

Abstract
Aminoglycosides are antibacterial compounds that act by binding to the A site of the small 30S bacterial ribosomal subunit and inhibiting protein translation. Clinical resistance to aminoglycosides is generally the result of the expression of enzymes that covalently modify the antibiotic, including phosphorylation, adenylylation, and acetylation. Bisubstrate analogs for the aminoglycoside N-acetyl-transferases are nanomolar inhibitors of Enterococcus faecium AAC(6′)-Ii. However, in the case of the Salmonella enterica aac(6′)-Iy-encoded aminoglycoside N-acetyltransferase, we demonstrate that a series of bisubstrate analogs are only micromolar inhibitors. In contrast to studies with AAC(6′)-Ii, the inhibition constants toward AAC(6′)-Iy are essentially independent of both the identity of the aminoglycoside component of the bisubstrate and the number of carbon atoms that are used to link the CoA and aminoglycoside components. The patterns of inhibition suggest that the CoA portion of the bisubstrate analog can bind to the enzyme–aminoglycoside substrate complex and that the aminoglycoside portion can bind to the enzyme–CoA product complex. However, at the high concentrations of bisubstrate analog used in crystallization experiments, we could crystallize and solve the three-dimensional structure of the enzyme–bisubstrate complex. The structure reveals that both the CoA and aminoglycoside portions bind in essentially the same positions as those previously observed for the enzyme–CoA–ribostamycin complex, with only a modest adjustment to accommodate the “linker”. These results are compared to previous studies of the interaction of similar bisubstrate analogs with other aminoglycoside N-acetyltransferases.
Aminoglycosides are hydrophilic molecules consisting of a central aminocyclitol ring linked to one or more amino sugars (Figure 1). These molecules are among the most important compounds used to treat serious infections caused by Gram-negative aerobic bacteria. Aminoglycosides bind to the small, 30S ribosomal subunit, at the tRNA acceptor site (A site) and inhibit bacterial translation by causing codon misreading and/or obstructing the translocation step (1). The use of these drugs in 1960s and 1970s led to the appearance of resistant clinical strains. The most common mechanism of clinical resistance is the modification of the drug resulting from the covalent transformations at specific amino or hydroxyl groups by the action of intracellular bacterial enzymes. Three types of aminoglycoside modifications have been demonstrated: O-phosphorylation, O-adenylylation, and N-acetylation. The structurally altered drug binds to the ribosome with substantially reduced affinity and is, therefore, an ineffective inhibitor (2). Aminoglycoside 6′-N-acetyl-transferases (AAC(6′)s) catalyze the transfer of an acetyl group from acetyl-CoA to the 6′-amino group of typical aminoglycoside molecules and are one of the most widespread determinants of clinical resistance to aminoglycosides (3–7).
Figure 1.

Proposed chemical mechanism for AAC(6′)-Iy (8). Both substrates bind to the enzyme active site, and catalysis occurs involving direct transfer of the acetyl group from acetyl-CoA to the amine substrate via the formation of a tetrahedral intermediate.
