

Abstract
The synthesis of γ-lactams that are unsubstituted at the 1-position (nitrogen) as well as their subsequent N-functionalization is reported. A recently discovered four-component reaction (4CR) is employed with either an ammonia precursor or a protected form of ammonia that can be deprotected in a subsequent synthetic step. These methods represent the first multicomponent assembly of complex lactam structures that are unsubstituted at nitrogen. In addition, two methods for the introduction of nitrogen substituents that are not possible through the original 4CR are reported. X-ray crystallographic analysis of representative structures reveals conformational changes in the core structure that will enable future deployment of this chemistry in the design and synthesis of diverse collections of lactams suitable for the discovery of new biological probes.
Keywords: multicomponent reaction, stereoselective
Five-membered ring lactams, which are known as γ-lactams or 2-oxopyrrolidines, are important structural motifs in biologically active natural products that are also found in medicinal leads and approved drugs (Fig. 1). Heliotropamide (1, 2) and bisavenanthramide B (3, 4) are examples of ferrulic acid amides that undergo biosynthetic dimerization to produce γ-lactams, whereas lactacystin (5) and salinosporamide (6) emanate from more complex biosyntheses. Although these compounds share the γ-lactam core, an important difference emerges in the substitution at nitrogen, in that the latter examples are sometimes described as “N-H lactams.” Among drug compounds that are approved or in development, N-H lactams are represented significantly more often as shown by UCB-2892, rolapitant, and salinosporamide, in which only UCB-2892 is substituted at nitrogen. A survey of γ-lactams of this general structure in preclinical and clinical development (based on substructure searches of the Thomson Pharma database) showed a nearly two-to-one ratio of N-H lactams over their substituted counterparts. Perhaps more telling is the overall occurrence of γ-lactams related to the structures in Fig. 1, which was quite low at just 89 out of 3 million. These examples highlight both the potential use of this substructure in the discovery of new biological probes and the fact that this core unit is underutilized, perhaps because of synthetic limitations.
Fig. 1.

Structures of γ-lactams from natural sources (green box) and as drug leads (red box). N-substituted lactams are shown in the Left column, and “N-H” lactams are shown in the Right column.
Importantly, some drugs in clinical development (6, 7, and four others not shown), and many of the compounds reported in hit-to-lead studies, are synthetic derivatives of pyroglutamic acid (5). Although this useful building block has found many applications as a member of the “chiral pool,” it is a poor starting point for the diverse synthesis of lactams given the limited opportunities for introducing substituents at the carbon centers of the ring. Recently reported methods address this gap in synthesis technology by allowing the rapid assembly of polysubstituted γ-lactams (7–9).
Multicomponent reactions (MCRs) are transformations involving the combination of three or more reagents in a single operation and are among the most powerful methods for the efficient generation of molecular complexity (10). Although many MCRs have been reported, only a small subset involve the simultaneous combination of reagents (i.e., without regard to the order of addition). Only a subset of these involve components that are variable. Typical variable building blocks include amines, aldehydes, ketoesters, isocyanides, etc, as seen in the Hantszch (11), Biginelli (12) and Ugi-type (13) MCRs. It can be argued that these processes and others that meet both of these criteria are the most powerful MCRs because they will generate maximum complexity with minimal effort (14). We discovered a mechanistically distinct four-component reaction (4CR, Fig. 2A) that enables the assembly of γ-lactams in a single synthetic step in high yield and diastereoselectivity (15). This method allows for the introduction of different substituents at various positions around the ring by the choice of building blocks that are employed or by subsequent functionalization. This reaction has recently proven useful in the preparation of large libraries of compounds for screening experiments that have, in one case, identified potential ligands (13, Fig. 2B) for the disruption of p53-HDMA (16). In addition, we have disclosed a synthesis of the natural product heliotropamide (1, Fig. 1) using this reaction to assemble the lactam core (2). In this report, we disclose methodology for the synthesis of structurally diverse γ-lactams using our previously described 4CR as a starting point. Specifically, we recognized the need for the synthesis of N-H lactams, which is a general problem facing many MCRs that typically employ amines. We demonstrate two methods for preparing these structures and then document subsequent N-functionalization reactions that allow for the divergent synthesis of lactams that are not currently available from the MCR directly. The functionalization of sterically hindered lactams is a problem of fundamental importance that is, in part, addressed by the reactions that we have surveyed and optimized for this process (Fig. 2C).
Fig. 2.

(A) Lactam-forming four-component reaction reported by our group in 2007. (B) 4CR product converted to the corresponding amide (two steps total) recently described as a potential inhibitor of the p53-HDMA interaction. (C) Summary of the transformations reported herein.
