

Abstract
Alkaloid and terpenoid natural products display an extensive array of chemical frameworks and biological activities. However such scaffolds remain underrepresented in current screening collections and are, thus, attractive targets for the synthesis of natural product-based libraries that access underexploited regions of chemical space. Recently, we reported a systematic approach to the stereoselective synthesis of multiple alkaloid/terpenoid-like scaffolds using transition metal-mediated cycloaddition and cyclization reactions of enyne and diyne substrates assembled on a tert-butylsulfinamide lynchpin. We report herein the synthesis of a 190-membered library of alkaloid/terpenoid-like molecules using this synthetic approach. Translation to solid-phase synthesis was facilitated by the use of a tert-butyldiarylsilyl (TBDAS) linker that closely mimics the tert-butyldiphenysilyl protecting group used in the original solution-phase route development work. Unexpected differences in stereoselectivity and regioselectivity were observed in some reactions when carried out on solid support. Further, the sulfinamide moiety could be hydrolyzed or oxidized efficiently without compromising the TBDAS linker to provide additional amine and sulfonamide functionalities. Principal component analysis of the structural and physicochemical properties of these molecules confirmed that they access regions of chemical space that overlap with bona fide natural products and are distinct from areas addressed by conventional synthetic drugs and drug-like molecules. The influences of scaffolds and substituents were also evaluated, with both found to have significant impacts on location in chemical space and three-dimensional shape. Broad biological evaluation of this library will provide valuable insights into the abilities of natural product-based libraries to access similarly underexploited regions of biological space.
Keywords: diversity-oriented synthesis, multiscaffold library, asymmetric synthesis, cheminformatics
A major goal in the field of diversity-oriented synthesis is the efficient production of small-molecule libraries that address underexploited regions of biologically relevant chemical space to enable the discovery of new biological probes and potential therapeutic lead compounds (1). A key approach to addressing this challenge is to emulate natural products and other biogenetic molecules, which have coevolved with macromolecular biological targets (2). Toward this end, a variety of natural product-based libraries have been synthesized, with promising early results (3). These libraries can be validated initially by evaluation of their structural and physicochemical properties using principal component analysis (PCA) to determine the regions of chemical space that are accessed. Subsequently, screening across a wide range of biological assays provides direct biological validation of the functional capabilities of these libraries.
Alkaloids and terpenoids have long served as important small-molecule drugs and leads for drug discovery (4, 5). Indeed, alkaloid and terpenoid cores are prevalent among privileged scaffolds able to bind multiple biological targets (6), and a variety of alkaloid- and terpenoid-based libraries have been reported (7–11).
We recently reported a systematic approach to the synthesis of multiple polycyclic scaffolds related to structures found in diverse alkaloid and terpenoid natural products (12). Our synthetic approach was designed to emulate divergent biosynthetic strategies used in nature by converting a small number of relatively simple, acyclic starting materials into a diverse array of polycyclic scaffolds (13). A series of enyne and diyne substrates is assembled using a tert-butylsulfinamide lynchpin that affords asymmetric induction, a uniquely suited reactivity pattern, and an unusual structural motif for biological evaluation (14). Transition metal-mediated cycloaddition and cyclization reactions are then used to convert these substrates to 10 distinct classes of polycyclic scaffolds that are primed for further functionalization. PCA of structural and physicochemical properties indicates that these scaffolds sample a distinct region of chemical space compared to drugs and drug-like libraries, overlapping with polycyclic alkaloid and terpenoid natural products.
Building upon this work, we envisioned the construction of a larger library of alkaloid/terpenoid-like small molecules for further chemical and biological evaluation. Importantly, our original solution-phase synthesis was designed to facilitate future translation to solid-phase parallel synthesis through the incorporation of a tert-butyldiphenysilyl (TBDPS)-protected primary alcohol. This moiety serves as a faithful surrogate for a tert-butyldiarylsilyl (TBDAS) linker that we have previously developed (15). This robust linker is known to be stable to a variety of acidic and basic reaction conditions, and was expected to be similarly compatible with the desired transition metal-mediated cycloaddition and cyclization reactions, many of which had not previously been carried out on solid support. Moreover, we envisioned that the versatile tert-butylsulfinamide moiety could be used to introduce additional diversity through its inherent stereochemical diversity, hydrolysis to afford the corresponding secondary amines, and oxidation to provide related tert-butylsulfonamide congeners.
Results and Discussion
Solid-Phase Synthesis of Enyne and Diyne Precursors.
Primary alcohols can be loaded onto solid support via the TBDAS linker at any of several stages in the synthesis. To minimize the number of solution-phase transformations, we elected to synthesize the R- and S-tert-butylsulfinimine precursors in solution, then load them onto TBDAS-polystyrene resin to afford 1 (Scheme 1, R-series shown). The yield of each solid-phase reaction was determined by cleavage of an aliquot of resin in the presence of an internal standard, followed by HPLC [evaporative light-scattering detector (ELSD)] and/or 1H-NMR analysis (Materials and Methods).
Scheme 1.

Solid-phase synthesis of enyne and diyne substrates (R-series shown). Yields determined by cleavage of an aliquot of resin in the presence of p-methoxybenzylalcohol internal standard, followed by HPLC (ELSD) analysis. (HMPA = hexamethylphosphoramide, LiHMDS = lithium hexa-methyldisilazide, TBS = tert-butyldimethylsilyl, TMS = trimethylsilyl)
Diastereoselective solid-phase alkyne additions were then carried out to introduce a diversity position (R2) in propargyl sulfinimines 2a–c (16, 17). We were gratified to find that these reactions proceeded with complete diastereoselectivity [≥95∶5 diastereomeric ratio (DR), 1H-NMR] and in yields comparable to those observed in the corresponding solution-phase reactions (2a, 91% vs. 85%; 2b, 69% vs. 80%; 2c, 85% vs. 77%) (12). Subsequent desilylation of 2c with K2CO3 in MeOH also provided the terminal alkyne 2d in 78% yield.
