridge upon Z-DNA binding (15). In the free hZβDAI, they are not salt-bridged but exposed to the solvent, as shown by their chemical-shift values, which are in the average range. Intriguingly, although the backbone signals were not perturbed significantly, the side chains of Y145 and W162 showed strong perturbation by the B-DNA binding as well as Z-DNA binding, which implies that these side chains are closely involved in the DNA contact before B–Z transition. This situation may facilitate the conformational change of Y145 in B–Z transition, resulting in the aromatic ring of Y145 positioned to have the CH-π interactions with both the edge of the W162 and guanosine base of Z-DNA.
Discussion
This study clearly demonstrated that hZβDAI undergoes conformational adaptations such as the movement of the wing and the recognition helix upon Z-DNA binding. Such changes were not expected because it was believed that ZBDs have a prefixed DNA binding interface, as found for hZαADAR1 (11). In addition, Y145 is not prepositioned in hZβDAI. In the free state, Y145 of hZβDAI interacts with the side chain of neighboring R146. Cζ of Y145 is located near Cγ of R146 with a distance of 3.7 Å. Upon Z-DNA binding, the aromatic ring of Y145 moves to become sandwiched between the guanidine base of Z-DNA and the aromatic ring of W162, forming two stabilizing CH-π interactions (17). This movement affects the side chains of W162 and M155, and, in turn, the backbone of the wing from D154 to T163. In addition, the side chain of H151 in the hinge region was affected because of the movement of the wing region, resulting in large CSPs of the NMR signals. A somewhat related conformational change might take place when vvZαE3L binds to Z-DNA, as its analogous tyrosine is flipped away from its binding position in the free state (12) (Fig. 2B).
Residues of hZβDAI involved in B-DNA binding were identified, and the mode of B-DNA binding was partially revealed. Based on the CSP experiment with B-DNA, its B-DNA binding mode may be related to the initial stage of the binding to Z-DNA forming sequences in the B-conformation. The charged residues in the N terminus of the α3 recognition helix may thus be important for the initial recognition of both B- and Z-conformations of DNA. However, there were subtle differences in the CSPs of B- and Z-DNA binding, which may be attributable to structural differences in the two DNA forms. These results support the idea that hZβDAI may have a unique B-DNA binding mode, distinct from the binding to Z-DNA. The wing, which is essential for Z-DNA binding, does not seem to be involved in binding B-DNA. Lack of additional interacting regions other than the recognition helix might explain the relatively low binding affinity and relative nonspecificity of hZβDAI toward B-DNA. Indeed, the measurements of binding affinities by isothemal titration calorimetry revealed significantly higher binding affinity for Z-DNA, with Kd values of 226 nM and 16 μM to CG6 and AT6, respectively (Fig. S7 A and B). This result is consistent with the binding affinities estimated by CSPs. Binding to Z-DNA shows a slow to medium exchange binding pattern, whereas binding to B-DNA shows a fast exchange pattern (Fig. S5 B and C). Considering the interacting residues involved in B-DNA binding, which were identified from CSP experiments, hZβDAI could bind to the minor groove in a nonspecific manner (Fig. S7C), whereas virtually all B-DNA binding proteins bind to major grooves of B-DNA. In this binding model, the residues with a large CSP are located close to the phosphate backbone of B-DNA, which means that hZβDAI can have access to DNAs in the B-conformation without interacting with anything in the major groove. In the binding model to the major groove (Fig. S7D), K160 and Y145 collide with B-DNA, but R135 does not make any contact with it.
In the conversion mechanism to Z-DNA, when hZβDAI binds to CG6 in the B-DNA conformation, it may play an active role in initiating its conversion to the Z-DNA conformation. This is supported by the recent biophysical study of hZαADAR1 (20). In that study, it was observed that the amounts of Zα-bound B-DNA and Zα-bound Z-DNA are the same and in equilibrium when the protein is limited. Then, the Z conformation of DNA is stabilized by additional binding of Zα to Z-DNA (20). A crystal structure of the B-DNA:Z-DNA junction revealed that at the junction one base pair was broken, and the two bases were extruded on either side of the DNA duplex (16). This base extrusion is likely to be central to initiating Z-DNA formation. Could that process be facilitated by B-DNA binding to ZBDs through its minor groove? Clearly, this is an avenue for further research.