Results and Discussion
γ-Lactams are produced by using ammonia or ammonium salts in the lactam-forming MCR. Although there was scant precedent on which to draw from related MCRs (17, 18), we examined the effectiveness of a series of ammonium salts and ammonia solutions (Table 1). Our first reaction employed aqueous ammonium hydroxide on the assumption that the azeotropic removal of water, which is necessary for the formation of the presumed iminium ion intermediate, would enable the net delivery of ammonia that would react with the anhydride. Although this reaction was low-yielding, the observance of a small amount of the desired product prompted us to explore a series of potential ammonia precursors. Various salts were employed, revealing that ammonium acetate (Table 1, entry 11) provided the highest yield (32%). Changes in reaction time or temperature failed to increase the yield significantly, as did increasing the amount of ammonium acetate. Ammonium trifluoroacetate provided the product in similar yield (Table 1, entry 20), whereas ammonium benzoate was relatively ineffective (Table 1, entry 21). In all cases, the yield reflects the isolated yield of the major diasteromer, thus representing a lower limit for the efficiency of this reaction. In two parallel runs of this reaction on identical scale, the products were isolated in yields of 39% and 35% with a diastereomeric ratio (dr) of 56∶44 for the first run and 78∶22 for the second. The relative configuration of the major diastereomer was established by chemical correlation to 4CR products that were deprotected and by X-ray crystallography (see below). We also explored the delivery of ammonia gas to the three solvents that have proven favorable for the 4CR (CHCl3, toluene, and acetonitrile) and found that none of these conditions was satisfactory, regardless of whether the ammonia was predissolved in the solvent or bubbled through the reaction mixture (Table 1, entries 26–29). Although the highest observed yield in this reaction is still rather low, the practical simplicity of this process, paired with the readily available and inexpensive reagents, makes it viable for the facile preparation of large quantities of N-H lactam 19.
Table 1.
Optimization of the 4CR using various ammonia sources
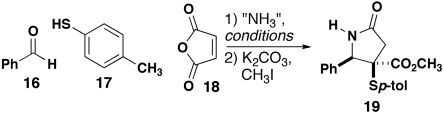
| Entry | Ammonia source, “NH3” | Conditions | Yield of 19, % |
| 1 | NH4OH (aq) | PhMe, 145 °C, 22 h | 15 |
| 2 | NH4OH (aq) | PhMe, 150 °C, 3 h | 22 |
| 3 | NH4HCO2 | PhMe, 150 °C, 3 h | 16 |
| 4 | NH4CO2NH2 | PhMe, 150 °C, 3 h | 17 |
| 5 | NH4Cl | PhMe, 150 °C, 1 h | — |
| 6 | NH4Cl + DIPEA | PhMe, 150 °C, 1 h | — |
| 7 | NH4OAc | PhMe, 150 °C, 0.5 h | 30 |
| 8 | NH4OAc | PhMe, 110 °C, 1 h | — |
| 9 | NH4OAc | PhMe, 130 °C, 1 h | 7* |
| 10 | NH4OAc | PhMe, 150 °C, 10 min | 11 |
| 11 | NH4OAc | PhMe, 150 °C, 1 h | 32 |
| 12 | NH4OAc | PhMe, 150 °C, 3 h | 23 |
| 13 | NH4OAc | CHCl3, 150 °C, 3 h | 12 |
| 14 | NH4OAc | PhMe, 170 °C, 3 h | 17 |
| 15 | NH4OAc | PhMe, 170 °C, 1 h | 13 |
| 16 | NH4OAc | PhMe, 145 °C, 22 h | 5 |
| 17 | NH4OAc (4 eq) | PhMe, 145 °C, 22 h | 10 |
| 18 | NH4OAc (4 eq) | PhMe, 150 °C, 1 h | 9 |
| 19 | NH4OAc + DMAP | PhMe, 150 °C, 1 h | 11 |
| 20 | NH4CF3CO2 | PhMe, 150 °C, 1 h | 32* |
| 21 | NH4OBz | PhMe, 150 °C, 1 h | 21 |
| 22 | (NH4)2CO3 | PhMe, 150 °C, 1 h | 18 |
| 23 | NH3 | MeOH, 150 °C, 1 h | — |
| 24 | NH3 | 1,4-dioxane, 150 °C, 1 h | 12 |
| 25 | NH4OH (aq) | CH3CN, 150 °C, 3 h | 3 |
| 26 | NH3 (g)† | PhMe, 145 °C, 22 h | 14 |
| 27 | NH3 (g)† | PhMe, 150 °C, 1 h | 6 |
| 28 | NH3 (g)† | CHCl3, 150 °C, 1 h | 4 |
| 29 | NH3 (g)† | CH3CN, 150 °C, 1 h | 4 |
*Refers to conversion observed by HPLC. In all other cases, isolated yields are listed.
†Ammonia gas was bubbled through the solvent before adding the other reagents. Changing the order of addition so that the ammonia was added last resulted in a slight (2%) reduction in isolated yield for entries 26 and 27.