Next, the reaction pathway was branched to form the enyne substrates 3 by N-allylation and the diyne substrates 4 by N-propargylation (18, 19). Notably, the TBDAS linker withstood these strongly basic reaction conditions, and the reactions proceeded in good yields, comparable to those previously obtained in solution phase (3a, 87% vs. 98%; 3b, 72% vs. 76%; 3c, 88% vs. 80%; 3d, 81%; 4a, 86% vs. 82%; 4b, 82% vs. 90%; 4c, 85% vs. 82%; 4d, 79%) (12). Because the terminal alkynes (3d, 4d) were more broadly effective substrates compared to the trimethylsilyl (TMS)-alkynes (3c, 4c) in the original solution-phase studies (12), and afford identical products after desilylation, the former were used in most of the subsequent transition metal-mediated cycloaddition and cyclization reactions.
Transition Metal-Mediated Cycloaddition and Cyclization Reactions.
With the enyne and diyne substrates 3 and 4 in hand, we were poised to investigate the key transition metal-mediated solid-phase cycloaddition and cyclization reactions (Scheme 2). Although there are several examples of such reactions being carried out successfully on solid phase (20), many of these specific reactions had not been carried out on solid support previously, and none had been explored in the context of the TBDAS linker. However, we hoped that this linker would continue to mirror faithfully the previously established reactivity profile of the TBDPS protecting group used in the original solution-phase experiments.
Scheme 2.

Solid-phase synthesis of a 190-membered library of alkaloid/terpenoid-like small molecules from enynes 3 and diynes 4 (R-series shown). (*) Diversification of the sulfinamide in each scaffold series as indicated by the arrows. (†) The sulfonamide 35b was not recovered. (‡) Cleavage of library members derived from 29c and 29′c concurrently removes the TMS group to provide 45d–50d. [BINAP = 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl; COD = 1,4-cyclooctadiene; Cp* = pentamethylcyclopentadienyl; m-CPBA = meta-chloroperbenzoic acid; DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone; DMAD = dimethylacetylene dicarboxylate; Grubbs I = benzylidene-bis(tricyclohexylphosphine)dichlororuthenium; Grubbs II = benzylidene[1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(tricyclohexylphosphine)ruthenium; IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazoly-2-ylidene; Tf = trifluoromethanesulfonate]
Indeed, we were delighted to find that translation to solid phase proved largely uneventful for the majority of these reactions. Consistent with our solution-phase results, Krische and coworkers’ Rh(I)–2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP) catalyzed reductive cyclization (21) proceeded with reagent-controlled diastereoselectivity for 3a,b, but not for 3d, affording access to both exo-pyrroline relative diastereomers 6a,b and 6′a,b and the major diastereomer 6d (Scheme 2). The yields and diastereoselectivities were comparable to those observed in the original solution-phase studies in this first application to solid-phase synthesis.
Enynes 3a,b,d also underwent Evans et al.’s diastereoselective Rh(I)-catalyzed [4 + 2 + 2] reaction with 1,3-butadiene successfully (22). This reaction has not previously been used in solid-phase synthesis and, based on our previous solution-phase studies, the sulfinamides were oxidized to the corresponding sulfonamides prior to cycloaddition, leading to cyclooctapyrrolidines 7a,b,d.
Ring-closing metathesis (23) of enynes 3a,b,d proceeded uneventfully to vinylpyrrolines 8a,b,d. We had previously found in our solution-phase studies that, although these products were unreactive to most dienophiles, oxidation to the corresponding sulfonamides afforded more reactive dienes. Thus, these Diels–Alder reactions (24, 25) provided tricyclic benzodipyrrolidines 9a,b,d and isoindoline dicarboxylates 10a,b,d.
In the diyne substrate series, Yamamoto et al.’s Ru(II)-catalyzed [2 + 2 + 2] cyclotrimerization reaction with benzyl isocyanate (26) was also translated effectively to solid-phase synthesis (27), using diyne substrates 4a,b,d. Some optimization of reaction conditions was required, because extended reaction times (12 h vs. 6 h) resulted in formation of an inseparable side product, apparently due to coupling of a second equivalent of benzyl isocyanate (M + 133). Under the optimized conditions, consistent with our solution-phase results, pyrrolopyridone products 27a,b were formed efficiently with complete regioselectivity and 27d/27′d were formed as a 1∶1 mixture of regioisomers that were separable after cleavage from the solid support.
Tanaka et al.’s Ru(I)-catalyzed [2 + 2 + 2] cyclotrimerization of ethyl cyanoformate (28, 29) also proceeded effectively for diynes 4a,b,d, to afford pyrrolopyridine carboxylates 28a,b,d regioselectively. Deiters and coworkers have carried out similar reactions previously on solid support (27, 30).
Similarly, Saito and coworkers’ Ni(0)-catalyzed [3 + 2 + 2] cycloaddition with ethyl cyclopropylidene acetate (31) was translated effectively to solid support-bound diynes 4a–c. The initial cycloheptapyrrolidine ester products were then converted to the corresponding Weinreb amides 29a–c/29′a–c (32) to facilitate downstream separation of E and Z isomers after cleavage from the solid support.
