

Abstract
The morphogenetic transition between yeast and filamentous forms of the human fungal pathogen Candida albicans is regulated by a variety of signaling pathways. How these pathways interact to orchestrate morphogenesis, however, has not been as well characterized. To address this question and to identify genes that interact with the Regulation of Ace2 and Morphogenesis (RAM) pathway during filamentation, we report the first large-scale genetic interaction screen in C. albicans. Our strategy for this screen was based on the concept of complex haploinsufficiency (CHI). A heterozygous mutant of CBK1 (cbk1Δ/CBK1), a key RAM pathway protein kinase, was subjected to transposon-mediated, insertional mutagenesis. The resulting double heterozygous mutants (6,528 independent strains) were screened for decreased filamentation on Spider Medium (SM). From the 441 mutants showing altered filamentation, 139 transposon insertion sites were sequenced, yielding 41 unique CBK1-interacting genes. This gene set was enriched in transcriptional targets of Ace2 and, strikingly, the cAMP-dependent protein kinase A (PKA) pathway, suggesting an interaction between these two pathways. Further analysis indicates that the RAM and PKA pathways co-regulate a common set of genes during morphogenesis and that hyper-activation of the PKA pathway may compensate for loss of RAM pathway function. Our data also indicate that the PKA–regulated transcription factor Efg1 primarily localizes to yeast phase cells while the RAM–pathway regulated transcription factor Ace2 localizes to daughter nuclei of filamentous cells, suggesting that Efg1 and Ace2 regulate a common set of genes at separate stages of morphogenesis. Taken together, our observations indicate that CHI–based screening is a useful approach to genetic interaction analysis in C. albicans and support a model in which these two pathways regulate a common set of genes at different stages of filamentation.
Author Summary
Candida albicans is the most common cause of fungal infections in humans. As a diploid yeast without a classical sexual cycle, many genetic approaches developed for large-scale genetic interaction studies in the model yeast Saccharomyces cerevisiae cannot be applied to C. albicans. Genetic interaction studies have proven to be powerful genetic tools for the analysis of complex biological processes. Here, we demonstrate that libraries of C. albicans strains containing heterozygous mutations in two different genes can be generated and used to study genetic interactions in C. albicans on a large scale. Double heterozygous mutants that show more severe phenotypes than strains with single heterozygous mutations are indicative of genetic interactions through a phenomenon referred to as complex haploinsufficiency (CHI). We applied this approach to the study of the RAM (Regulation of Ace2 and Morphogenesis) signaling network during the morphogenetic transition of C. albicans from yeast to filamentous growth. Among the genes that interacted with CBK1, the key signaling kinase of the RAM pathway, were transcriptional targets of the RAM pathway and the protein kinase A pathway. Further analysis supports a model in which these two pathways co-regulate a common set of genes at different stages of filamentation.
Introduction
Candida albicans is a member of the resident flora of the gastrointestinal tract and is the most common fungal pathogen in humans. The most severe manifestations of candidiasis occur in immunocompromised patients and include debilitating mucosal disease such as oropharyngeal candidiasis as well as life-threatening disseminated infections of the bloodstream and major organ systems [1]. Animal studies have shown that the pathogenic potential of C. albicans is associated with its ability to transition between three morphological states: yeast, pseudohyphae, and hyphae [2], [3]. Further insights into the contributions of the different morphotypes to pathogenesis have emerged from elegant studies with C. albicans strains that allow the conditional induction of filamentation in vivo [4]. For example, C. albicans genetically restricted to the yeast form by constitutive expression of NRG1 are able to establish infection in mice but no disease results until the expression of NRG1 is repressed and the organism is able to form filaments.
The relationship between morphogenesis and virulence in C. albicans is, however, not a simple one. Many mutants that are unable to undergo morphogenesis also display other phenotypes. For example, many transcription factors that are required for morphogenesis regulate a host of other genes and display pleiomorphic phenotypes. The complicated nature of the relationship between morphogenesis has been further highlighted by the elegant study recently reported by Noble et al. [5]. Noble et al. generated a bar-coded collection of homozygous deletion mutants and used it in a signature-tagged mutagenesis study of infectivity in a mouse model [5]. Mutants with defects in morphogenesis were more likely to have decreased infectivity; however, a significant portion of mutants with severe morphogenesis defects retained the ability to cause infection. It is important to note that Noble et al. assayed for infection and not for disease. Thus, their results are not necessarily in conflict with studies discussed above that indicate that morphogenesis is required for disease progression in animal models [4]. Furthermore, their work serves to highlight the fact that additional studies will be required to fully understand the complex relationship between morphogenesis and pathogenesis in C. albicans.
Given the close association of morphogenesis with C. albicans pathogenesis, the genetic and cell biologic analysis of this process has been the subject of intensive study [6]. Consequently, many genes have been shown to affect filamentation, and, correspondingly, a number of regulatory pathways have been shown to play a role in the orchestration of the morphogenetic program in C. albicans [7]. The PKA, CPH1, HOG1, RIM101, CHK1, and CBK1/RAM pathways are among those that regulate morphogenesis under a variety of conditions [6], [7]. Although much remains to be learned about how individual pathways and genes contribute to morphogenesis, an important question that has not been extensively studied is how these various pathways interact to regulate morphogenesis.
In the model yeast S. cerevisiae, relationships between regulatory pathways can be readily characterized using recently developed systematic, genome-wide genetic interaction strategies [8]–[10]. These approaches have yielded a wealth of information regarding the mechanisms through which cells regulate complex biological processes [11]. However, because C. albicans is diploid and lacks a classical meiotic cycle, the mating-based genetic strategies used to create genome-wide libraries of double mutant strains in S. cerevisiae are not applicable. Consequently, genetic interaction studies in C. albicans have been limited to gene-by-gene analyses. Despite these limitations, such studies have proven quite informative and suggest that large scale interaction studies could represent a powerful approach to studying regulatory networks in C. albicans. For example, Braun et al. carried out a thorough, systematic epistasis analysis of three transcriptional regulators (EFG1, TUP1 and CPH1) and showed that each played a distinct role in the regulation of filamentation [12].