The modest yield of the ammonia-based MCR prompted us to consider a two-step process using an ammonia equivalent in analogy to the Gabriel amine synthesis and other reactions using synthetic equivalents of ammonia. In this process, we envisioned using a primary amine in the 4CR and then cleaving the attached group to reveal the N-H lactam product. The second step amounts to a deprotection, which immediately revealed the general problem of protecting groups for amides. Unlike amines, alcohols, and other functional groups for which dozens of common protecting groups have been developed, there are few amide protecting groups, and only a subset of these can be cleaved efficiently in a one-step process (18). Fewer still would represent stable species when appended to an NH2 group, as would be necessary in the 4CR. A thorough survey of the literature revealed several possibilities (Table 2): p-methoxyphenyl, p-methoxybenzyl, 2,4-dimethoxybenzyl, allyl, and diphenylmethyl (DPM) (19). In addition, several additional options emerged in the form of dianisylmethyl (DAM) (20), which should be more easily cleaved with acid than the analogous DPM group, and N,N-dimethylamino, which can be cleaved oxidatively (21) or photolytically (22). Finally, we drew inspiration from the work of Buchwald and Seeberger, who demonstrated that alcohol protection could be controlled by the use of different p-halobenzyl groups. The aromatic halide was then converted to a substituted aniline that could be cleaved under Lewis or Brønsted acidic conditions (23).
Table 2.
Use of synthetic equivalents of ammonia in the 4CR
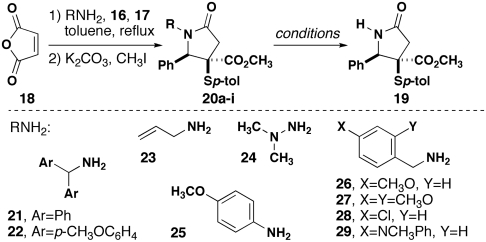
| Entry | RNH2 | Product (yield; dr) | Conditions for removal of R | Yield of 19, % |
| 1 | 21 | 20a (70%, 92:8) | TFA (neat), reflux, 48 h | 62 |
| 2 | 22 | 20b (77%, 80∶20) | TFA (neat), reflux, 48 h | 91 |
| 3 | 23 | 20c (52%, 90∶10) | RhCl3 (2 mol %), EtOH, 2 h; HOAc/H2O, reflux, 22 h | 65 |
| 4 | 24 | 20d (NR) | MMPP•H2O, MeOH, RT | — |
| 5 | 25 | 20e (66%, 92∶8) | CAN, CH3CN/H2O, 0 °C, 30 min | 30 |
| 6 | 26 | 20f (65%, 98∶2) | CAN, CH2Cl2/H2O, 0 °C, 16 h | 40 |
| 7 | 27 | 20g (66%, 92∶8) | CAN, CH3CN, RT, 16 h | 29 |
| 8 | 28 | 20h (62%, 92∶8) | Pd (1 mol %), PhNHCH3; Lewis acid, CH2Cl2, RT | — |
| 9 | 29 | 20i (67%, 95∶5) | CAN, CH3CN/H2O, RT, 16 h | 29 |
MMPP, magnesium monoperoxyphthalate; CAN, ceric ammonium nitrate; NR, no reaction.
Each of the protecting groups was evaluated, and nearly all provided access to the desired product. In each case, we determined the efficiency and diastereoselectivity of the 4CR and the highest-yielding conditions for cleavage (Table 2). Diphenylmethylamine (21) underwent the 4CR with good dr, and its cleavage was effected in modest (62%) yield using TFA. Attempted cleavage by hydrogenolysis was ineffective (24). The introduction of p-methoxy groups to both phenyl rings in the form of dianisylmethylamine (DAM-NH2, 22) (25) resulted in higher yield and lower diastereoselectivity for the 4CR as well as high yield for the cleavage reaction (91%). The utility of this sequence is reduced somewhat by the harsh conditions for cleavage. Allylamine (23) exhibited a lower yield in the 4CR and offered the mildest possible conditions for cleavage by rhodium-catalyzed rearrangement and acidic cleavage of the resultant enamide (26). N,N-dimethylhydrazine failed to produce any 4CR product. p-Anisidine (25), p-methoxybenzylamine (26), and 2,4-dimethoxybenzylamine (27) all provided satisfactory yields of 4CR product and low yields of 19 under the oxidative conditions needed for cleavage (27–31). Resubjection of 19 to the oxidative conditions revealed that the product was stable to the reaction medium, suggesting that aberrant oxidative processes were destroying the starting material. Although p-chlorobenzylamine (28) provided the 4CR product in decent yield, the conditions needed for amination with N-methylaniline (23, 32) seemed to destroy the starting material. We could escape this liability by preparing the requisite amine 29 before executing the 4CR, which proceeded in good yield. The cleavage of this group from the lactam was predictably more difficult than for an alcohol and was never higher than 30%. All things considered, use of dianisylmethylamine (22) gives the highest yield of the final product (70%, two steps). Although the other methods in Table 2 might find specialized applications for certain substrates, none provides a significantly higher yield of 19 over the two steps when compared to ammonia 4CR (32%, Table 1).