Considering the function of DAI in the recognition of foreign DNA to boost the innate immune response (5), the conformational diversity and flexibility of hZβDAI may be an integral feature in recognizing foreign DNAs in various conformations. Because the domain on the C-terminal side of hZβDAI also undergoes a conformational change on binding DNA (5), these changes may be correlated. The current study contributes to understating the role of DAI at the molecular level in the innate immune system.
Materials and Methods
Protein Preparation.
The Zβ domain (103–166) of human DAI (hZβDAI) and the double-stranded DNAs were prepared as described previously (15). Five extra amino residues (98-Ser-His-Met-Ala-Ser-102) were inserted during cloning. To produce isotopic labeled hZβDAI, bacteria were grown in M9 medium containing 1 g L-1 of 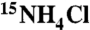 and 2 g L-1 of 13C-glucose (Cambridge Isotope Laboratories). After purification using Ni-NTA and HiTrap SP columns (GE Healthcare), double-labeled hZβDAI was dialyzed and concentrated in the NMR buffer [10 mM 2-(N-morpholino) ethanesulfonic acid (MES) pH 5.5, 10 mM NaCl]. The 15N-labeled hZβDAI P122A mutant was also prepared in the same manner as the wild-type hZβDAI.
and 2 g L-1 of 13C-glucose (Cambridge Isotope Laboratories). After purification using Ni-NTA and HiTrap SP columns (GE Healthcare), double-labeled hZβDAI was dialyzed and concentrated in the NMR buffer [10 mM 2-(N-morpholino) ethanesulfonic acid (MES) pH 5.5, 10 mM NaCl]. The 15N-labeled hZβDAI P122A mutant was also prepared in the same manner as the wild-type hZβDAI.
NMR Spectroscopy.
NMR experiments were carried out at 35 °C on a 2 mM 13C, 15N-labeled protein sample in the NMR buffer containing 10% D2O on Avance 900 MHz (Bruker). 1H, 15N, and 13C-resonance assignments were obtained from the following three-dimensional heteronuclear correlation experiments: CBCA(CO)NH, HNCACB, HN(CA)CO, HNCO, HN(CO)CA, HNCA, HBHA(CO)NH, H(CCO)NH, C(CCO)NH, HCCH–correlation spectroscopy, and HCCH–total correlation spectroscopy (TOCSY). Distance restraints were derived from 15N- or 13C-resolved 3D NOESY. Aromatic side chains were assigned with aromatic 1H-13C HSQC and 2D 1H-1H TOCSY. Aromatic 3D 13C-NOESY was used to produce the distance restraint. The dipolar coupling constants were obtained by taking the difference in the corresponding J-couplings measured in aligned hexanol-polyethylene glycol system wt = 4%, r = 0.85 and with splitting of D2O 20.8 Hz (21, 22) and isotropic (water) media.  -dipolar coupling constants were measured by recording 2D in-phase/antiphase 1H-15N transverse relaxation optimized spectroscopy spectra and
-dipolar coupling constants were measured by recording 2D in-phase/antiphase 1H-15N transverse relaxation optimized spectroscopy spectra and  in IPAP-J-HNCO-based three-dimensional experiment (23, 24). NMR spectra were processed with NMRPipe (25) and analyzed with SPARKY 3.110 (26). For 1D imino proton NMR, samples were prepared by adding hZβDAI to 0.15 mM CG6 or AT6 dissolved in 10 mM MES pH 5.5, 50 mM NaCl to the indicated molar ratios of protein to DNA (P/N). The NMR spectra was taken at 500 MHz NMR (Bruker), and processed by SpinWorks 3.1.7 (27).
in IPAP-J-HNCO-based three-dimensional experiment (23, 24). NMR spectra were processed with NMRPipe (25) and analyzed with SPARKY 3.110 (26). For 1D imino proton NMR, samples were prepared by adding hZβDAI to 0.15 mM CG6 or AT6 dissolved in 10 mM MES pH 5.5, 50 mM NaCl to the indicated molar ratios of protein to DNA (P/N). The NMR spectra was taken at 500 MHz NMR (Bruker), and processed by SpinWorks 3.1.7 (27).
Structure Determination.