The chromosomally encoded AAC(6′)-Iy was identified in clinical isolates of aminoglycoside-resistant strains of Salmonella enterica (4). The kinetic characterization of AAC(6′)-Iy suggested that catalysis occurs through a sequential random mechanism involving the direct transfer of the acetyl group from acetyl-CoA to the amine substrate via the formation of a ternary enzyme–AcCoA–aminoglycoside complex (Figure 1) (8). Such enzyme systems, where catalysis occurs via the formation of a ternary complex, can be strongly inhibited by analogues where both substrates are covalently linked to one another. The covalent coupling of the two substrates can potentially increase the affinity of the bisubstrate by the product of the respective association constants (9). In some cases, this rationale has led to the development of compounds with powerful therapeutic properties, as in the case of mupirocin, a femtomolar, bisubstrate inhibitor of bacterial leucyltRNA synthetase that is used as topical antibiotic (10). Bisubstrate analogue inhibitors have also been shown to be probes of the kinetic mechanisms of enzymes, including aminoglycoside N-acetyltransferases. The first aminoglycoside–acetyl-CoA bisubstrate analogue reported was gentamycin–acetyl-CoA prepared enzymatically using AAC(3)-I, which inhibits gentamycin N- acetyltransferase with nanomolar affinity (11). Unfortunately, the inhibitor does not potentiate the action of aminoglycosides when added to cultures of resistant strains, presumably because it cannot pass through the cell wall. Recently, Gao et al. have generated a series of 6′-N-acetyl-CoA bisubstrate analogues with nanomolar affinity for Enterococcus faecium AAC(6′)-Ii (12). The sequence identity between AAC(6′)-Ii and AAC(6′)-Iy is only 14%, and AAC(6′)-Ii utilizes a sequential, ordered kinetic mechanism with acetyl-CoA binding first followed by the antibiotic (13). The compounds varied in the nature of the aminoglycoside molecule (neamine, kanamycin, or ribostamycin) as well as in the linker length (1–4 carbons) (Scheme 1). A second generation of smaller size inhibitors was prepared more recently to determine structure–activity relationships. Interestingly, one of these bisubstrate analogues was able to attenuate aminoglycoside resistance in cells (14).
Scheme 1.

Structures of Bisubstrate Inhibitors Used in This Study
Here, we have tested the first generation of aminoglycoside–CoA bisubstrate analogues as inhibitors of the S. enterica AAC(6′)-Iy. The patterns of inhibition versus AcCoA and aminoglycosides suggests that these compounds bind to different enzyme–substrate and enzyme–product complexes than reported for the related AAC(6′)-Ii.
MATERIALS AND METHODS
Measurement of Enzyme Activity
AAC(6′)-Iy was purified as previously described (15). Aminoglycoside-dependent acetyltransferase activity was monitored spectrophotometrically by following the increase in absorbance at 324 nm due to the reaction between the sulfhydryl group of the product CoASH and 4,4′-dithiodipyridine (DTDP), releasing 4-thiopyridone (ε324 = 19 800 M−1 cm−1). Reactions were monitored continuously on a UVIKON 943 spectrophotometer, and enzyme activities were calculated from the initial (<10% completion) rates. Assay mixtures contained 50 mM HEPES, pH 7.2, and 0.5 mM DTDP, in addition to enzyme, substrate, and inhibitors in a final volume of 1 mL. Reactions were initiated by the addition of enzyme and followed at 25°C.
Bisubstrate Analog Inhibition Patterns
A series of bisubstrates analogs of acetyl-CoA and aminoglycosides (neamine, kanamycin, or ribostamycin) were synthesized as previously described (12). Inhibition constants for bisubstrate analog inhibitors were determined by measuring initial velocities where either (a) the concentration of acetyl-CoA remained fixed (10 μM; 10 × Km) and the concentration of tobramycin was varied or (b) the concentration of tobramycin remained fixed (70 μM, 35 × Km) and the concentration of acetyl-CoA was varied using four different fixed levels of the inhibitor. Equations 1 and 2 were used to fit linear, noncompetitive, and uncompetitive data, respectively.
| (1) |
| (2) |
The data was also globally fitted to the rate eq 3:
| (3) |
In eqs 1–3, v is the measured reaction velocity, V is the maximal velocity, [A] and [B] are the concentrations of the substrates A and B, respectively, Ka and Kb are the corresponding Michaelis–Menten constants, [IA–B] is the concentration of bisubstrate inhibitor, and Kis and Kii are the slope and intercept inhibition constants, respectively.