The optimized conditions for the formation of 19 were employed in the synthesis of related substrates (Fig. 3). A series of p-substituted benzaldehydes suggests that the electronic nature of this substituent significantly impacts their reactivity and, as such, the yield of final product. p-Anisaldehyde provided the lactam in almost double the yield (59%) of benzaldehyde, and p-bromobenzaldehyde was higher as well. An electron-deficient aldehyde (p-trifluoromethylbenzaldehyde) was slightly lower yielding. Replacement of the phenyl ring of benzaldehyde with a furan resulted in a comparable yield of the product, which is one of the few successful reactions that we have observed with this substrate. Finally, use of two different thiols, one that is unsubstituted (thiophenol) and one that is electron-rich (p-methoxythiophenol), provided the 4CR products in lower (30e) and higher (30f) yields, respectively. In all cases, the diasteromer ratio hovered around that of 19. Control experiments revealed that extended heating of 19 caused the dr to erode to 50∶50 over time, whereas a similar experiment with 30e showed no change. It is clear from these results that the electronic natures of both the aldehyde and the thiol impact the efficiency of the 4CR.
Fig. 3.

Substrate scope of 4CR with ammonium acetate, maleic anhydride, and various aldehydes and thiols.
Having established unique routes to N-H lactams, we set out to use these compounds in reactions that would provide access to N-substituted products not currently available from our 4CR. The high propensity for related compounds to be derived from pyroglutamic acid (5), or derivatives in which the exocyclic carboxy group has been reduced, results in many γ-lactam functionalization reactions that are not suitable for larger substituents at the 5-position, such as phenyl rings. We ultimately focused on two types of functionalization: acylation (including phosphonylation and sulfonylation) and arylation. Additional possible N-functionalization reactions of a related bicyclic substrate have been summarized in the work of Borthwick et al. (33).
The key to effective acylation was the choice of base that was used. In our substrate, there is a risk of equilibration between the amidate and enolate ions, the latter of which can spontaneously decompose by β-elimination of the thioaryl group. After surveying various bases including lithium bis(trimethylsilyl)amide, potassium bis(trimethylsilyl)amide, and NaH, we found that n-BuLi provided the highest yields of products derived from the resultant anion. Acylation reactions, in analogy to the work of Gage and Evans (34), proceed in modest yields with a variety of acyl chlorides (Fig. 4) (35). Additionally, ethyl chloroformate and phenyl isocyanate (36) provided acceptable yields of the corresponding acyl-carbamate and urea products, respectively. Finally, dimethyl chlorophosphate chloride (33) and p-toluenesulfonyl chloride (37) were also effective electrophiles for reactions with the lactam-derived anion.
Fig. 4.

Acylation, sulfonylation, and phosphonylation of 19.
Arylation of lactam 19 is achieved in high yields using cross-coupling reactions that employ stoichiometric quantities of copper. Although many methods, employing catalytic quantities of either copper (38–40) or palladium (41), have been described, these are often limited to substrates with little steric occlusion of the amide nitrogen. Use of these conditions with our substrate resulted in little or no observed product. A complement to these methods is found in the use of boronic acids to functionalize amines, amides, alcohols, and phenols in the presence of stoichiometric quantities of a copper (II) salt, often Cu(OAc)2 as originally described by Chan et al. (42) and Evans et al. (43). Since these initial studies, this reaction has been adopted widely and used in the syntheses of many complex molecules. The popularity is, in part, derived from the very mild temperatures and wide compatibility with other functional groups that might be present. In spite of the wide use of this reaction for amide functionalization, few reports subsequent to the original work of Chan document success on substrates with substitution similar to 19. Specific examples are found in the work of Ishikura and coworkers (44) and Bolshan and Batey (45), the latter of which replaces the boronic acid with the analogous aryl or alkenyl trifluoroborate salt. Extensive exploration of conditions revealed that boronic acids functioned well for the arylation of 19, that Cu(OAc)2 was the optimal reagent, and that triethylamine was the optimal base (Fig. 5). Electron-rich aromatic groups were introduced in high yield (32a, 32b) unless the electron-donating substituent was in the ortho position (32c). Aromatic groups lacking an electron-donating substituent worked less well with a similar trend regarding ortho substitution (32d, 32j). Electron-poor aromatic rings were introduced in generally poorer yields (32g, 32h, 32o). A similar reactivity range was observed for heterocycles; i.e., an electron-rich indole ring was added in high yield (32k), whereas electron-poor pyridylboronic acids were unreactive (32m, 32n). Copper-mediated direct functionalization of heterocycles with amides and the cross-coupling of bromides using the protocols of Wang and Schreiber (46) and Borthwick et al. (47), respectively, were unsuccessful.