Distance restraints were mainly derived from 3D 15N- or 13C-edited and 2D 1H-1H NOESY experiments. NOE assignment algorithm of CYANA 2.1 (28) performed both the structure calculation and the assignment of NOE peaks automatically. A total of 1,897 NOE-based upper distance restraints were gained. TALOS program (29) derived 110 backbone torsion angle restraints from chemical shifts. We obtained hydrogen bond restraints by H–D exchange experiment, and with these restraints, a total of 100 structures without significant violations were generated by CYANA 2.1. The structures were further refined by AMBER 10 (30) using an all-atom force field (ff99SB). The AMBER refinement consisted of three stages: 1,500 steps of energy minimization, 20-ps molecular dynamics, and 1,500 steps of energy minimization. The best 20 structures were selected as a final ensemble. A structure closest to the lowest energy was employed as representative. Final structures were well refined with a backbone and heavy atom RMSD of 0.13 and 0.69 Å, respectively, for residues 98–166. The structures also have good geometry with 92.3% of residues in the most favored region of the Ramachandran plot (Table S1) examined by PROCHECK (31). The N-terminal 10 residues were not well converged in the ensemble because of absence of long-range NOE.
Isothermal Titration Calorimetry.
The CG6 and AT6 were dialyzed in 10 mM MES pH 5.5, 50 mM NaCl prior to the isothermal titration calorimetry experiments. The dissociation constant (Kd) for CG6 or AT6 was measured using a VP-ITC (MicroCal) at 25 °C.
CD Spectroscopy.
The conversion of B- to Z-DNA was monitored by CD spectroscopy as described in the previous work (15) with slight modifications in 10 mM MES pH5.5, 50 mM NaCl. The B-to-Z conversion kinetics of the wild-type and P122A mutant were monitored using CG6 for 50 min at 255 nm at 25 °C in 10 mM MES pH 5.5, 150 mM NaCl.
CSP Experiment.
We titrated 0.6 mM 15N-labeled hZβDAI with CG6 and AT6 in a concentration of 0.035 mM, 0.075 mM, and 0.15 mM, respectively. 1H-15N HSQC spectra were collected for the backbone CSPs and 1H-13C constant time version HSQC spectra for aliphatic side chain CSPs. Both of them were performed in 10 mM MES pH 5.5, 50 mM NaCl at 35 °C. NMR resonances of 15N, 13C hZβDAI were affected along with increasing CG6 and AT6, respectively, in the following ways: (i) none or minimal change in chemical shift, (ii) vanished signal through medium exchange binding pattern, and (iii) major perturbation of chemical shift. Chemical-shift differences were calculated using the following equations:  for backbone amide signals, and
for backbone amide signals, and  for aliphatic side chain signals.
for aliphatic side chain signals.
Supplementary Material
Acknowledgements
This work was supported by National Research Laboratory Program (NRL-2006-02287), Korea Research Foundation Grant (KRF-2008-220-C00040), 21C Frontier Functional Proteomics Program (FPR08B2-270), Korea Healthcare Technology Research and Development Project (A092006), and Ubiquitome Research Program (M105 33010001-05N3301-00100) to K.K.K. and Bio-MR research program and Global Frontier Project Grant NRF-M1AXA002-2010-0029765 to Y.H.J.
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates and NMR constraints have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2L4M).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1014898107/-/DCSupplemental.