Recent advances in the genetic analysis of C. albicans have greatly facilitated the development of innovative approaches to the study of this important human pathogen [13]. Among these important developments is the application of transposon-based mutagenesis strategies [14] to the creation and study of large-scale libraries of heterozygous [15], and homozygous [17], [18] C. albicans mutants. Similarly, large collections of homozygous null [19] and conditional mutants [20] have been created in a targeted manner and analyzed for a variety of phenotypes including morphogenesis, virulence and drug susceptibility. To our knowledge, one area that has not been explored is the development of approaches to large-scale synthetic genetic interaction analysis in C. albicans.
Here, we describe the first large-scale synthetic genetic interaction screen in C. albicans. Our strategy builds on pioneering yeast genetics approaches developed in both S. cerevisiae and C. albicans and is based on the concept of complex haploinsufficiency (CHI). CHI is a special case of a genetic phenomenon referred to as unlinked non-complementation in the context of yeast genetics and as dominant enhancers or dominant modifiers when applied to Drosophila [21]. Unlinked non-complementation occurs when a cross between two haploid strains containing single recessive mutations located in separate loci results in a diploid strain (complex heterozygote) that retains the phenotype of a parental strain. In yeast, the construction of such mutants was used to great advantage in the genetic analysis of cytoskeletal genes such as tubulin [22] and actin [23]. CHI, which is a special case of unlinked non-complementation, occurs when strains containing heterozygous mutations at two separate loci display a more severe phenotype than strains that contain heterozygous mutations at the single loci alone [21]. In essence, CHI can also be called synthetic haploinsufficiency. Recently, a genome-wide CHI-based strategy was developed in S. cerevisiae and successfully used to create a genetic interaction network for the essential gene, ACT1 [21].
As described in the seminal work of Uhl et al. [15], large scale haploinsufficiency-based screening was first applied to C. albicans in the transposon-mediated, insertional mutagenesis analysis of filamentation and, thus, haploinsufficiency-based screening has excellent precedence in this system. Whereas Uhl et al. carried out their haploinsufficiency screen starting with a “wild type” strain [15], we reasoned that transposon mutagenesis of a parental strain containing a heterozygous mutation at a locus of interest would represent an expedient approach to the generation of a large library of complex heterozygotes that could then be the basis of a CHI screen for genes that interact with the parental mutant.
In principle, CHI-based screening has a number of attractive features. First, CHI allows one to identify genes that function within the pathway affected by the parental or query mutation including upstream and downstream components of the pathway, transcriptional outputs of the pathway, and substrates of pathway enzymes. Second, CHI-based screening can also identify genes or pathways that function in parallel with the query pathway and, therefore, allow one to identify pathways that co-regulate a given process. Third, CHI is ideal for the study of essential genes because only heterozygous mutations are generated.
We developed a CHI-based screening strategy (Figure 1) and applied it to the identification of genes that interact with the RAM signaling network during C. albicans filamentation [24]–[27]. The RAM network has been extensively studied in S. cerevisiae [28] and is required for a variety of cellular processes in both S. cerevisiae and C. albicans including polarity, cell wall synthesis, cell separation and filamentous growth. Cbk1 is the key serine/threonine protein kinase [24], [27] that mediates many of the functions of the RAM network through its regulation of the transcription factor Ace2 [24], [27]. RAM pathway mutants in C. albicans show two distinct filamentation phenotypes: CBK1 null mutants are unable to form filaments on Spider Medium (SM) or serum-containing medium [24], [27] while ACE2 null mutants are constitutively pseudohyphal and form true hyphae on serum [25]. Although our understanding of the RAM network in C. albicans has increased in recent years [24]–[27], many questions remain, including: how does it interact with the many other regulatory pathways during morphogenesis and what genes and proteins are regulated by Cbk1 and/or its downstream transcription factor Ace2?
Figure 1. Schematic of screening strategy.

In vitro mutagenesis of C. albicans genomic library WO-1 using a Tn7-based transposon containing the CaURA3-dpl200 auxotrophic marker [29] yielded a library of plasmids from which genomic inserts were released by restriction endonuclease digestion and transformed into a cbk1Δ/CBK1 heterozygote strain. The resulting library was screened on SM for altered filamentation relative to the parental strain.
Through this novel application of a CHI-based screening strategy, we have identified RAM/Ace2 transcriptional targets and generated genetic evidence for an interaction between the RAM and PKA pathways during morphogenesis. Follow-up studies of the screening results further suggest that a balance between RAM and PKA-pathway activity is required for cells to establish a normal distribution of morphotypes during nutrient-induced filamentation. Taken together with previous work on these two pathways, our observations support a model where PKA-regulated transcriptional activity is most important in the transcription of RAM/PKA co-regulated genes early in morphogenesis, while the RAM pathway is more important as daughter nuclei accumulate within the hyphal structure.
Results
Construction of the insertional library and CHI screening strategy
An outline of the CHI-based screening strategy is presented in Figure 1. In preparation for the CHI screen, we first constructed a transposon suitable for large-scale insertional mutagenesis in C. albicans. To enable efficient mutagenesis with limited transposition bias, we generated a donor plasmid derived from the bacterial element Tn7. The Tn7 system has been used extensively for in vitro mutagenesis [29], [30] with low reported insertion site specificity [31]. For purposes of this screen, the Tn7 element was modified to contain a recyclable allele of the CaURA3 gene; specifically, we inserted the URA3-dpl200 allele into Tn7 sequence encoded in the donor plasmid pGPS3. The URA3-dpl200 allele was designed by Wilson et al. [32] to allow recombinational excision of the URA3 gene under counter-selection with 5-fluoro-orotic acid (5-FOA). Subsequently, we performed in vitro mutagenesis of the genomic library pEMBLY23 (Materials and Methods) derived from C.albicans strain WO-1. Non-specific Tn7 transposition was achieved using the TnsA, TnsB, and TnsC* proteins paired with the TnsAB transposase and appropriate cofactors [29]. The genomic library was mutagenized to yield an estimated 20,000 independent insertions. The resulting insertional library was recovered in E. coli, and genomic DNA inserts were released by enzyme digestion for introduction into the C. albicans Ura- parental strain, cbk1Δ/CBK1 (CAMM292, see Table S1 for strain table). By homologous recombination, the mutagenized genomic DNA fragment will replace its native chromosomal locus, thereby generating a heterozygous insertion mutant in the parent cbk1Δ/CBK1 strain. DNA transformations were performed nine times, yielding a total of 6528 independent Ura+ transformants. The C. albicans double heterozygotes were isolated and screened for decreased filamentation as follows.