Fig. 5.

Arylation of lactam 19 by copper-mediated reactions of arylboronic acids.
X-Ray crystallographic analysis of several representative structures reveals changes to the lactam conformation that result from the changes in substitution at nitrogen (Fig. 6). Unsubstituted lactam 19 sits in a nearly flat five-membered ring conformation with a slight upward envelope that disposes the phenyl ring and carbomethoxy groups in pseudoequatorial positions with a dihedral angle of 100°. When nitrogen is substituted, with an acyl group, the conformation flips to a more puckered downward envelope in which the phenyl ring and the carbomethoxy groups of 31e and 31d now adopt a dihedral angle of 153°. In addition, the two acyl substituents adopt different conformations. Coplanarity of the two imide carbonyls minimizes 1,2-allylic strain with the benzylic hydrogen of the lactam ring. Although the carboethoxy group of 31e can adopt a conformation in which the carbonyl group aligns with that of the lactam, the cinnamoyl group of 31d is in an antiparallel conformation due to the decreased steric demand of the carbonyl group when compared to the benzylidene carbons. N-Aryl lactam 32g adopts a conformation similar to that of the two acyl compounds in which the aromatic ring is almost coplanar with the lactam carbonyl. Although conformational information derived from the solid state is likely to be less reliable for the peripheral substituents of these molecules, knowledge of the core conformations should be helpful in assessing how functionality might be presented by one of these γ-lactams when it interacts with a biological target.
Fig. 6.

X-Ray crystal structures of 19 (A, B), 31e (C, D), 31d (E, F), and 32g (G, H).
In summary, we have demonstrated that our lactam-forming 4CR is useful for the preparation of unsubstituted γ-lactams and that these substrates can be further functionalized through acylation and arylation. The diverse products are useful starting points for the discovery of biological probes and medicinal leads. The synthetic advances reported will help pave the way for increased use of this important scaffold in the discovery and development of biologically active molecules.
Materials and Methods
A subset of experimental procedures is provided. A detailed description of materials, methods, and full characterization data for all previously undescribed compounds is provided in SI Appendix. Most of the compounds reported herein will be deposited in the National Small Molecule Repository (National Institutes of Health) and made available for screening experiments.
Lactam 19.
The following were combined in a microwave vial: maleic anhydride (0.098 g, 1.0 mmol), p-thiocresol (1.0 mL, 1.0 mmol, 1.0 M in toluene), ammonium acetate (0.077 g, 1.0 mmol), and benzaldehyde (0.10 mL, 1.0 mmol). The reaction mixture was stirred at room temperature (RT) for 5 min, then heated in the microwave at 150 °C for 1 h. After cooling to RT, the solvent was evaporated under reduced pressure, and the reaction mixture was dissolved in acetone (20 mL). Potassium carbonate (0.55 g, 4.0 mmol) and methyl iodide (0.25 mL, 4.0 mmol) were then added, and the reaction was stirred for 16 h. The solvent was removed under reduced pressure, and the residue was partitioned with CH2Cl2 and water. The aqueous layer was extracted twice with CH2Cl2. The combined organic was washed with brine, dried over Na2SO4, filtered, and concentrated. Purification was accomplished using 20–80% EtOAc:hexanes, which gave the product as a white solid (0.11 g, 32%).
Lactam 20b.
A mixture of maleic anhydride (0.098 g, 1.0 mmol), p-thiocresol (1.0 mL, 1.0 mmol, 1 M in toluene), diphenylmethylamine (0.243 g, 1.0 mmol), and benzaldehyde (0.10 mL, 1.0 mmol) in toluene (5 mL) were heated to 145 °C with a Dean Stark trap under a reflux condenser for 16 h. After cooling to RT, the reaction mixture was concentrated under reduced pressure and was dissolved in acetone (20 mL). K2CO3 (0.55 g, 4.0 mmol) and methyl iodide (0.25 mL, 4.0 mmol) were then added, and the reaction was stirred for 16 h. The solvent was removed under reduced pressure, the residue was taken up in CH2Cl2 and water, and the layers were separated. The aqueous layer was extracted twice more with CH2Cl2. The combined organics was washed with brine, dried over Na2SO4, filtered, and concentrated. Purification by column chromatography afforded the desired product (0.44 g, 77%).
Deprotection of 20b.
TFA (3 mL) was added to 20b (0.198 g, 0.35 mmol), and the mixture was stirred for 48 h at reflux. The excess TFA was removed under reduced pressure, and the residue was purified by column chromatography (50 to 70% EtOAc:hexanes) to afford 19 (0.11 g, 91%).
Lactam 31d.