Crystal Structure of the AAC(6′)-Iy–CoA-S-monomethyl-N-6′-acetylneamine Complex
Prior to crystallization AAC-(6)′-Iy (15 mg/mL, 10 mM TEA pH 8.0, 100 mM ammonium sulfate) was incubated with 2 mM inhibitor (CoA-S-monomethyl-N-6′-acetylneamine) for 2 h on ice. The inhibitor complex was crystallized by vapor diffusion under oil in which 2 μL of inhibitor complex was combined with 2 μL of precipitant (20% PEG 6000, 100 mM MES pH 5.75) under 150 μL of FISHER silicon oil and incubated at 18 °C open to room humidity. Crystals were briefly placed in 20% PEG 6000, 100 mM MES pH 5.75, 25% glycerol prior to vitrification by immersion in liquid nitrogen. X-ray diffraction data were collected on an R-Axis IV++ image plate detector using Cu Kα radiation from a Rigaku RU-H3R X-ray generator. The data was processed with MOSFLM (16) and scaled with SCALA (17). The inhibitor complex crystals belong to space group C2 with unit cell dimensions of a = 85.0, b = 44.6, c = 88.4, β = 93.2 and are isomorphous with the crystals of the AAC(6′)-Iy–ribostamycin complex (PDBID = 1S3Z) (15). Graphical structural manipulations were performed in COOT (18), and the structure was refined against the data using REFMAC (19). Stereochemical constraints for the inhibitor were generated by PRODRG2 (20). Statistics for the data collection and refinement are presented in Table 2.
Table 2.
Data Collection and Refinement Statisticsa
| Data Collection | |
| resolution (Å) | 25–2.0 (2.11–2.0) |
| completeness (%) | 95.9 (92.3) |
| redundancy | 2.4 (2.4) |
| I/σ(I) | 16.9 (4.5) |
| Rmerge | 0.044 (0.193) |
| Refinement | |
| resolution (Å) | 25–2.0 (2.05–2.0) |
| Rfactor (%) | 18.1 (22.1) |
| Rfree (%) | 23.0 (26.7) |
| residues fit, small molecules | A (−2 to 145), B (2 to 145) 2 inhibitors, 1 glycerol, 1 Ni2+ |
| no. of atoms | |
| protein | 1143 |
| solvent/inhibitor | 219/74 |
| av B-factors (Å2) | |
| protein | 22.5 |
| solvent/inhibitor | 28.3/21.3 |
| rms deviations | |
| bonds (Å) | 0.014 |
| angles (deg) | 1.562 |
Statistics in parentheses are for the highest resolution shell.
RESULTS AND DISCUSSION
The aac(6′)-Iy-encoded 6′-N-acetyltransferase was identified in an aminoglycoside-resistant clinical isolate of S. enterica (4). The aac(6′)-Iy gene is chromosomally encoded, and aminoglycoside resistance is the result of a chromosomal deletion that led to gene expression by transcriptional fusion (4); the physiological role of AAC(6′)-Iy is still unknown. AAC(6′)-Iy exhibits very broad specificity with respect to aminoglycosides containing a 6′-amino functionality. Initial velocity patterns indicated that both substrates must bind to the enzyme before catalysis occurs, and a number of lines of evidence suggested that the order of substrate binding is random (8, 21). The structural characterization of this enzyme confirmed that AAC(6′)-Iy is a member of the GNAT superfamily and revealed strong structural similarities with the Sacharomyces cerevisiae Hpa2-encoded histone acetyl-transferase (15). The structure of the enzyme–CoA–ribostamycin structure complex (15) allowed for the analysis of the interactions that generated the regioselectivity for the 6′-amino group and the identification of Asp115 as the general base. Additionally, the extremely tight binding of the product CoA was demonstrated, suggesting that the dissociation of CoA was a rate-limiting step, at least in vitro (8, 15).
Recently, the detailed theory for predicting the expected patterns of bisubstrate inhibition against the two substrates in bireactant kinetic mechanisms has been presented (9). The steady-state equations for ordered and random kinetic mechanisms were developed, and two appropriate examples were considered. In the simplest and most frequent case, bisubstrate IA–B, will bind only to free enzyme E, occupying the A and B subsites of the active site and precluding binding of either A or B. In these situations, ordered mechanisms show linear competitive inhibition versus one substrate and noncompetitive inhibition versus the second substrate. In the random mechanism, IA–B shows competitive inhibition against both substrates (9). In some other less frequent cases, IA–B can bind to the free enzyme but also can bind to EA (at the B subsite) and/or EB (at the A subsite). In such cases, random mechanisms can show noncompetitive inhibition when A or B is varied.