Although translation to solid-phase synthesis was gratifyingly straightforward for most of these reactions, interesting differences from the original solution-phase results were noted in certain cases. For example, application of the Pauson–Khand reaction (19, 33, 34) to solid support-bound enynes 3 showed a slight trend toward higher yields and diastereoselectivities for the cyclopentapyrrolidinone products 5 in most cases (Scheme 3). This trend was particularly evident in the case of the phenyl-substituted product 5b (74% vs. 64% yield, 70∶30 vs. 50∶50 DR). Recognizing that the choice of solvent is known to affect this reaction in solution (35), we hypothesized that the polystyrene matrix might provide a more hydrophobic reaction milieu than suggested by the CH3CN solvent used to induce the cycloaddition reaction. Consistent with this idea, when the corresponding solution-phase reaction was carried out in benzene or toluene, similarly improved diastereoselectivities were observed. The diastereoselectivity of the solid-phase reaction could also be further improved by changing the solvent to benzene or toluene. This finding highlights the influence of the solid support in “solvent effects” on some reactions.
Scheme 3.

Resin and solvent effects in the solid-phase Pauson–Khand reaction. Yields and DR shown for reactions carried out on solid phase (R1 = TBDAS) and in solution phase (parentheses, R1 = TBDPS, ref. 12 and data herein). Solid-phase reactions analyzed by cleavage of an aliquot of resin in the presence of p-methoxybenzylalcohol internal standard, followed by HPLC (ELSD) and 1H-NMR analysis.
In the [2 + 2 + 2] cyclotrimerization (36–38) of diynes 4a,b,d using Grubbs’ first-generation catalyst and propargyl alcohol (39), one significant difference in regioselectivity was noted in comparison to our original solution-phase results. While isoindolines 26a,d were formed in the expected regioselectivities (26a, 100∶0; 26d/26′d, 50∶50), the corresponding phenyl-substituted product 26b/26′b was formed in significantly lower regioselectivity on solid support (67∶33 vs. 91∶9). We again postulate that this difference may result from interactions of this aromatic substrate with the polystyrene matrix in the solid-phase reaction. Notably, however, this decreased selectivity was advantageous in this particular context, because it afforded access to the minor regioisomer 26′b that was not accessible in useful quantities via the solution-phase reaction.
Diversification of Sulfinamide Moiety and Library Cleavage.
With these 10 classes of scaffolds in hand, we next investigated diversification of the tert-butylsulfinamide moieties. We had originally selected this lynchpin in part because of its known acid-lability (40), and were pleased to find that the support-bound sulfinamides could be cleaved with HCl [1 M in dioxane/THF, room temperature (RT)] to afford the corresponding secondary amines without compromising the TBDAS linker (Scheme 2). This result opens the door for additional diversification of this position in the future (N-acylation, N-alkylation, etc.). Alternatively, treatment of the sulfinamides with meta-chloroperbenzoic acid (m-CPBA) (CH2Cl2, 0 °C) also provided the corresponding tert-butylsulfonamides efficiently.
Accordingly, seven scaffolds (5, 6, 8, 26–29) were produced with three variations at this position (amine, sulfinamide, sulfonamide). Three scaffolds required prior oxidation to the tert-butylsulfonamide to enable the cycloaddition reactions (7, 9, 10) and were limited to this functionality because it could not be cleaved effectively under a variety of conditions (e.g., PhSH, K2CO3; TFA, anisole; TfOH, anisole).
Finally, all library members were cleaved from the solid support using HF·pyridine (pyridine, THF, 50 °C) and purified by preparative reverse-phase HPLC to afford the final library of 190 alkaloid/terpenoid-like compounds (both enantiomeric series), comprised of 18 cyclopentapyrrolidinones (11–13), 30 exo-pyrrolines (14–19), 6 cyclooctapyrrolidines (20), 18 vinylpyrrolines (21–23), 6 benzodipyrrolidines (24), and 6 isoindoline dicarboxylates (25) derived from the enyne substrates and 28 isoindoline alcohols (30–38), 24 pyrrolopyridones (36–41), 18 pyrrolopyridine carboxylates (42–44), and 36 cycloheptapyrrolidine amides (45–50) derived from the diyne substrates. The products were isolated in 20.2% average yield over 5–8 solid-phase steps, at 74.0% average yield per step (SI Appendix, Table S1).
Chemical Space Analysis of Small-Molecule Library.
We have previously analyzed a subset of 32 sulfinamides from this alkaloid/terpenoid-like library for 20 structural and physicochemical parameters to define their positions in chemical space in comparison to natural products, drugs, and drug-like libraries using our established PCA protocol (3, 12). Importantly, the alkaloid/terpenoid-like scaffolds were found to overlap with related polycyclic alkaloid and terpenoid natural products (SI Appendix, Fig. S1), sampling regions of chemical space distinct from those accessed by drugs and drug-like libraries. To delve more deeply into the impacts of various structural features upon positioning in chemical space, we expanded our PCA-based analysis to the complete 190-membered alkaloid/terpenoid library synthesized herein (Fig. 1A and Dataset S1). In this analysis, the first two principal components represented 81% of the variance in the total data set. Analysis of component loadings (SI Appendix, Fig. S2) indicated hydrophobicity (positive) and multiple factors correlating with size (negative) influenced positioning on the x axis (PC1), while aromatic ring count (positive) and stereochemical density (negative) impacted positioning on the y axis (PC2).
Fig. 1.