CHI screening of the cbk1Δ/CBK1 mutant
The cbk1Δ/CBK1 mutant was originally studied in C. albicans by McNemar and Fonzi [24] and was found to be haploinsufficient with respect to filamentation on a variety of media. Uhl et al. also isolated a heterozygous cbk1 insertion mutant in their haploinsufficiency screen [15]. As shown in Figure 2A, cbk1Δ/CBK1 colonies show a decreased area of central wrinkling and a more prominent ring of peripheral filamentation on SM at 37°C. The haploinsufficiency of this parental strain was advantageous for two reasons. First, it provided increased sensitivity in that the strain was already deficient for filamentation. Second, it could also improve specificity because weak phenotypes of non-interacting, transposon-derived mutants would not be apparent due to masking by the cbk1Δ/CBK1 phenotype.
Figure 2. CHI–based screening identifies synthetic genetic interactions with CBK1 during morphogenesis.

(A) Examples of primary screening data for complex heterozygotes showing synthetic genetic interactions with CBK1; each strain was spotted on SM and incubated at 37°C for 3 days. Mutants with decreased peripheral invasion and decreased central wrinkling were selected. Representative positive scoring mutants from the primary screen are shown. An example of a strain complemented by re-integration of plasmid-borne CBK1 is shown. (B) Representative examples of independently constructed complex heterozygote strains showing complex haploinsufficient genetic interactions with cbk1Δ. (C) The ratio of pseudohyphal∶hyphal cells for the indicated strains was determined by light microscopy after 3 hours incubation in liquid SM at 37°C. The bars indicate mean values of two-three independent replicates of at least 100 cells. Error bars indicate standard deviation. Brackets indicate the results of Student's t test evaluation of differences between the indicated mutants; p<0.05 indicates a statistically significant difference. (D) Micrograph of filaments isolated from colonies of the parental cbk1Δ/CBK1 strain and the complex heterozygote cbk1Δ/CBK1 snz1Δ/SNZ1. Arrowheads indicate areas of hyphal-like morphology in the cbk1Δ/CBK1 mutant and pseudohyphae-like morphology in the cbk1Δ/CBK1 snz1Δ/SNZ1 double mutant.
As described above [24], [25], [27], RAM pathway mutants show two distinct phenotypes depending on the conditions used to induce filamentation, but both phenotypes are apparent on solid Spider Medium (SM). In order to identify mutations that potentially interacted with both general functions of the pathway, we, therefore, screened for decreased filamentation on SM at 37°C. All subsequent experiments were conducted under these conditions unless otherwise indicated.
The library of 6528 complex heterozygous mutants was spotted in 96-well format and scored for decreased peripheral invasion and altered colony wrinkling relative to a Ura+ derivative of the parental cbk1Δ/CBK1 strain (11, Figure 2A). Clones showing both phenotypes were re-tested using two independent colonies. A total of 441 complex heterozygous mutants with decreased peripheral invasion and altered colony wrinkling were re-confirmed on both SM and SM containing uridine to control for positional effects of the URA3 gene (Figure 2A). We specifically selected mutants with decreased zones of peripheral filamentation and less pronounced central wrinkling relative to the parental strain (Figure 2A and 2B). All mutants showed some degree of peripheral filamentation. The most common composite phenotype indicated a small zone of peripheral agar invasion with a broad region of moderate wrinkling (Figure 2B).
The transposon insertion sites for approximately one-third of the mutants (139 strains) showing potential synthetic genetic interactions were identified using a 3′-RACE/sequencing approach (see Materials and Methods), yielding 42 unique transposon-derived mutations as putative CBK1-interactors. Since 8 insertion sites were identified in at least 5 separate clones (Figure 3A and 3B), the screen appeared to be saturated to the limits of the library and the mutagenesis technique. Therefore, we did not sequence the remaining two-thirds of the mutants and focused on evaluating the initial set of 42 mutants. It is, however, important to note that the screen itself is unlikely to be saturated for all possible CBK1 interactors, as the library almost certainly did not contain insertions in all predicted C. albicans genes.
Figure 3. Summary and bioinformatic analysis of screening data.

(A) Summary of screening results and list of CBK1-interacting genes. (B) List of CBK1-synthetic genetic interactions during morphogenesis grouped according to three most common GO terms. Colors indicate the number of times each insertion was isolated. (C) Venn diagram depicting the number of genes putatively co-regulated by the RAM and PKA pathways. (D) List of CBK1-interacting genes with Ace2 and both Ace2/Efg1 consensus binding sites within the region 1000 bp upstream of the start codon.
The URA3 marker was recycled from the heterozygotes by 5-FOA mediated recombinational excision [32]. Following phenotypic re-testing to confirm that homozygosis was not responsible for curing the URA3 marker, CBK1 was re-integrated at its chromosomal position using plasmid pMM4 [24]. The phenotypes of 41 of 42 candidate CHI strains were modified by re-integration of CBK1 (Figure 2A), indicating that the observed phenotypes were dependent on the cbk1 mutation and were likely due to a synthetic genetic interaction between cbk1Δ/CBK1 and the transposon insertion. The high percentage of CBK1-dependent phenotypes may be due to the fact that the parental cbk1Δ heterozygote is itself haploinsufficient on SM and most non-interacting insertion mutations that are themselves haploinsufficient do not have sufficiently strong phenotypes to appreciably change the phenotype of the double heterozygote relative to the parental strain. To confirm these interactions further, a subset of ten complex heterozygous mutants was independently constructed from CAMM-292 by single gene-replacement [33]. All ten double mutants recapitulated phenotypes displayed by the transposon-derived mutants and showed distinct phenotypes relative to strains with single deletions of the interacting genes. Representative images from this analysis are shown in Figure 2B.