The enzymatic synthesis of bisubstrate analogs of aminoglycoside N-acetyltransferases has been reported (9, 11). More recently, the chemical synthesis of bisubstrate analogs in which the 6′-amino group of various aminoglycosides were regioselectively coupled to CoA has been reported, and those compounds with the shortest linkers were shown to be 40–70 nM inhibitors of the E. faecium AAC(6′)-Ii (12). All inhibitors tested were shown to exhibit competitive inhibition versus AcCoA. To investigate the influence of the carbon linker and the aminoglycoside moiety of the bisubstrate analogs on the strength of inhibition, we have tested the series of compounds used previously in the case of the AAC(6′)-Ii with AAC(6′)-Iy (Scheme 1). Inhibition patterns for the bisubstrate analogue inhibitors (IA–B) were tested varying either the aminoglycoside or acetyl-CoA at fixed, saturating concentrations of the other substrate (Table 1). Although we had expected to observe competitive inhibition versus both substrates since the kinetic mechanism is random, all inhibitors tested in this study exhibited linear noncompetitive inhibition versus acetyl-CoA (Figure 2A) and linear uncompetitive inhibition versus the aminoglycoside tobramycin (Figure 2B). Remarkably, the slope and intercept inhibition constants for the various bisubstrate analogs are nearly the same versus AcCoA, whereas more significant differences in the intercept inhibition constants are observed for the various bisubstrate analogs versus tobramycin.
Table 1.
Bisubstrate Inhibition Constants
| inhibitor | inhibition pattern | Km AcCoA (μM) | Kis (μM) | Kii (μM) | kcat (s−1) |
|---|---|---|---|---|---|
| 1Aa | NC | 0.93 ± 0.3 | 3.7 ± 0.9 | 41 ± 33 | 6.0 ± 0.3 |
| 1Ba | NC | 0.30 ± 0.04 | 3.4 ± 0.7 | 42 ± 12 | 3.8 ± 0.1 |
| 1Da | NC | 0.49 ± 0.06 | 2.8 ± 0.5 | 41 ± 17 | 4.8 ± 0.1 |
| 2Aa | NC | 0.99 ± 0.15 | 2.9 ± 0.8 | 10 ± 2 | 4.9 ± 0.2 |
| 3Aa | NC | 0.5 ± 0.1 | 1.9 ± 0.7 | 53 ± 39 | 7.3 ± 0.4 |
| inhibitor | inhibition pattern | Km Tob (μM) | Kii (μM) | kcat (s−1) |
|---|---|---|---|---|
| 1Ab | UC | 1.7 ± 0.1 | 30 ± 2 | 4.0 ± 0.2 |
| 1Bb | UC | 2.1 ± 0.3 | 16 ± 2 | 3.6 ± 0.1 |
| 1Cb | UC | 2.1 ± 0.4 | 8.2 ± 0.8 | 3.9 ± 0.2 |
| 1Db | UC | 1.6 ± 0.5 | 7.2 ± 1.1 | 3.6 ± 0.3 |
| 2Ab | UC | 8.2 ± 0.36 | 13.4 ± 0.5 | 2.9 ± 0.1 |
| 3Ab | UC | 1.6 ± 0.3 | 11.5 ± 0.9 | 4.1 ± 0.2 |
AcCoA was the varied substrate at saturating concentrations of tobramycin.
Tobramycin was the varied substrate at saturating concentrations of AcCoA.
Figure 2.

Bisubstrate inhibition studies of AAC(6′)-Iy. (A) Plot of 1/kcat vs 1/[AcCoA] at varying concentrations of 1B [(●) 0, (○) 2, (▼) 4, and (▽) 6 μM]. (B) Plot of 1/kcat vs 1/[tobramycin] at varying concentrations of 1B [(●) 0, (○) 4, (▼) 8, and (▽) 16 μM]. The symbols represent the experimentally determined values, whereas the solid lines are the best fit of the data to eq 1.