Chemical space analysis of alkaloid/terpenoid-like library. (A) Principal component analysis of 20 structural and physicochemical descriptors of the 40 top-selling drugs (red circles), 60 diverse natural products (open blue triangles), 20 polycyclic alkaloids and terpenoids (filled blue triangles), 20 ChemBridge and ChemDiv library members (crosses), and 190 multiscaffold library members (green diamonds). (B) Influence of the scaffold: enyne-derived scaffolds (filled diamonds), diyne-derived scaffolds (filled squares). (C) Influence of the R2 substituent: CH2CH2OH (green fill), Ph (yellow fill), H (pink fill). (D) Influence of the R3 N capping group: sulfinamides (green fill), free amines (pink fill), sulfonamides (yellow fill). See SI Appendix for full details and high-resolution images.
Overall, the complete alkaloid/terpenoid library again overlapped with bona fide alkaloid and terpenoid natural products, while also covering a larger area of the plot than the original 32 sulfinamides (Fig. 1A). Notably, the individual scaffolds had a significant influence on positioning, with many of the enyne-derived products appearing more natural product-like, because of their higher stereochemical content and lower aromatic ring content compared to the diyne-derived products (Fig. 1B). The nature of the alkyne substituent (R2 = CH2CH2OH, Ph, or H) also had a strong influence on position, with the phenyl-substituted library members overlapping partially with drugs and drug-like libraries whereas the other two nonaryl substituents led to more natural product-like structures (Fig. 1C). In contrast, the nature of the N-capping group (R3 = sulfinamide, free amine, or sulfonamide) had relatively less influence, although a slight bias toward drug-like chemical space could be observed in the sulfonamide series (Fig. 1D).
We also carried out a shape-based analysis of the library based on normalized principal moment of inertia (PMI) ratios (SI Appendix, Figs. S7–S9) (41–45). In this analysis, the library members exhibited a diverse range of rod-like and disc-like shapes, with more limited sphere-like character. Individual scaffolds were biased toward slightly different regions of the plot, whereas the nature of the alkyne substituent (R2) and N-capping group (R3) did not result in obvious trends. Thus, the shapes of the library members were dependent upon the combination of both scaffold and substituents.
Conclusions
We have synthesized a library of 190 polycyclic, alkaloid/terpenoid-like small molecules using solid-phase transition metal-mediated cycloaddition and cyclization reactions of relatively simple enyne and diyne substrates. The key sulfinamide lynchpin was also diversified to the corresponding free amines and sulfonamides. Translation to solid-phase synthesis using a TBDAS linker was greatly facilitated by the use of a TBDPS surrogate during earlier solution-phase studies. Principal component analysis of structural and physicochemical parameters indicates that this library samples regions of chemical space that overlap with bona fide natural products and are distinct from those areas accessed by conventional drugs and drug-like libraries. Aromatic ring content and stereochemical complexity were identified as two major factors that distinguish natural products from synthetic drugs. The complete set of 190 compounds is now being screened in a variety of assays at The Rockefeller University High-Throughput Screening Resource Center. In addition, 94 compounds have been submitted for broad screening in the National Institutes of Health (NIH) Molecular Libraries Program (PubChem Substance search term: DST_AT1_*, http://pubchem.ncbi.nlm.nih.gov). These efforts will provide critical data to evaluate the biological properties of this library and to assess the abilities of natural product-based libraries to address challenging biological targets that remain intractable to conventional drug-like molecules.
Materials and Methods
See SI Appendix for detailed methods and complete analytical data.
General Procedure for Monitoring Solid-Phase Reactions.
An aliquot of vacuum-dried resin (4 mg) was swollen with THF (5 mL) for 10 min. A solution of 4-methoxybenzylalcohol (internal standard) (25 mM in THF, 1 eq) was added, followed by tetra-n-butylammonium fluoride (0.1 M in THF, 3 eq). After stirring for 2 h, the THF solution was recovered from the resin, and the solution concentrated by rotary evaporation. The residue was taken up in CH3CN (300 μL) and filtered through a Pasteur pipette with a 0.5-inch plug of normal phase silica gel over a 0.5-inch plug of reverse-phase silica gel. The plug was rinsed carefully with 3 mL CH3CN, and the crude cleavage products were recovered by rotary evaporation. HPLC analysis (ELSD) was used to determine the ratio of cleaved scaffold to internal standard, and the yield was calculated based on a calibration curve. Analysis by 1H-NMR was used to determine product ratios where appropriate.
Sulfinimine Alcohol Loading (1).
The (R,E)- and (S,E)-N-(3-hydroxypropylidene)-2-methylpropane-2-sulfinamide (SI Appendix) were loaded onto TBDAS resin (800 mg, 1.53 meq/g, where meq is milliequivalent) by the previously described general procedure (12) to afford sulfinimine resins R-1 and S-1.
General Procedure for Diastereoselective Alkyne Addition (2).
Lithium hexa-methyldisilazide (LiHMDS) (1 M in THF, 4.4 mmol, 4.0 eq) was dissolved in anhydrous hexanes (20 mL) at -78 °C, the appropriate alkyne (4.4 mmol, 4.0 eq) was added, and the reaction was removed from the cold bath and stirred for 10 min, then recooled to -78 °C. Vacuum-dried resin 1 (800 mg, 1.11 meq, 1.0 eq) was swollen with THF (20 mL) for 10 min, then cooled to -78 °C and the lithium acetylide solution was added via cannula. The reaction was allowed to warm to RT with stirring over 24 h. The reaction was quenched with water (100 mL), then the resin was washed and dried to afford propargyl sulfinamides 2.
General Procedure for Sulfinamide N-Alkylation (3, 4).