To further characterize the morphologies of the mutants, we determined the proportion of yeast, pseudohyphae and hyphae after 3 hours induction in liquid SM at 37°C. The interacting mutants consistently showed an increased proportion of pseudohyphal cells relative to wild type and cbk1Δ/CBK1 strains (Figure 2C). Similarly, examination of cells scraped from SM plates showed that the filaments of double mutants had constricted septal areas characteristic of pseudohyphae (Figure 2D). Importantly, all of the mutants were indistinguishable from wild type and the parental strain when serum was used as the inducer of filamentation (data not shown). Since ace2Δ/Δ mutants also show decreased peripheral invasion, decreased central wrinkling, increased levels of pseudohyphae, and normal filamentation on serum (25), we conclude that the majority of the CBK1-interacting genes isolated in the screen appear to affect the Ace2-dependent functions of the RAM pathway.
The set of CBK1-interactors contains genes related to Ace2 function
Literature analysis of the set of CBK1-interactoring genes revealed that approximately one-half are involved in glycolysis/respiration, biosynthesis, or cell wall metabolism (Figure 3B), cell processes consistent with established functions of the RAM pathway [24]–[28]. An important interactor in terms of validating the screen is SSD1 because it is a likely Cbk1 substrate in S. cerevisiae [34], displaying well-characterized genetic interactions with CBK1 in both S. cerevisiae [35] and C. albicans [27]. Comparison of our dataset with that generated by the haploinsufficiency screen of Uhl et al. revealed no overlap [15]. As discussed above, we suspect that this lack of overlap is also related to the fact that our parental strain is haploinsufficient for filamentation and, thus, non-interacting transposon-derived mutations causing simple haploinsufficiency were, in effect, masked by the phenotype of the parental strain.
In principle, the Ace2-deficient phenotypes displayed by the double heterozygous mutants could result from mutations that interfere with the activation of Ace2 or from mutations that affect a key transcriptional target of Ace2. We isolated three mutants that could cause a CHI-interaction with CBK1 through the former mechanism. First, we isolated orthologs of two genes that regulate mitotic exit in S. cerevisiae, CDH1 [36] and SLK19 [37]. Ace2 is well known to localize to the nuclei of daughter cells in both S. cerevisiae [38] and C. albicans [25], [39]. Since Cdh1 and Slk19 regulate mitotic exit, the point in the cell cycle when Ace2 localizes to the nuclei [38], we suggest that disruption of mitotic exit through the loss of these proteins may further decrease the overall activity of Ace2. In addition, NSP1, a key component of the nuclear import machinery, was isolated. Studies in S. cerevisiae [40] have indicated that decreased NSP1 gene dosage leads to inhibition of nuclear import, and it seems plausible that a strain lacking an allele of NSP1 could have decreased nuclear import of Ace2 which would further decrease the overall Ace2-mediated transcriptional activity of the cbk1Δ/CBK1 mutant.
The larger class of CBK1-interacting mutants that relate to Ace2 function is the set of genes that appear to be part of the transcriptional output of the RAM pathway (Figure 3C and 3D). To identify such genes in our data set, we searched the promoter regions of CBK1-interactors and found 22 genes that contain a C. albicans Ace2-consensus binding sequence [MMCCASC, 26]. Of these genes, 11 have been shown to display decreased expression in ace2Δ/Δ mutants during hyphal induction as reported in a recent transcriptional profiling study [26]. To further confirm that our screen identified genes regulated by Ace2, we examined the binding of Ace2 to the promoters of 5 CBK1-interactors with consensus binding sites (ACT1, ADH1, ENO1, HGT6, & RGD3) during both yeast and hypha-phase growth by chromatin immunoprecipitation (ChIP). Consistent with ChIP data for Ace2 reported by Wang et al. [39], the absolute enrichment was relatively low, most likely due to its cell cycle regulation and our non-synchronous experiments (Figure 4). Nevertheless, all five promoters were bound by Ace2 at levels comparable to those observed for the well-established Ace2 target CHT3 and to those reported by Wang et al. [39] during yeast growth. In addition, three promoters were bound in hyphal phase (Figure 4). Taken together, the presence of Ace2 binding sites, the transcriptional profiling data, and ChIP data support the notion that many of the CBK1-interacting genes are transcriptional targets of Ace2.
Figure 4. The set of CBK1-interacting genes includes transcriptional targets of Ace2.

The binding of Ace2-TAP to the promoter regions of 5 CBK1-interacting genes was assessed by ChIP in yeast and hyphae-phase cells (SM, 3 h, 37°C) containing a TAP-tag fused to the C-terminus of Ace2. Bars indicate the ratio of promoter DNA (determined by PCR) in tagged extracts relative to un-tagged extracts (error bars indicate SD of three replicates). Grey bars show promoters with increased abundance in tagged extracts, suggesting they are bound by Ace2. P cht3, a known target of Ace2 [13], [21], and primers to a coding sequence (ORF cds) serve as positive and negative controls. A persistent contaminating band prevented accurate assessment of ACT1 in hyphae.
The CHI screen of cbk1Δ/CBK1 reveals potential interaction between the RAM and PKA pathways during morphogenesis
Comparison of the set of CBK1-interactors with data from a variety of transcriptional profiles of C. albicans morphogenesis indicated that a substantial subset of CBK1-interactors (14 interactors, 34%) are regulated by the cAMP/PKA pathway through the transcription factor Efg1 [41]. Indeed, 10 CBK1-interactors contain consensus binding sites for both Ace2 and Efg1 (Figure 3C and 3D), suggesting that these two transcription factors may regulate a common set of genes. Further supporting this notion are previous studies indicating that both Ace2 and Efg1 induce glycolytic genes and repress genes involved in oxidative respiration [26], [41]. Indeed, we searched the C. albicans genome and found that the promoters of 384 genes contain consensus binding sites for both Ace2 and Efg1 (Table S2). Consistent with previous studies of the two pathways, the set of putatively co-regulated genes is enriched for genes contributing to glycolysis, biosynthesis, and cellular stress responses. Recently, Wang et al. have also shown that the promoters of Ace2-regulated cell wall and cell separation genes are bound by both Efg1 and Ace2 during morphogenesis [39]. Taken together our genetic data strongly support the notion that genes regulated by the PKA pathway may also be important components of the transcriptional output of the RAM pathway during morphogenesis.