Lactam 19 (0.044 g, 0.129 mmol) dissolved in 2 mL of dry THF was added to a flame-dried flask. The solution was cooled to -78 °C and allowed to stir for 10 min. At this time n-BuLi (0.082 mL, 0.129 mmol, 1.58 M in hexanes) was added, and the solution was stirred at -78 °C for 2 h; 3-Phenylacryloyl chloride (0.032 g, 0.194 mmol) was then added, and the solution was stirred at -78 °C for an additional 1 h. The reaction was warmed to 0 °C, stirred for 20 min, and then warmed to room temperature and stirred for an additional hour. Finally, the reaction was quenched with 5 mL of saturated NaHCO3 solution and allowed to stir for 20 min. After quenching, THF was removed in vacuo, and the remaining solution of water and oily solid was partitioned between 5 mL of water and 10 mL of CH2Cl2. The layers were separated, and the aqueous layer was extracted with 3 × 5 mL of CH2Cl2. The combined organic layers were washed with 3 × 5 mL of brine, dried over NaSO4, filtered, and concentrated to give a yellow solid. Purification by flash chromatography (20 to 80% EtOAc/hexanes) afforded the title compound (0.046 g, 76%) as a white solid.
Lactam 32a.
A flask with 3 Å molecular sieves and a stir bar was flame dried under vacuum and allowed to cool to RT under argon. Lactam 19 (0.027 g, 0.08 mmol), 4-chlorophenylboronic acid (0.049 g, 0.32 mmol), and Cu(OAc)2 (0.029 g, 0.32 mmol) were weighed in air combined in the flask. Dry acetonitrile (0.3 M) and triethylamine (0.044 mL, 0.32 mmol) were added through the septa. The reaction mixture was stirred at RT for 48 h then filtered through a pad of Celite. The solvent was removed under reduced pressure, and the resulting oil was purified by column chromatography (20–50% EtOAc:hexanes) to afford product as a solid (0.03 g, 84%).
Supplementary Material
Acknowledgments.
This work was supported by funding from the National Science Foundation (CAREER award to J.T.S.) and the National Institutes of Health (NIGMS/P41GM089153 and NCI/R01CA131458). D.Q.T. acknowledges support in the form of a Bradford Borge Graduate Research Fellowship from University of California, Davis.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: X-ray crystallographic data in the form of crystallographic information files have been deposited in the Cambridge Structural Database, Cambridge Crystallographic Data Centre, Cambridge CB2 1EZ, United Kingdom [CSD reference nos. 805345 (19), 805485 (31d), 805347 (31e), and 805346 (32g)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1015261108/-/DCSupplemental.
References
- 1.Guntern A, et al. Heliotropamide, a novel oxopyrrolidine-3-carboxamide from Heliotropium ovalifolium. J Nat Prod. 2003;66:1550–1553. doi: 10.1021/np0302495. [DOI] [PubMed] [Google Scholar]
- 2.Younai A, Chin GF, Fettinger JC, Shaw JT. Diastereoselective synthesis of (±)-heliotropamide by a one-pot, four-component reaction. J Org Chem. 2010;75:8333–8337. doi: 10.1021/jo1019317. [DOI] [PubMed] [Google Scholar]
- 3.Okazaki Y, Ishihara A, Nishioka T, Iwamura H. Identification of a dehydrodimer of avenanthramide phytoalexin in oats. Tetrahedron. 2004;60:4765–4771. [Google Scholar]
- 4.Okazaki Y, Ishizuka A, Ishihara A, Nishioka T, Iwamura H. New dimeric compounds of avenanthramide phytoalexin in oats. J Org Chem. 2007;72:3830–3839. doi: 10.1021/jo0701740. [DOI] [PubMed] [Google Scholar]
- 5.Omura S, et al. Structure of lactacystin, a new microbial metabolite which induces differentiation of neuroblastoma cells. J Antibiot. 1991;44:117–118. doi: 10.7164/antibiotics.44.117. [DOI] [PubMed] [Google Scholar]
- 6.Feling RH, et al. Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angew Chem Int Edit. 2003;42:355–357. doi: 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]
- 7.Lettan RB, Galliford CV, Woodward CC, Scheidt KA. Amide enolate additions to acylsilanes: In situ generation of unusual and stereoselective homoenolate equivalents. J Am Chem Soc. 2009;131:8805–8814. doi: 10.1021/ja808811u. [DOI] [PubMed] [Google Scholar]