Propargyl sulfinamide resin 2 (800 mg, 0.92 meq, 1.0 eq) was swollen with THF (20 mL) for 10 min, then the mixture was cooled to -78 °C and a solution of n-BuLi (2.4 M in hexanes, 2.02 mmol, 2.2 eq) was added by syringe. The reaction was stirred for 45 min, then hexamethylphosphoramide (640 μL, 3.68 mmol, 4.0 eq) was added. After stirring for an additional 30 min, allyl or propargyl bromide (9.2 mmol, 10 eq) was added. The mixture was stirred at -78 °C for 6 h, then allowed to warm to RT overnight. The reaction was quenched with water (100 mL), then the resin was washed and dried to afford enynes 3 or diynes 4.
General Procedure for Pauson–Khand Reaction (5).
Vacuum-dried enyne resin 3 (800 mg, 0.86 meq, 1.0 eq) was swollen with CH2Cl2 (20 mL) for 10 min, then Co2(CO)8 (1.18 g, 3.44 mmol, 4.0 eq) was added and the slurry was stirred at RT for 3 h. The solution was decanted from the flask under positive Ar pressure and the resin was washed with anhydrous CH3CN (2 × 25 mL). The resin was swollen with anhydrous CH3CN (20 mL) and heated to 75 °C for 12 h, then allowed to cool to RT. The resin was washed and dried to afford cyclopentapyrrolidinones 5.
General Procedure for Krische Reductive Cyclization (6).
Vacuum-dried resin 3 (800 mg, 0.86 meq, 1.0 eq) was swollen with 1,2-dichloroethane (DCE) (20 mL) for 10 min, then a stock solution of Rh(COD)2OTf, where COD is 1,4-cyclooctadiene, and (R)- or (S)-BINAP (1∶1 molar ratio, 0.1 M in DCE, 3.4 mL, 0.34 mmol, 0.4 eq) was added via cannula. The reaction atmosphere was flushed with H2, and the mixture was stirred under a balloon of H2 at RT for 12 h. The resin was washed and dried to afford exo-pyrrolines 6 or 6′.
General Procedure for Evans Butadiene [4 + 2 + 2] Cycloaddition (7).
Vacuum-dried resin 3 (800 mg, 0.86 meq, 1.0 eq) was swollen with CH2Cl2 (20 mL) for 10 min, then cooled to 0 °C. Solid m-CPBA (580 mg, 2.58 mmol, 3.0 eq) was added and the reaction was stirred at 0 °C for 1 h, then allowed to warm to RT and stirred for 6 h. The resin was washed and dried, then swollen with toluene (20 mL) for 10 min. A stock solution of Rh(IMes)(COD)Cl4, where IMes is 1,3-bis(2,4,6-trimethylphenyl)imidazoly-2-ylidene, and AgOTf (1∶2 molar ratio, 0.1 M Rh in degassed toluene, 3.44 mL, 0.34 mmol Rh, 0.4 eq) was added to the resin, and the reaction atmosphere was flushed with 1,3-butadiene gas, then the mixture was stirred under a balloon of 1,3-butadiene at RT for 2 h, refilling the balloon when empty. The balloon was removed and the mixture was heated to 108 °C for 24 h. The resin was cooled, washed, and dried to afford cyclooctapyrrolidines 7.
General Procedure for Enyne Metathesis (8).
Vacuum-dried resin 3 (800 mg, 0.86 meq, 1.0 eq) was swollen with degassed toluene (200 mL) for 10 min, then a stock solution of Grubbs’ second-generation catalyst (0.1 M in degassed toluene, 5.1 mL, 0.51 mmol, 0.6 eq) was added via syringe and the mixture was stirred at 60 °C overnight. The resin was cooled, washed, and dried to afford vinylpyrrolines 8.
General Procedure for Diels–Alder Reactions (9, 10).
Vacuum-dried resin 3 (800 mg, 0.86 meq, 1.0 eq) was swollen with CH2Cl2 (20 mL) for 10 min, then cooled to 0 °C. Solid m-CPBA (580 mg, 2.58 mmol, 3.0 eq) was added and the reaction was stirred at 0 °C for 1 h, then allowed to warm to RT and stirred for 6 h. The resin was washed and dried, then swollen with CH2Cl2 (200 mL) for 10 min. A stock solution of Grubbs’ second-generation catalyst (0.1 M in degassed toluene, 5.1 mL, 0.51 mmol, 0.6 eq) was added and the mixture stirred at 80 °C overnight. The resin was cooled, washed, and dried, then swollen in toluene (20 mL). For 9, N-phenylmaleimide (1.14 g, 6.6 mmol, 7.7 eq) was added and the mixture was stirred at 80 °C for 36 h. The resin was cooled, washed, and dried to afford benzodipyrrolidines 9. For 10, dimethyl acetylenedicarboxylate (650 μL, 5.16 mmol, 6.0 eq) was added and the mixture was stirred at 100 °C for 48 h. The resin was cooled, washed quickly with benzene, and swollen in benzene (20 mL) for 10 min. A stock solution of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (1.0 M in benzene, 3.44 mL, 3.44 mmol, 4.0 eq) was added and the mixture was stirred at 80 °C for 48 h. The resin was cooled, washed, and dried to afford isoindoline dicarboxylates 10.
General Procedure for Propargyl Alcohol [2 + 2 + 2] Cyclotrimerization (26).
Vacuum-dried resin 4 (800 mg, 0.86 meq, 1.0 eq) was swollen with degassed toluene (20 mL) for 10 min. A stock solution of Grubbs’ first-generation catalyst (0.1 M in degassed toluene, 3.4 mL, 0.34 mmol, 0.4 eq) and propargyl alcohol (500 μL, 8.6 mmol, 10.0 eq) were added via syringe and the mixture was stirred at 90 °C overnight. The resin was cooled, washed, and dried to afford isoindolines 26/26′.