In addition to transcriptional targets of the PKA pathway, three other CBK1 interactors (MAF1, SLF1 & ACT1) have connections to the PKA pathway (Figure 3A). MAF1 and SFL1 are both orthologs of PKA-regulated transcriptional regulators in S. cerevisiae [42], [43], suggesting that proper PKA-mediated transcriptional control is important in the absence of full RAM pathway activity. Further suggesting that the activity of the PKA pathway is important in RAM pathway mutants, we isolated ACT1 as a CBK1-interactor. Although ACT1 is, of course, a crucial part of the cell cytoskeleton, it also plays an important role in activation of the cAMP/PKA pathway. The Sundstrom lab has shown that actin dynamics regulate PKA activity [44] and, recently, Zou et al. have elegantly demonstrated that actin functions as part of a PKA sensor/activator complex during hyphal development [45]. Indeed, decreased G-actin levels lead to decreased PKA pathway activity and, in turn, decreased filamentation in C. albicans [45]. As such, one explanation for the interaction between ACT1 and CBK1 is that the lowered ACT1 gene dosage in the act1Δ/ACT1 cbk1Δ/CBK1 mutant exacerbates the filamentation defects of decreased RAM pathway activity by concomitantly limiting PKA activity. This explanation also implies that the PKA pathway may compensate for decreased RAM pathway activity during morphogenesis.
RAM pathway mutants show evidence of increased PKA pathway activity
To test the hypothesis that the RAM and PKA pathways regulate a common set of genes during morphogenesis, we examined the expression of two CBK1-interacting genes containing both Ace2 and Efg1 binding sites in ace2Δ/Δ and efg1Δ/Δ mutants after 3 hours of hyphal induction with SM. As shown in Figure 5A, the expression of the transcripts increased in both strains relative to wild type by quantitative RT-PCR. These observations suggest either that Efg1 and Ace2 are functioning as transcriptional repressors or that compensatory responses are occurring to maintain expression of these genes during morphogenesis when one of the two pathways is disabled.
Figure 5. The PKA pathway compensates for decreased RAM pathway activity during morphogenesis.
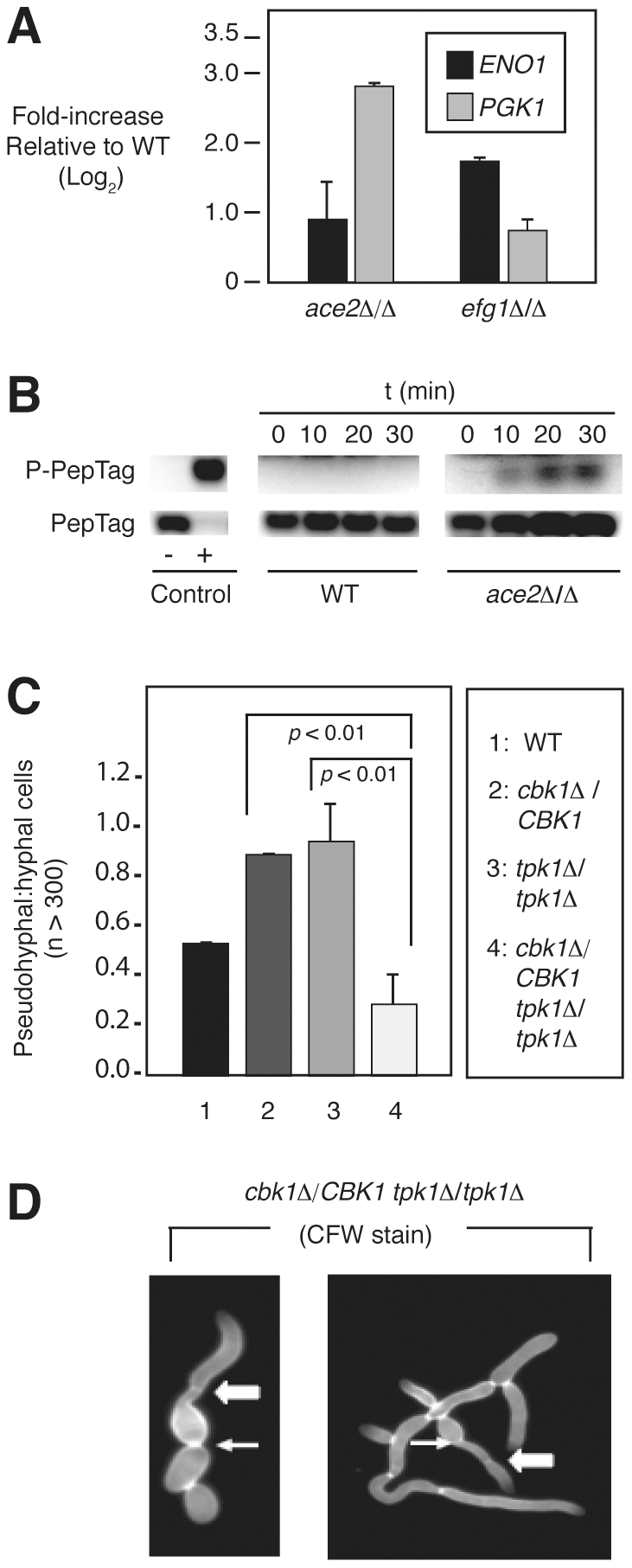
(A) Transcript levels of ENO1 and PGK1 in each mutant were compared to wild type by qRT-PCR using the 2−ΔΔCt method and are graphed as Log2 change over wild type (three independent experiments performed in triplicate). Bars indicate mean value and error bars indicate standard deviation. The observed elevation in levels of each transcript in the indicated mutants relative to wild type were statistically significant by Student's t test (p<0.05). (B) Phosphorylation of fluorescent PKA substrate (PepTag, Promega) by cell extracts (10 µg protein) derived from wild type (WT) and ace2Δ/2Δ cells harvested after incubation in SM for 3 h at 37°C. The indicated time points represent PKA reaction time. (C) The ratio of pseudohyphal∶hyphal cells for the indicated strains was determined by light microscopy after 3 h incubation in liquid SM at 37°C. The bars indicate mean values of two-three independent replicates of at least 100 cells. Error bars indicate standard deviation. Brackets indicate the results of Student's t test evaluation of differences between the indicated mutants; p<0.05 indicates a statistically significant difference. (D) Hybrid pseudohyphae/hyphae cells of cbk1Δ/CBK1 tpk1Δ/1Δ following staining with Calcofluor white. Arrows indicate budneck localized septa (pseudohyphae-like) and block arrows indicate distal septa (hyphae-like).