We have used the kinetic model shown in Scheme 2 to analyze and interpret these data. The substrates bind randomly to the free enzyme, and the release of CoA is the slow step in the overall reaction (8, 15, 22). When tobramycin is varied at a saturating concentration of AcCoA, the observed uncompetitive pattern observed suggests that the bisubstrate analogs are binding to one of the product complexes, and the most likely is the E–CoA product complex. If the aminoglycoside portion of the bisubstrate analogs were binding to the enzyme–AcCoA substrate and enzyme–CoA product complexes, increasing concentrations of tobramycin would displace the bound bisubstrates giving rise to noncompetitive inhibition (9). The observed uncompetitive inhibition pattern indicates that the aminoglycoside and linker portion of the bisubstrate analog binds only to the E–CoA complex, and the intercept inhibition constants vary depending on the linker length.
Scheme 2.
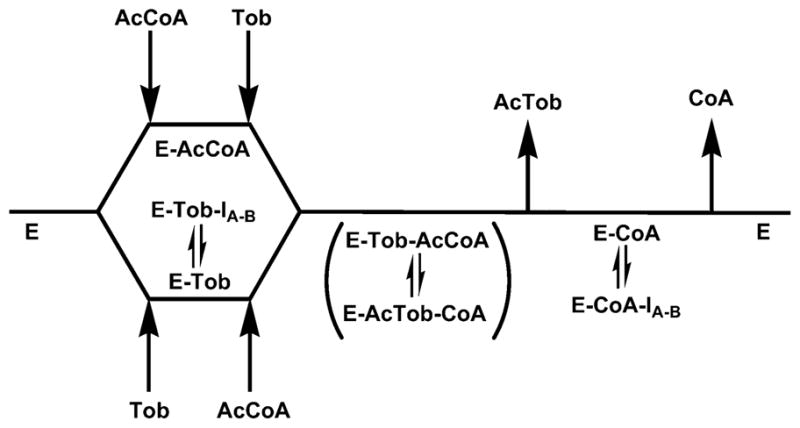
Kinetic Model Used to Evaluate Bisubstrate Inhibition Dataa
a AcCoA, acetyl-coenzyme A; Tob, tobramycin; AcTob, 6′-N-acetyltobramycin; CoA, coenzyme A; IA–B, bisubstrate inhibitor.
When AcCoA is the varied substrate and tobramycin is saturating, the enzyme forms that predominate are the E–CoA and E–tobramycin complexes. The CoA portion of the bisubstrate analogs can compete with AcCoA for binding in the structurally well-defined CoA subsite, leading to an effect on the slopes of the reciprocal plot. Binding of the bisubstrate to the E–CoA product complex (vide supra) generates effects on the intercepts of the reciprocal plot, resulting in the observed noncompetitive inhibition pattern. This interpretation is supported by the significantly lower values of the slope versus intercept inhibition constants, reflecting the stronger binding of the CoA portion of the bisubstrate to the E–tobramycin complex.
By comparison, the E. faecium AAC(6)-Ii shows an ordered binding of AcCoA and aminoglycoside substrate and an ordered release of acetylated aminoglycoside and CoA (13). The rate-limiting steps are physical and include aminoglycoside binding and product release steps. When the inhibitory strength of the series of bisubstrate analogs studied here were determined against AAC(6′)-Ii, there was a clear dependence of the competitive inhibition constants and linker length. For example, the reported Ki values versus AcCoA of compounds 1A, 1B, 1C, and 1D were 70, 40, 160, and 8000 nM, respectively. In a follow up study of truncated bisubstrate analogs lacking the AMP portion of CoA, similar inhibitors were shown to exhibit competitive inhibition versus AcCoA. This is exactly what would be predicted for an ordered mechanism with AcCoA binding first (14).