General Procedure for Yamamoto Benzyl Isocyanate [2 + 2 + 2] Cyclotrimerization (27).
Vacuum-dried resin 4 (800 mg, 0.86 meq, 1.0 eq) was swollen with degassed DCE (20 mL) for 10 min. A stock solution of benzyl isocyanate and RuCp*(COD)Cl (40∶1 molar ratio, 0.5 M isocyanate in DCE, 7 mL, 3.5 mmol isocyanate, 4.0 eq) was added and the slurry was stirred at 90 °C for 6 h. The resin was cooled, washed, and dried to afford pyrrolopyridones 27/27′.
General Procedure for Tanaka Ethyl Cyanoformate [2 + 2 + 2] Cyclotrimerization (28).
Vacuum-dried resin 4 (800 mg, 0.86 meq, 1.0 eq) was swollen with degassed DCE (20 mL) for 10 min. A stock solution of ethyl cyanoformate, Rh(COD)2BF4, and rac-BINAP (50∶1.5∶2 molar ratio, 0.5 M nitrile in DCE, 8 mL, 4.0 mmol, 4.65 eq) was added and the mixture was stirred at 80 °C for 12 h. The resin was cooled, washed, and dried to afford pyrrolopyridine carboxylates 28.
General Procedure for Saito [3 + 2 + 2] Cyclotrimerization (29).
Vacuum-dried resin 4 (800 mg, 0.86 meq) was swollen with degassed DCE (20 mL) for 10 min. A stock solution of ethyl cyclopropylidineacetate, Ni(COD)2 and PPh3 (30∶3.5∶7 molar ratio, 0.5 M cyclopropylidine in degassed toluene, 8 mL, 4.0 mmol cyclopropylidine, 4.65 eq) was added and the mixture was stirred at 70 °C for 36 h. The resin was cooled, washed, and swollen in THF, then cooled to -10 °C. Solid HN(Me)OMe·HCl (850 mg, 8.6 mmol, 10.0 eq) and cyclopentyl-MgCl (2.0 M in Et2O, 13 mL, 26 mmol, 30 eq) were added, and the resulting mixture was stirred at -10 °C for 4 h. The reaction was quenched with water and the resin was washed and dried to afford cycloheptapyrrolidines 29/29′.
General Procedure for Hydrolysis of Sulfinimides to Amines.
The vacuum-dried resin (400 mg, 0.42 meq, 1.0 eq) was swollen with THF (16 mL) for 10 min. HCl (4 M in dioxane, 0.42 mL, 1.68 mmol, 4.0 eq) was added and the mixture was stirred at RT for 4 h. The resin was washed and dried to afford the corresponding free secondary amines.
General Procedure for Oxidation of Sulfinamides to Sulfonamides.
The vacuum-dried resin (400 mg, 0.42 meq, 1.0 eq) was swollen with CH2Cl2 (10 mL) for 10 min, then cooled to 0 °C. Solid m-CPBA (150 mg, 0.63 mmol, 1.5 eq) was added and the mixture was stirred at 0 °C for 1 h, then allowed to warm to RT and stirred for 6 h. The resin was washed and dried to afford the corresponding tert-butylsulfonamides.
General Procedure for Product Cleavage from the TBDAS Resin.
Vacuum-dried resin (400 mg, 0.42 meq, 1.0 eq) was swollen with THF (10 mL) for 10 min. A stock solution of HF·pyridine and pyridine (1∶2 molar ratio, 0.5 M HF·pyridine in THF, 5.1 mL, 2.5 mmol, 6 eq) was added and the resulting mixture was stirred at 50 °C for 3 h. The reaction was quenched with methoxytrimethylsilane (0.35 mL, 2.52 mmol, 6.0 eq), and the supernatant was recovered. The resin was extracted with additional THF (2 × 50 mL), and the combined supernatants were concentrated by rotary evaporation. Initial purification was carried out on an ISCO Optix 10 CombiFlash system, and the products were further purified by preparative reverse-phase HPLC. The purified library members were analyzed by HPLC (ELSD/MS) and 1H-NMR and submitted for biological screening at The Rockefeller University High-Throughput Screening Resource Center and NIH Molecular Libraries Program.
Supplementary Material
Acknowledgments.
We thank Dr. Lakshmi B. Akella (Broad Institute) for carrying out the PMI analysis and Dr. George Sukenick, Dr. Hui Liu, Hui Fang, and Dr. Sylvi Rusli for mass spectral analyses. D.S.T. is an Alfred P. Sloan Research Fellow. JChem for Excel was generously provided by ChemAxon. Financial support from the National Institutes of Health (R21 GM104685, P41 GM076267, T32 CA062948-Gudas) is gratefully acknowledged.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1015268108/-/DCSupplemental.