References
- 1.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 2.Ishii KJ, et al. A toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol. 2006;7:40–48. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- 3.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-β produced in mouse embryos carrying undigested DNA. Nat Immunol. 2005;6:49–56. doi: 10.1038/ni1146. [DOI] [PubMed] [Google Scholar]
- 5.Takaoka A, et al. DAI (DLM-1/DAI) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–506. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z, et al. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc Natl Acad Sci USA. 2008;105:5477–5482. doi: 10.1073/pnas.0801295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim YG, et al. A role for Z-DNA binding in vaccine virus pathogenesis. Proc Natl Acad Sci USA. 2003;100:6974–6979. doi: 10.1073/pnas.0431131100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartz T, Rould M, Lowenhaupt K, Herbert A, Rich A. Crystal structure of the Zalpha domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science. 1999;284:1841–1845. doi: 10.1126/science.284.5421.1841. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz T, Behlke J, Lowenhaupt K, Heinemann U, Rich A. Structure of the DLM-1-Z-DNA complex reveals a conserved family of Z-DNA-binding proteins. Nat Struct Biol. 2001;8:761–765. doi: 10.1038/nsb0901-761. [DOI] [PubMed] [Google Scholar]
- 10.Ha SC, et al. A poxvirus protein forms a complex with left-handed Z-DNA: Crystal structure of a Yatapoxvirus Zalpha bound to DNA. Proc Natl Acad Sci USA. 2004;101:14367–14372. doi: 10.1073/pnas.0405586101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schade M, et al. The solution structure of the Zα domain of the human RNA editing enzyme ADAR1 reveals a prepositioned binding surface for Z-DNA. Proc Natl Acad Sci USA. 1999;96:12465–12470. doi: 10.1073/pnas.96.22.12465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kahmann JD, et al. The solution structure of the N-terminal domain of E3L shows a tyrosine conformation that may explain its reduced affinity to Z-DNA in vitro. Proc Natl Acad Sci USA. 2004;101:2712–2717. doi: 10.1073/pnas.0308612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Athanasiadis A, et al. The crystal structure of the Zb domain of the RNA-editing enzyme ADAR1 reveals distinct conserved surface among Z-domains. J Mol Biol. 2005;351:496–507. doi: 10.1016/j.jmb.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 14.Schade M, Turner CJ, Lowenhaupt K, Rich A, Herbert A. Structure-function analysis of the Z-DNA-binding domain Zalpha of dsRNA adenosine deaminase type I reveals similarity to the (α + β) family of helix-turn-helix proteins. EMBO J. 1999;18:470–479. doi: 10.1093/emboj/18.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ha SC, et al. The crystal structure of the second Z-DNA binding domain of human DAI (ZBP1) in complex with Z-DNA reveals an unusual binding mode to Z-DNA. Proc Natl Acad Sci USA. 2008;105:20671–20676. doi: 10.1073/pnas.0810463106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ha SC, Lowenhaupt K, Rich A, Kim YG, Kim KK. Crystal structure of a junction between B-DNA and Z-DNA reveals two extruded bases. Nature. 2005;437:1183–1186. doi: 10.1038/nature04088. [DOI] [PubMed] [Google Scholar]
- 17.Brandl M, Weiss MS, Jabs A, Sühnel J, Hilgenfeld R. CH-π interactions in proteins. J Mol Biol. 2001;30:357–377. doi: 10.1006/jmbi.2000.4473. [DOI] [PubMed] [Google Scholar]
- 18.Ha SC, et al. Biochemical characterization and preliminary X-ray crystallographic study of the domains of human ZBP1 bound to left-handed Z-DNA. Biochim Biophys Acta. 2006;1764:320–323. doi: 10.1016/j.bbapap.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 19.Quyen DV, et al. Characterization of DNA-binding activity of Z alpha domains from poxviruses and the importance of the beta-wing regions in converting B-DNA to Z-DNA. Nucleic Acids Res. 2007;35:7714–7720. doi: 10.1093/nar/gkm748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang YM, et al. NMR spectroscopic elucidation of the B-Z transition of a DNA double helix induced by the Z alpha domain of human ADAR1. J Am Chem Soc. 2009;131:11485–11491. doi: 10.1021/ja902654u. [DOI] [PubMed] [Google Scholar]
- 21.Ruckert M, Otting G. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J Am Chem Soc. 2000;122:7793–7797. [Google Scholar]
- 22.Bax A, Kontaxis G, Tjandra N. Dipolar couplings in macromolecular structure determination. Method Enzymol. 2001;339:127–174. doi: 10.1016/s0076-6879(01)39313-8. [DOI] [PubMed] [Google Scholar]
- 23.Nietlispach D. Suppression of anti-TROSY lines in a sensitivity enhanced gradient selection TROSY scheme. J Biomol NMR. 2005;31:161–166. doi: 10.1007/s10858-004-8195-7. [DOI] [PubMed] [Google Scholar]
- 24.Permi P, Rosevear PR, Annila A. A set of HNCO-based experiments for measurement of residual dipolar couplings in 15N, 13C, (2H) labeled proteins. J Biomol NMR. 2000;17:43–54. doi: 10.1023/a:1008372624615. [DOI] [PubMed] [Google Scholar]
- 25.Delaglio F, et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 26.Goddard TD, Kneller DG. San Francisco: University of California; 2002. SPARKY 3. [Google Scholar]
- 27.Marat Kirk. Winnipeg, Manitoba, Canada: University of Manitoba; SpinWorks. Version 3.1.7. [Google Scholar]
- 28.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 29.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 30.Case DA, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.