To test the latter hypothesis, total cell lysates of the RAM pathway mutant ace2Δ/Δ were prepared and the level of PKA enzymatic activity determined after 3 hours exposure to hypha-inducing conditions (Figure 5B). At this time point, PKA activity has reduced to low levels in wild type cells [45], but there is clearly increased PKA activity in the ace2Δ/Δ mutant. This suggests that the PKA pathway is hyperactive in RAM pathway mutants and is consistent with the hypothesis that the PKA pathway may compensate for decreased RAM pathway activity. To further test the interaction between the RAM and PKA pathways, we deleted one allele of CBK1 in strains containing homozygous null mutations in one of the catalytic subunits of the PKA enzyme [46] to yield the mutants cbk1Δ/CBK1 tpk1Δ/Δ and cbk1Δ/CBK1 tpk2Δ/Δ. The two triple mutants along with wild type and the parental mutants were incubated in SM for 3 hours at 37°C to induce filamentation. As shown in Figure 5C, deletion of TPK1 in the cbk1Δ/CBK1 background decreases the proportion of pseudohyphae formed by the cbk1Δ/CBK1 mutant, while deletion of TPK2 has no effect (data not shown), suggesting that the increased proportion of pseudohyphae formed by cbk1Δ/CBK1 is dependent on TPK1. The phenotypic differences evident upon deleting the two isoforms of PKA are consistent with previous data indicating that they have distinct and redundant roles in filamentation [46].
Interestingly, cultures of cbk1Δ/CBK1 tpk1Δ/Δ in SM contained significant numbers of filaments that showed characteristics of both pseudohyphae and hyphae (Figure 5D). This hybrid morphology was not observed in cultures of wild type, cbk1Δ/CBK1, or tpk1Δ/Δ cells. Similar hyphae-pseudohyphae hybrid morphologies were recently observed by Carlisle et al. in cells expressing an intermediate level of UME6 [47], suggesting that concurrent disruption of both RAM and PKA pathways interferes with the ability of the cell to commit to one morphotype. These observations also suggest that a balance between the activities of the PKA and RAM pathway is required for normal morphogenesis.
Increased and/or dysregulated PKA pathway activity has been linked previously to increased pseudohyphae formation. For example, Tebarth et al. have shown that overexpression of EFG1 induces constitutive pseudohyphae [48]. We, therefore, hypothesized that elevated PKA activity might be responsible for the constitutively pseudohyphal phenotype displayed by ace2Δ/Δ as well as the increased proportion of pseudohyphae observed with cbk1Δ/CBK1 heterozygotes showing CHI. Three observations support this hypothesis. First, treatment of ace2Δ/Δ cells with the substrate-based PKA inhibitor MyrPKI [49], under non-inducing conditions, significantly increased the number of yeast-like cells and decreased the number of mature pseudohyphae (Figure 6A), strongly supporting the notion that increased PKA activity is involved in the constitutive pseudohyphal phenotype of ace2Δ/Δ. Second, EFG1 expression is elevated in both RAM pathway mutants and cbk1Δ heterozygotes relative to wild type over the time course of hyphal induction (Figure 6B). Densitometric analysis of three replicates of the 180 min time point indicates that the EFG1 levels are 2–4 fold higher in each of the mutants relative to wild type (p<0.02, Student's t test). To further confirm this elevation, we compared the levels of EFG1 in wild type and the double heterozygote cbk1Δ/CBK1 pgk1Δ/PGK1. Consistent with the semi-quantitative data, EFG1 is elevated in cbk1Δ/CBK1 pgk1Δ/PGK1 relative to wild type (4.8 log2, std. dev. 0.9, p = 0.01, Student's t test). Third, deletion of both alleles of EFG1 in the cbk1Δ/CBK1 background decreases expression of ENO1 by a modest 1.5-fold and PGK1 a more significant 8-fold relative to the parental strain (Figure 6C), indicating that at least a portion of the increased expression of putatively co-regulated genes in RAM mutants is mediated by the PKA-Efg1 pathway. Taken together, these experiments suggest that some of the CBK1-interacting genes isolated in our screen are part of the transcriptional output of both the PKA and RAM pathways and that decreased RAM function in the CBK1 double heterozygotes leads to a compensatory increase in PKA pathway activity which, in turn, manifests as a phenotype of increased pseudohyphal growth due to increased EFG1 levels [45].
Figure 6. Elevated PKA activity accounts for increase pseudohyphae in RAM mutants.