The previously determined structures of AAC(6′)-Iy were solved as both a binary complex with CoA (PDBID = 1S5K/1S60) and a ternary complex with CoA and ribostamycin (PDBID = 1S3Z) (14). The structure of the complex of AAC(6′)-Iy with the bisubstrate inhibitor CoA-S-monomethyl-N-6′ acetylneamine (1A) was pursued to examine the ability of both the aminoglycoside and CoA portions of the bisubstrate to access the active site and the effect of the carbon linker region to their binding. Crystals of the 1A complex were isomorphous with the ribostamycin complex. AAC(6′)-Iy is a dimer in solution, and there is a molecular dimer in the asymmetric unit of the E–1A complex crystal form (Figure 3A). Clear electron density was observed in each of the subunits for both the CoA and neamine portions of the inhibitor (Figure 3B). In the final model the thermal factors for the neamine and CoA moieties are roughly similar to each other and to the surrounding protein residues suggesting that the inhibitor, in the crystallization conditions, has displaced the CoA molecule from the enzyme active site. In contrast the linker region has elevated thermal factors suggesting either enhanced mobility or multiple conformations. Indeed, the two subunits display distinct conformations for the linker region (Figure 3C). In subunit B the linker region is kinked in such a fashion as to bury the methyl linker within a hydrophobic crevice at the bottom of the active site, in a location similar to where the acetyl group of acetyl-CoA is proposed to bind. In subunit A, the linker region projects outward from the crevice and is solvent exposed. Interestingly, in both conformations the carbonyl group of the linker is hydrogen bonded to the backbone amide of I81 on β-strand 4, mimicking the proposed interaction of the acetyl carbonyl of the acetyl group of AcCoA (14). The binding of the inhibitor does not lead to any large structural rearrangements when compared to the ternary complex with ribostamycin (rmsd of 0.21 Å, 291 common Cα atoms). The majority of CoA, and the 2-deoxystreptamine ring of neamine and ribostamycin, are identical in position and hydrogen-bonding patterns to the enzyme–CoA–ribostamycin complex (Figure 3D). In contrast, the primed ring has rotated slightly toward β-strand 4, breaking the hydrogen bond between the 6′-amino group and the main chain carbonyl of D115. In addition, the proposed base, D115, whose side chain was previously interacting with the 6′-amino group through a water molecule, has rotated (Chi2 −17 to 60) to instead form hydrogen bonds to the 3′ (2.7 Å) and 4′ (2.7 Å) hydroxyl substituents of the inhibitor. These movements are presumably due to the altered nature of the 6′ substituent of the inhibitor compared to ribostamycin and the interaction of the linker carbonyl group with β-strand 4. The structure of AAC(6′)-Iy in complex with 1A demonstrates that both the aminoglycoside and the CoA moieties of the inhibitor can occupy their respective binding sites at the same time and are minimally affected by the linker region. In addition the structure demonstrates two conformations for the linker for a single methyl linker and sufficient flexibility and space to accommodate longer linkers.
Figure 3.
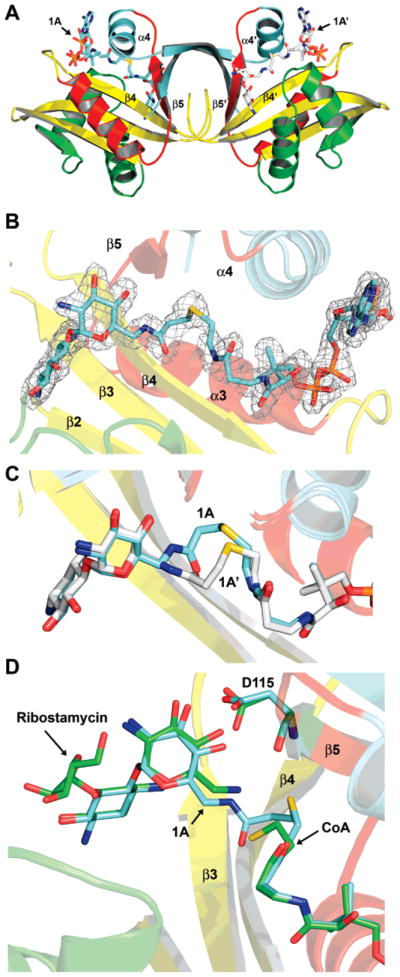
Structure of AAC(6)-Iy with bound inhibitor. (A) Ribbon diagram of the AAC(6)-Iy–1A binary complex. Bisubstrate 1A is shown as a stick model colored by atom type. AAC(6)-Iy secondary structure is colored according to Vetting et al. (22). (B) Final 2Fo−Fc electron density for the inhibitor contoured at the 1σ level. For the map, bisubstrate 1A was omitted for a round of refinement prior to map calculation. Bisubstrate 1A is shown as a stick model colored by atom type. (C) Superposition of the two conformations displayed by 1A around the linker region for subunit A (cyan carbons) and subunit B (white carbons). (D) Superposition of the AAC(6)-Iy/ribostamycin/CoA ternary complex (green carbons) with the AAC-(6)-Iy–1A binary complex (cyan carbons).