- 8.Raup DEA, Cardinal-David B, Holte D, Scheidt KA. Cooperative catalysis by carbenes and Lewis acids in a highly stereoselective route to γ-lactams. Nat Chem. 2010;2:766–771. doi: 10.1038/nchem.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang Y, Fettinger JC, Shaw JT. One-step synthesis of complex nitrogen heterocycles from imines and alkyl-substituted maleic anhydrides. Org Lett. 2009;11:3802–3805. doi: 10.1021/ol901018k. [DOI] [PubMed] [Google Scholar]
- 10.Zhu J, Bienaymé H, editors. Multicomponent Reactions. Weinheim, Germany: Wiley–VCH Verlag; 2005. p. 468. [Google Scholar]
- 11.Simon C, Constantieux T, Rodriguez J. Utilisation of 1,3-dicarbonyl derivatives in multicomponent reactions. Eur J Org Chem. 2004;2004:4957–4980. [Google Scholar]
- 12.Kappe CO, Stadler A. The Biginelli dihydropyrimidine synthesis. Org Reactions. 2004;63:1–116. [Google Scholar]
- 13.Doemling A. Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem Rev. 2006;106:17–89. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]
- 14.Biggs-Houck JE, Younai A, Shaw JT. Recent advances in multicomponent reactions for diversity-oriented synthesis. Curr Opin Chem Biol. 2010;14:371–382. doi: 10.1016/j.cbpa.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Wei J, Shaw JT. Diastereoselective synthesis of γ-lactams by a one-pot, four-component reaction. Org Lett. 2007;9:4077–4080. doi: 10.1021/ol701911u. [DOI] [PubMed] [Google Scholar]
- 16.Burdack C, Kalinski C, Ross G, Weber L, Khazak V. EPO 2009-EP6670 2010028862. European Patent Office. 2010 Mar 25;
- 17.Kazmaier U, Hebach C. Peptide syntheses via Ugi reactions with ammonia. Synlett. 2003:1591–1594. [Google Scholar]
- 18.Thompson MJ, Chen B. Ugi reactions with ammonia offer rapid access to a wide range of 5-aminothiazole and oxazole derivatives. J Org Chem. 2009;74:7084–7093. doi: 10.1021/jo9014529. [DOI] [PubMed] [Google Scholar]
- 19.Wuts PGM, Greene TW. Greene's Protective Groups in Organic Synthesis. 4th Ed. Hoboken, NJ: John Wiley & Sons, Inc.; 2007. p. 1082. [Google Scholar]
- 20.Carlier PR, Zhao H, MacQuarrie-Hunter SL, DeGuzman JC, Hsu DC. Enantioselective synthesis of diversely substituted quaternary 1,4-benzodiazepin-2-ones and 1,4-benzodiazepine-2,5-diones. J Am Chem Soc. 2006;128:15215–15220. doi: 10.1021/ja0640142. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez R, et al. A practical oxidative method for the cleavage of hydrazide N-N bonds. Chem Eur J. 2004;10:737–745. doi: 10.1002/chem.200305501. [DOI] [PubMed] [Google Scholar]
- 22.Lebrun S, Couture A, Deniau E, Grandclaudon P. A practical photochemically induced method for N-N bond cleavage of N,N-disubstituted hydrazides. Synlett. 2009:2621–2624. [Google Scholar]
- 23.Plante OJ, Buchwald SL, Seeberger PH. Halobenzyl ethers as protecting groups for organic synthesis. J Am Chem Soc. 2000;122:7148–7149. [Google Scholar]
- 24.Effenberger F, Mueller W, Keller R, Wild W, Ziegler T. Amino acids. 8. A novel synthesis of γ-carboxy-L-glutamic acid from L-5-oxoproline esters. J Org Chem. 1990;55:3064–3067. [Google Scholar]
- 25.Ito Y, Kobayashi Y, Kawabata T, Takase M, Terashima S. Novel syntheses of the carbapenem key intermediates, (3R,4R)-4-acetoxy-3-[(R)-1-(tert-butyldimethylsilyloxy)ethyl]-2-azetidinone and (3S,4R)-3-[(R)-1-(tert-butyldimethylsilyloxy)ethyl]-4-carboxymethyl-2-azetidinone, from (S)-ethyl lactate. Tetrahedron. 1989;45:5767–5790. [Google Scholar]
- 26.Kanno O, Miyauchi M, Kawamoto I. Efficient syntheses of (S)-4-hydroxy-2-pyrrolidinone derivatives. Heterocycles. 2000;53:173–181. [Google Scholar]
- 27.Wee AGH, McLeod DD. Studies on the Rh(II)-catalyzed C-H insertion reaction of some derivatives of N-{4-[(S)-1,2-dihydroxybutyl]} alpha -diazo anilides: Site-selectivity. Heterocycles. 2000;53:637–655. [Google Scholar]
- 28.Dai C-F, Cheng F, Xu H-C, Ruan Y-P, Huang P-Q. Diversity-oriented asymmetric synthesis of hapalosin: Construction of three small C9/C4/C3-modified hapalosin analogue libraries. J Comb Chem. 2007;9:386–394. doi: 10.1021/cc060166h. [DOI] [PubMed] [Google Scholar]