(A) ace2Δ/Δ cells were incubated in YPD at 30°C for 3 h −/+ PKA inhibitor MyrPKI (10 µM) and examined by light microscopy. (B) EFG1 expression was determined by semi-quantitative RT-PCR for each strain at the indicated time after transfer to SM at 37°C. ACT1 levels were used as loading control. The graph indicates the fold change in EFG1 levels for the mutant strains relative to wild type at the 180 min time point. The bars indicate the mean fold change in EFG1 relative to wild type for three independent replicates and the error bars indicate standard deviation. The brackets indicate that the difference between EFG1 transcript levels was statistically significant for each mutant relative to wild type (Student's t test, p<0.02). (C) The expression of ENO1 and PGK1 were examined in the indicated strains as described in Figure 5A. The brackets indicate that the difference between PGK1 transcript levels was statistically significant for the two mutants (Student's t test, p<0.02).
A balance between RAM and PKA pathwzay activity is required for normal morphogenesis
Although our results strongly suggest that the RAM and PKA pathways interact during morphogenesis and that the PKA pathway may be hyper-activated in the absence of RAM activity, it remained to be determined how these pathways interact during normal morphogenesis. As discussed above, one of the best characterized functions of Ace2 in both S. cerevisiae and C. albicans is as a daughter cell-specific transcription factor [26], [38], [39]. Two other laboratories [26], [39] have previously shown that in C. albicans, Ace2 localizes to daughter nuclei in actively dividing yeast-phase cells as well as in serum-induced filaments; our results confirm those findings in SM (Figure 7A). We, therefore, hypothesized that the relative contributions of Ace2 and Efg1 to gene regulation during the course of hyphal development may correspond to the timing of their nuclear localization. To our knowledge, the nuclear localization of Efg1 during filamentation had not been described previously.
Figure 7. Efg1 and Ace2 are present in the nuclei at different time points during morphogenesis.

(A) The localization of Ace2-GFP was determined in stationary phase cells prior to initiation of hyphal induction (yeast form) and after 3 hr hyphal induction in SM. DAPI staining was used to identify the nuclei. (B) The localization of Efg1-Myc was determined by indirect immunofluorescence under conditions identical to those described for Ace2-GFP. (C) The binding of Efg1-Myc to promoter regions of ENO1 and PGK1 was examined by ChIP for cells corresponding to the time points examined in A and B. (D) ACE2 expression in wild type (WT) cells compared to ACT1 by RT-PCR at the indicated times after hyphal induction in SM at 37°C. (E) ACE2 expression was determined in wild type cells −/+ PKA inhibitor (MyrPKI, 10 µM) after 3 h induction in SM at 37°C.
To test this hypothesis, we used indirect immunofluorescence to compare the proportion of cells with nuclear Efg1 at the initiation of hyphal development to the proportion in hyphal cell nuclei. As shown in Figure 7B, Efg1 is present in the nuclei of 50–60% (n = 100 cells) of cells prior to shifting to SM. In contrast, Efg1 is detectable in only ∼10% of hyphal nuclei. Correspondingly, Efg1 occupancy of the promoter regions of ENO1 and PGK1 is also higher at the initiation of hyphal development by ChIP analysis (Figure 7C). This suggests that Efg1 may be more important at the onset of, or early in, the filamentous transition, while Ace2 contributes to Efg1/Ace2 co-regulated gene transcription as daughter cell nuclei accumulate within the hyphal structure.
Consistent with this model, ACE2 expression increases over the 3-hour time course of hyphal induction (Figure 7D); this finding is also consistent with its role in gene expression within daughter cell nuclei. Interestingly, the promoter region of ACE2 has five Efg1 consensus binding sites, suggesting that the PKA pathway may contribute to the regulation of ACE2 expression. However, treatment with the PKA inhibitor MyrPKI reduced levels of ACE2 expression only modestly after 3 hours in SM (Figure 7E). Although this observation supports a possible direct link between the PKA and RAM pathways, it suggests that PKA-Efg1 is not the sole, or even dominant, regulator of ACE2 expression.
As a whole, these data support a model in which Efg1 plays a more important role at the initiation of hyphal development in SM, and Ace2 plays a more important role once daughter nuclei accumulate within the hyphal structure. Since EFG1 expression is maintained throughout the time course of hyphal development (Figure 6B) and Efg1 is present in some hyphal nuclei (Figure 7A), it is unlikely that the relationship between Ace2 and Efg1 represents an “either/or” type of scenario. Instead, it seems more likely that a balance exists between the relative contributions of the two transcription factors to gene expression and that this balance varies during hyphal development.
Discussion
Methods for the large-scale genetic analysis of Candida albicans have advanced tremendously in recent years, leading to a number of important and informative studies [14]–[20]. To our knowledge, however, no large-scale synthetic genetic analyses have yet been reported. Here, we present the first such screen. Our approach was based on a CHI strategy, and, like other large-scale genetic analyses of C. albicans, we employed transponson-mediated insertional mutagenesis to generate a large collection of double heterozygous mutants derived from a parental strain containing a heterozygous null mutation of the RAM pathway kinase CBK1. This library was then used to screen for genes that interacted with CBK1 during SM-induced morphogenesis.
First and foremost, our data establishes that CHI-based genetic interaction screening is a useful method for the genetic analysis of the obligate diploid yeast C. albicans. A priori, CHI-based genetic screening of a signaling network such as the RAM pathway would be expected to identify genes that interact with the query gene through a variety of mechanisms. Inspection of our dataset confirms these expectations in that it includes transcriptional targets of the RAM pathway (e.g., ENO1, PGK1), genes that likely affect the function of pathway components (e.g., NSP1, SLK19), and genes that function in parallel pathways (e.g., MAF1, SLF1). In the specific case of screening a protein kinase mutant, it should also be possible to identify substrates of that kinase. Although no bona fide substrate of Cbk1 has been confirmed in C. albicans, our screen identified a very likely candidate in Ssd1. Ssd1 is a well characterized Cbk1 substrate in S. cerevisiae [34] and has been shown previously to interact genetically with CBK1 in both S. cerevisiae [35] and C. albicans [27]. A consensus Cbk1 phosphorylation sequence has recently been identified in S. cerevisiae [34]. Supporting the possibility that CaSsd1 is a substrate of CaCbk1 is the presence of this consensus phosphorylation sequence. Of the remaining CBK1-interactors, RGD3, an uncharacterized potential Rho GTPase, and VPS13, a protein involved in vacuolar protein sorting, also have sequences that match the consensus phosphorylation sequence for ScCbk1 (data not shown). Studies directed towards confirming these putative Cbk1 substrates are in progress.