Given the nanomolar inhibition of these bisubstrate analogs against E. faecium AAC(6′)-Ii (12), one would have expected comparable tight binding to AAC(6′)-Iy. In contrast, these compounds inhibit AAC(6′)-Iy in the micromolar range. The fact that CoA release is rate-limiting in vitro and that only the aminoglycoside part of the bisubstrate might bind to the E–CoA complex can explain such low affinity constants, comparable to the aminoglycoside Km values. The high affinity of CoA for E (8, 14, 20) may lead to the near absence of free E in steady-state experiments. Additionally, Ki decreases as the linker length increases (Table 1), when the aminoglycoside is varied. Accordingly, as the linker between the CoA and aminoglycoside moieties becomes larger, the two components of the bisubstrate analogs become more flexible, allowing the aminoglycoside portion of the molecule to behave similarly to a “free aminoglycoside” molecule while its Ki value approaches the aminoglycoside Km values.
We have shown here that the first generation 6′-N-derivatized bisubstrate analogues produced by Gao et al. represent an important tool for kinetic studies of aminoglycoside-6′-N-acetyltransferases. In contrast to the competitive nanomolar tight-binding inhibition of AAC(6′)-Ii, the bisubstrate analogues inhibited the S. enterica AAC(6′)-Iy in the micromolar range. We were able, however, to explain such low affinity constants, as well as the unexpected inhibition patterns, based on the high affinity of the enzyme for its product: the CoA molecule in these in vitro studies. In vivo, the rapid conversion of CoA into AcCoA would relieve this effect, and the bisubstrate could potentially provide powerful inhibition of the substrate-free form of the enzyme. The use of these bisubstrate analogues should provide valuable guidance for the study of other members of the N-acetyl-transferase family of enzymes.
Footnotes
This work was supported by NIH Grant AI60899 (to J.S.B.) and the Natural Science and Engineering Research Council of Canada (NSERC) (to K.A). F.G. and L.F. were supported by scholarship awards from the Chemical Biology Strategic Training Initiative of the Canadian Institute of Health Research (CIHR).
Coordinates have been deposited with the Protein Data Bank as 2VBQ.pdb.
References
- 1.Magnet S, Blanchard JS. Molecular insights into aminoglycoside action and resistance. Chem Rev. 2005;105:477–498. doi: 10.1021/cr0301088. [DOI] [PubMed] [Google Scholar]
- 2.Llano-Sotelo B, Azucena EF, Jr, Kotra LP, Mobashery S, Chow CS. Aminoglycosides modified by resistance enzymes display diminished binding to the bacterial ribosomal aminoacyl-tRNA site. Chem Biol. 2002;9:455–463. doi: 10.1016/s1074-5521(02)00125-4. [DOI] [PubMed] [Google Scholar]
- 3.Okamoto S, Suzuki Y. Chloramphenicol-, dihydrostreptomycin-, and kanamycin-inactivating enzymes from multiple drug-resistant Escherichia coli carrying episome ‘R’. Nature. 1965;208:1301–1303. doi: 10.1038/2081301a0. [DOI] [PubMed] [Google Scholar]
- 4.Magnet S, Courvalin P, Lambert T. Activation of the cryptic aac(6′)-Iy aminoglycoside resistance gene of Salmonella by a chromosomal deletion generating a transcriptional fusion. J Bacteriol. 1999;181:6650–6655. doi: 10.1128/jb.181.21.6650-6655.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wright GD, Thompson PR. Aminoglycoside phosphotransferases: proteins, structure, and mechanism. Front Biosci. 1999;4:D9–D21. doi: 10.2741/wright. [DOI] [PubMed] [Google Scholar]