- 29.Huang P-Q, Guo Z-Q, Ruan Y-P. A versatile approach for the asymmetric syntheses of (1R,9aR)-epiquinamide and (1R,9aR)-homopumiliotoxin 223G. Org Lett. 2006;8:1435–1438. doi: 10.1021/ol0602203. [DOI] [PubMed] [Google Scholar]
- 30.Meng W-H, Wu T-J, Zhang H-K, Huang P-Q. Asymmetric syntheses of protected (2S,3S,4S)-3-hydroxy-4-methylproline and 4′-tert-butoxyamido-2′-deoxythymidine. Tetrahedron Asymmetry. 2004;15:3899–3910. [Google Scholar]
- 31.Liu G, et al. Design, synthesis, and biological evaluation of caprolactam-modified bengamide analogues. ChemMedChem. 2008;3:74–78. doi: 10.1002/cmdc.200700214. [DOI] [PubMed] [Google Scholar]
- 32.Maes BUW, Loones KTJ, Hostyn S, Diels G, Rombouts G. Rapid palladium-catalyzed aminations of aryl chlorides with aliphatic amines under temperature-controlled microwave heating. Tetrahedron. 2004;60:11559–11564. [Google Scholar]
- 33.Borthwick AD, et al. Design and synthesis of pyrrolidine-5,5-trans-lactams (5-oxohexahydropyrrolo[3,2-b]pyrroles) as novel mechanism-based inhibitors of human cytomegalovirus protease. 2. Potency and chirality. J Med Chem. 2002;45:1–18. doi: 10.1021/jm0102203. [DOI] [PubMed] [Google Scholar]
- 34.Gage JR, Evans DA. Diastereoselective aldol condensation using a chiral oxazolidinone auxiliary. Org Synth. 1990;68:83–91. [Google Scholar]
- 35.Sibi MP, Gorikunti U, Liu M. Temperature dependent reversal of stereochemistry in enantioselective conjugate amine additions. Tetrahedron. 2002;58:8357–8363. [Google Scholar]
- 36.Dieltiens N, et al. The pyroglutamate hydantoin rearrangement. Eur J Org Chem. 2006;2006:2649–2660. [Google Scholar]
- 37.Occhiato EG, Prandi C, Ferrali A, Guarna A. Remote stereocontrol in the Nazarov reaction: A new approach to the core of roseophilin. J Org Chem. 2005;70:4542–4545. doi: 10.1021/jo0504058. [DOI] [PubMed] [Google Scholar]
- 38.Ghinet A, et al. Studies on pyrrolidinones. On the application of copper-catalyzed arylation of methyl pyroglutamate to obtain a new benzo[de]quinoline scaffold. Tetrahedron. 2010;66:215–221. [Google Scholar]
- 39.Phillips DP, et al. Copper-catalyzed C-N coupling of amides and nitrogen-containing heterocycles in the presence of cesium fluoride. Tetrahedron Lett. 2009;50:7293–7296. [Google Scholar]
- 40.Yamada K, Kubo T, Tokuyama H, Fukuyama T. A mild copper-mediated intramolecular amination of aryl halides. Synlett. 2002:231–234. [Google Scholar]
- 41.Yin J, Buchwald SL. Palladium-catalyzed intermolecular coupling of aryl halides and amides. Org Lett. 2000;2:1101–1104. doi: 10.1021/ol005654r. [DOI] [PubMed] [Google Scholar]
- 42.Chan DMT, Monaco KL, Wang R-P, Winters MP. New N- and O-arylation with phenylboronic acids and cupric acetate. Tetrahedron Lett. 1998;39:2933–2936. [Google Scholar]
- 43.Evans DA, Katz JL, West TR. Synthesis of diaryl ethers through the copper-promoted arylation of phenols with arylboronic acids. An expedient synthesis of thyroxine. Tetrahedron Lett. 1998;39:2937–2940. [Google Scholar]
- 44.Abe T, Takeda H, Yamada K, Ishikura M. Copper-catalyzed N-arylation reaction of 2-azabicyclo[2.2.1]hept-5-en-3-one with arylboronic acids under microwave irradiation. Heterocycles. 2008;76:133–136. [Google Scholar]
- 45.Bolshan Y, Batey RA. Enamide synthesis by copper-catalyzed cross-coupling of amides and potassium alkenyltrifluoroborate salts. Angew Chem Int Ed. 2008;47:2109–2112. doi: 10.1002/anie.200704711. [DOI] [PubMed] [Google Scholar]
- 46.Wang Q, Schreiber SL. Copper-mediated amidation of heterocyclic and aromatic C-H bonds. Org Lett. 2009;11:5178–5180. doi: 10.1021/ol902079g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borthwick AD, et al. Design and synthesis of pyrrolidine-5,5′-trans-lactams (5-oxo-hexahydropyrrolo[3,2-b]pyrroles) as novel mechanism-based inhibitors of human cytomegalovirus protease. 4. Antiviral activity and plasma stability. J Med Chem. 2003;46:4428–4449. doi: 10.1021/jm030810w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.