References
- 1.Tan DS. Diversity-oriented synthesis: Exploring the intersections between chemistry and biology. Nat Chem Biol. 2005;1:74–84. doi: 10.1038/nchembio0705-74. [DOI] [PubMed] [Google Scholar]
- 2.Hert J, Irwin JJ, Laggner C, Keiser MJ, Shoichet BK. Quantifying biogenic bias in screening libraries. Nat Chem Biol. 2009;5:479–483. doi: 10.1038/nchembio.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bauer RA, Wurst JM, Tan DS. Expanding the range of “druggable” targets with natural product-based libraries: An academic perspective. Curr Opin Chem Biol. 2010;14:308–314. doi: 10.1016/j.cbpa.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganesan A. The impact of natural products upon modern drug discovery. Curr Opin Chem Biol. 2008;12:306–317. doi: 10.1016/j.cbpa.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 5.Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981–2002. J Nat Prod. 2003;66:1022–1037. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- 6.Welsch ME, Snyder SA, Stockwell BR. Privileged scaffolds for library design and drug discovery. Curr Opin Chem Biol. 2010;14:347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boldi AM. Libraries from natural product-like scaffolds. Curr Opin Chem Biol. 2004;8:281–286. doi: 10.1016/j.cbpa.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 8.Taylor SJ, Taylor AM, Schreiber SL. Synthetic strategy toward skeletal diversity via solid-supported, otherwise unstable reactive intermediates. Angew Chem Int Ed. 2004;43:1681–1685. doi: 10.1002/anie.200353466. [DOI] [PubMed] [Google Scholar]
- 9.Lo MMC, Neumann CS, Nagayama S, Perlstein EO, Schreiber SL. A library of spirooxindoles based on a stereoselective three-component coupling reaction. J Am Chem Soc. 2004;126:16077–16086. doi: 10.1021/ja045089d. [DOI] [PubMed] [Google Scholar]
- 10.Oguri H, Schreiber SL. Skeletal diversity via a folding pathway: Synthesis of indole alkaloid-like skeletons. Org Lett. 2005;7:47–50. doi: 10.1021/ol047945w. [DOI] [PubMed] [Google Scholar]
- 11.Díaz-Gavilán M, Galloway WRJD, O’Connell KMG, Hodkingson JT, Spring DR. Diversity-oriented synthesis of bicyclic and tricyclic alkaloids. Chem Commun. 2010;46:776–778. doi: 10.1039/b917965h. [DOI] [PubMed] [Google Scholar]
- 12.Bauer RA, DiBlasi CM, Tan DS. The tert-butylsulfinamide lynchpin in transition metal-mediated multiscaffold library synthesis. Org Lett. 2010;12:2084–2087. doi: 10.1021/ol100574y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ortholand JY, Ganesan A. Natural products and combinatorial chemistry: Back to the future. Curr Opin Chem Biol. 2004;8:271–280. doi: 10.1016/j.cbpa.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 14.Ferreira F, Botuha C, Chemla F, Pérez-Luna A. tert-Butanesulfinimines: Structure, synthesis, and synthetic applications. Chem Soc Rev. 2009;38:1162–1186. doi: 10.1039/b809772k. [DOI] [PubMed] [Google Scholar]
- 15.DiBlasi CM, Macks DE, Tan DS. An acid-stable tert-butyldiarylsilyl (TBDAS) linker for solid-phase organic synthesis. Org Lett. 2005;7:1777–1780. doi: 10.1021/ol050370y. [DOI] [PubMed] [Google Scholar]
- 16.Ding CH, Chen DD, Luo ZB, Dai LX, Hou XL. Highly diastereoselective synthesis of N-tert-butylsulfinylpropargylamines through direct addition of alkynes to N-tert-butanesulfinimines. Synlett. 2006:1272–1274. [Google Scholar]
- 17.Chen BL, Wang B, Lin GQ. Highly diastereoselective addition of alkynylmagnesium chlorides to N-tert-butanesulfinyl aldimines: A practical and general access to chiral α-branched amines. J Org Chem. 2010;75:941–944. doi: 10.1021/jo902424m. [DOI] [PubMed] [Google Scholar]
- 18.Kuduk SD, Marco CND, Pitzenberger SM, Tsou N. Asymmetric addition reactions of Grignard reagents to chiral 2-trifluoromethyl tert-butyl (Ellman) sulfinimine-ethanol adducts. Tetrahedron Lett. 2006;47:2377–2381. [Google Scholar]
- 19.Hiroi K, Watanabe T. Asymmetric Pauson–Khand reactions of chiral sulfinamides: Asymmetric synthesis of 3-azabicyclo[3.3.0]oct-5-en-7-one derivatives. Heterocycles. 2001;54:73–76. [Google Scholar]
- 20.Nandy JP, et al. Advances in solution- and solid-phase synthesis toward the generation of natural product-like libraries. Chem Rev. 2009;109:1999–2060. doi: 10.1021/cr800188v. [DOI] [PubMed] [Google Scholar]
- 21.Jang HY, et al. Enantioselective reductive cyclization of 1,6-enynes via rhodium-catalyzed asymmetric hydrogenation: C-C bond formation precedes hydrogen activation. J Am Chem Soc. 2005;127:6174–6175. doi: 10.1021/ja042645v. [DOI] [PubMed] [Google Scholar]
- 22.Evans PA, Robinson JE, Baum EW, Fazal AN. Intermolecular transition metal-catalyzed [4 + 2 + 2] cycloaddition reactions: A new approach to the construction of eight-membered rings. J Am Chem Soc. 2002;124:8782–8783. doi: 10.1021/ja026351q. [DOI] [PubMed] [Google Scholar]
- 23.Diver ST, Giessert AJ. Enyne metathesis (enyne bond reorganization) Chem Rev. 2004;104:1317–1382. doi: 10.1021/cr020009e. [DOI] [PubMed] [Google Scholar]
- 24.Schurer SC, Blechert S. Sequences of yne-ene cross metathesis and Diels–Alder cycloaddition reactions—Modular solid-phase synthesis of substituted octahydrobenzazepinones. Synlett. 1999:1879–1882. [Google Scholar]
- 25.Schurer SC, Blechert S. Synthesis of pseudo-oligosaccharides by a sequence of yne-ene cross metathesis and Diels–Alder reaction. Chem Commun. 1999:1203–1204. [Google Scholar]
- 26.Yamamoto Y, et al. Cp*RuCl-catalyzed [2 + 2 + 2] cycloadditions of α,ω-diynes with electron-deficient carbon-heteroatom multiple bonds leading to heterocycles. J Am Chem Soc. 2005;127:605–613. doi: 10.1021/ja045694g. [DOI] [PubMed] [Google Scholar]
- 27.Young DD, Deiters A. A general approach to chemo- and regioselective cyclotrimerization reactions. Angew Chem Int Ed. 2007;46:5187–5190. doi: 10.1002/anie.200700802. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka K, Suzuki N, Nishida G. Cationic rhodium(I)/modified-BINAP catalyzed [2 + 2 + 2] cycloaddition of alkynes with nitriles. Eur J Org Chem. 2006:3917–3922. [Google Scholar]
-
29.Otake Y, Tanaka R, Tanaka K.