- 6.Culebras E, Martinez JL. Aminoglycoside resistance mediated by the bifunctional enzyme 6′-N-aminoglycoside acetyl-transferase-2″-O-aminoglycoside phosphotransferase. Front Biosci. 1999;4:D1–D8. doi: 10.2741/culebras. [DOI] [PubMed] [Google Scholar]
- 7.Costa Y, Galimand M, Leclercq R, Duval J, Courvalin P. Characterization of the chromosomal aac(6′)-Ii gene specific for Enterococcus faecium. Antimicrob Agents Chemother. 1993;37:1896–1903. doi: 10.1128/aac.37.9.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magnet S, Lambert T, Courvalin P, Blanchard JS. Kinetic and mutagenic characterization of the chromosomally encoded Salmonella enterica AAC(6′)-Iy aminoglycoside N-acetyltransferase. Biochemistry. 2001;40:3700–3709. doi: 10.1021/bi002736e. [DOI] [PubMed] [Google Scholar]
- 9.Yu M, Magalhaes ML, Cook PF, Blanchard JS. Bisubstrate inhibition: Theory and application to N-acetyltransferases. Biochemistry. 2006;45:14788–14794. doi: 10.1021/bi061621t. [DOI] [PubMed] [Google Scholar]
- 10.Brown MJ, Mensah LM, Doyle ML, Broom NJ, Osbourne N, Forrest AK, Richardson CM, O’Hanlon PJ, Pope AJ. Rational design of femtomolar inhibitors of isoleucyl tRNA synthetase from a binding model for pseudomonic acid-A. Biochemistry. 2000;39:6003–6011. doi: 10.1021/bi000148v. [DOI] [PubMed] [Google Scholar]
- 11.Williams JW, Northrop DB. Synthesis of a tight-binding, multisubstrate analog inhibitor of gentamicin acetyltransferase I. J Antibiot (Tokyo) 1979;32:1147–1154. doi: 10.7164/antibiotics.32.1147. [DOI] [PubMed] [Google Scholar]
- 12.Gao F, Yan X, Baettig OM, Berghuis AM, Auclair K. Regio- and chemoselective 6′-N-derivatization of aminoglycosides: bisubstrate inhibitors as probes to study aminoglycoside 6′-N-acetyltransferases. Angew Chem, Int Ed. 2005;44:6859–6862. doi: 10.1002/anie.200501399. [DOI] [PubMed] [Google Scholar]
- 13.Draker KA, Northrop DB, Wright GD. Kinetic mechanism of the GCN5-related chromosomal aminoglycoside acetyltransferase AAC(6′)-Ii from Enterococcus faecium: evidence of dimer subunit cooperativity. Biochemistry. 2003;42:6565–6574. doi: 10.1021/bi034148h. [DOI] [PubMed] [Google Scholar]
- 14.Gao F, Yan X, Shakya T, Baettig OM, Ait-Mohand-Brunet S, Berghuis AM, Wright GD, Auclair K. Synthesis and structure–activity relationships of truncated bisubstrate inhibitors of aminoglycoside 6′-N-acetyltransferases. J Med Chem. 2006;49:5273–5281. doi: 10.1021/jm060732n. [DOI] [PubMed] [Google Scholar]
- 15.Vetting MW, Magnet S, Nieves E, Roderick SL, Blanchard JS. A bacterial acetyltransferase capable of regioselective N-acetylation of antibiotics and histones. Chem Biol. 2004;11:565–573. doi: 10.1016/j.chembiol.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 16.Leslie AG. The integration of macromolecular diffraction data. Acta Crystallogr, Sect D: Biol Crystallogr. 2006;62:48–57. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- 17.Evans P. Scaling and assessment of data quality. Acta Crystallogr, Sect D: Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 18.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 19.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr, Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 20.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 21.Hegde SS, Dam TK, Brewer CF, Blanchard JS. Thermodynamics of aminoglycoside and acyl-coenzyme A binding to the Salmonella enterica AAC(6′)-Iy aminoglycoside N-acetyltransferase. Biochemistry. 2002;41:7519–7527. doi: 10.1021/bi020190l. [DOI] [PubMed] [Google Scholar]
- 22.Vetting MW, de Carvalho LPS, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys. 2005;433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]