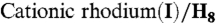 -BINAP complex catalyzed [2 + 2 + 2] cycloadditions of 1,6- and 1,7-diynes with carbonyl compounds. Eur J Org Chem. 2009:2737–2747. [Google Scholar]
-BINAP complex catalyzed [2 + 2 + 2] cycloadditions of 1,6- and 1,7-diynes with carbonyl compounds. Eur J Org Chem. 2009:2737–2747. [Google Scholar] - 30.Senaiar RS, Young DD, Deiters A. Pyridines via solid-supported [2 + 2 + 2] cyclotrimerization. Chem Commun. 2006:1313–1315. doi: 10.1039/b515901f. [DOI] [PubMed] [Google Scholar]
- 31.Maeda K, Saito S. Nickel-catalyzed [3 + 2 + 2] cycloaddition of ethyl cyclopropylideneacetate and diynes. Synthesis of 7,6- and 7,5-fused bicyclic compounds. Tetrahedron Lett. 2007;48:3173–3176. [Google Scholar]
- 32.Williams JM, et al. A new general method for preparation of N-methoxy-N-methylamides. Application in direct conversion of an ester to a ketone. Tetrahedron Lett. 1995;36:5461–5464. [Google Scholar]
- 33.Schore NE. The Pauson–Khand cycloaddition reaction for synthesis of cyclopentenones. Org React. 1991;40:1–90. [Google Scholar]
- 34.Kubota H, Lim J, Depew KM, Schreiber SL. Pathway development and pilot library realization in diversity-oriented synthesis. Exploring Ferrier and Pauson–Khand reactions on a glycal template. Chem Biol. 2002;9:265–276. doi: 10.1016/s1074-5521(02)00099-6. [DOI] [PubMed] [Google Scholar]
- 35.Suh WH, Choi M, Lee SI, Chung YK. Rh(I)-Catalyzed asymmetric intramolecular Pauson–Khand reaction in aqueous media. Synthesis. 2003;2003:2169–2172. [Google Scholar]
- 36.Sun Q, Zhou XM, Islam K, Kyle DJ. Solid-phase synthesis of isoindolines via a rhodium-catalyzed [2 + 2 + 2] cycloaddition. Tetrahedron Lett. 2001;42:6495–6497. [Google Scholar]
- 37.Young DD, Senaiar RS, Deiters A. Solid-supported [2 + 2 + 2] cyclotrimerizations. Chem—Eur J. 2006;12:5563–5568. doi: 10.1002/chem.200501360. [DOI] [PubMed] [Google Scholar]
- 38.Senaiar RS, Teske JA, Young DD, Deiters A. Synthesis of indanones via solid-supported [2 + 2 + 2] cyclotrimerization. J Org Chem. 2007;72:7801–7804. doi: 10.1021/jo7013565. [DOI] [PubMed] [Google Scholar]
- 39.Witulski B, Stengel T, Fernández-Hernández JM. Chemo- and regioselective crossed alkyne cyclotrimerisation of 1,6-diynes with terminal monoalkynes mediated by Grubbs’ catalyst or Wilkinson’s catalyst. Chem Commun. 2000:1965–1966. [Google Scholar]
- 40.Robak MT, Herbage MA, Ellman JA. Synthesis and applications of tert-butanesulfinamide. Chem Rev. 2010;110:3600–3740. doi: 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]
- 41.Sauer WHB, Schwarz MK. Molecular shape diversity of combinatorial libraries: A prerequisite for broad bioactivity. J Chem Inf Comput Sci. 2003;43:987–1003. doi: 10.1021/ci025599w. [DOI] [PubMed] [Google Scholar]
- 42.Akella LB, DeCaprio D. Cheminformatics approaches to analyze diversity in compound screening libraries. Curr Opin Chem Biol. 2010;14:325–330. doi: 10.1016/j.cbpa.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 43.Marcaurelle LA, et al. An aldol-based build/couple/pair strategy for the synthesis of medium- and large-sized rings: Discovery of macrocyclic histone deacetylase inhibitors. J Am Chem Soc. 2010;132:16962–16976. doi: 10.1021/ja105119r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pizzirani D, Kaya T, Clemons PA, Schreiber SL. Stereochemical and skeletal diversity arising from amino propargylic alcohols. Org Lett. 2010;12:2822–2825. doi: 10.1021/ol100914b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rolfe A, Lushington GH, Hanson PR. Reagent based DOS: A “Click, Click, Cyclize” strategy to probe chemical space. Org Biomol Chem. 2010;8:2198–2203. doi: 10.1039/b927161a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.