The CBK1-derived double heterozygous mutants isolated in our screen displayed phenotypes indicative of defects in the Ace2-dependent functions of the RAM pathway in that they were only observed on SM [25]; mutations in genes affecting Ace2-independent functions would be expected to display filamentation defects on both SM and serum [27]. Since many of the interacting genes appear to be transcriptional targets of Ace2, we propose that the effect of partially disabling the RAM pathway by deletion of one allele of CBK1 is exacerbated by further deletion of one allele of a gene regulated by the CBK1-dependent transcription factor Ace2. The cumulative effect of these two mutations results in phenotypes (increased proportion of pseudohyphae) consistent with a further decrease in Ace2-mediated RAM transcriptional activity. By this analysis, Ace2-transcriptional targets that display CHI interactions with CBK1 would, therefore, appear to be particularly important components of the transcriptional output of the RAM pathway during morphogenesis on SM.
A particularly powerful feature of synthetic genetic analysis is the ability to identify interactions between regulatory pathways and, in this regard, our CHI screen of cbk1Δ/CBK1 was quite informative, highlighting the interplay between the RAM and PKA pathways during morphogenesis. Although no components of the PKA signaling pathway were identified as CBK1-interactors, analysis of the dataset revealed that many of the interactors were regulated by the PKA pathway. Indeed, the similar transcriptional characteristics of the PKA-regulated transcription factor Efg1 and Ace2 in C. albicans have been previously noted [50] and, while our work was in progress, Wang et al. reported that Efg1 and Ace2 bound to the promoters of C. albicans genes involved in cell separation [39]. In addition, the PKA and RAM pathways have been linked genetically in S. cerevisiae through experiments showing that ectopic over-expression of the PKA kinase subunit TPK1 suppresses growth and budding defects of RAM pathway mutants in an Ace2-independent manner [51]. Our data suggest that the PKA and RAM pathway interact in C. albicans with respect to Ace2-dependent functions.
Consistent with this model, consensus binding sites for both Efg1 and Ace2 are located in the promoter regions of a significant proportion of CBK1-interactors. A genome-wide search identified 384 putative Efg1/Ace2 co-regulated genes, suggesting that the two pathways interact to modulate the expression of a substantial subset of genes. The interaction of these two pathways is further supported by our isolation of two PKA-regulated transcriptional modulators (MAF1 & SLF1) as CBK1 interactors as well as by the synthetic genetic interactions between CBK1 and TPK1 observed in our follow-up studies.
The simplest manifestation of a model in which the PKA and RAM pathways co-regulate a set of genes would be that deletion of either ACE2 or EFG1 results in the decreased expression of co-regulated genes. As shown in Figure 5A, this is not the case as the expression of putatively co-regulated genes is increased in both ace2Δ/Δ and efg1Δ/Δ mutants. This suggested that the two pathways may compensate for one another when the other pathway is disabled. Supporting this notion, the activity of the PKA pathway is increased in RAM pathway mutants (Figure 5B), and EFG1 mediates a substantial portion of the increased expression of co-regulated genes in the absence of full RAM pathway activity (Figure 6B). Accordingly, the level of EFG1 expression is also increased (Figure 6B) and, since inappropriately high levels of EFG1 promote pseudohyphal growth (48), this observation provides an explanation for the increased amounts of pseudohyphae displayed by RAM pathway mutants.
We, therefore, propose that the increased PKA activity in RAM pathway mutants represents a compensatory response that maintains expression of Ace2/Efg1 co-regulated genes in the absence of a fully functional RAM pathway. However, constitutively elevated levels of PKA activity represent a dysregulated state and, consequently, the expression levels of the genes are not returned to normal but are elevated. Thus, it appears that a balance between the activity of the PKA and RAM pathways is required to maintain properly regulated expression of co-regulated genes. Maintaining a balance between the activities of the two pathways appears to be required for normal hyphal development because: 1) loss of EFG1 leads to a failure to form filaments; 2) loss of ACE2 leads to the accumulation of pseudohyphae; and 3) concurrent partial disruption of both pathways leads to the formation of filaments with characteristics of both hyphae and pseudohyphae (Figure 5D).
If, as our results suggest, a balance between PKA and RAM pathway-mediated transcription is required for the cell to normally undergo filamentation, then how is this balance established and maintained? Although further work will be required to determine the molecular mechanism of this interaction, the cell cycle-regulated nature of both Efg1 and Ace2 suggests that the pathways might be active at different times during morphogenesis. Ace2, for example, localizes to the nuclei of daughter cells in both yeast and filamentous C. albicans [26], [39]. Efg1, on the other hand, has been shown to be rapidly down-regulated soon after hyphal induction in some conditions [48]. These considerations led us to propose that Efg1 may be more important in the expression of co-regulated genes earlier in morphogenesis, while Ace2 is the dominant regulator later in morphogenesis when daughter nuclei appear within the filament.
Consistent with that model, we showed that more nuclei contain Efg1 at the initiation of morphogenesis than later in the process. Ace2, on the other hand, is absent from the vast majority of nuclei at the initiation of morphogenesis but is found in daughter nuclei as they accumulate within the filament (Figure 7A). Consistent with its role later in morphogenesis, overall expression of ACE2 also increases as the cells are exposed to inducing condition for longer periods of time (Figure 7D). Since Efg1 remains detectable in hyphal nuclei (Figure 7B), it is unlikely that Ace2 replaces Efg1 entirely but rather Ace2 may become relatively more important as daughter nuclei accumulate within the filament and undergo mitosis. Thus, it seems that a balance between the PKA and RAM pathways exists and that this balance is important for smooth morphogenesis. A potential illustration of the importance of this balance is provided by the morphologies displayed by the tpk1Δ/Δ cbk1Δ/CBK1 mutant in which single filaments show characteristics of both hyphae and pseudohyphae.
This model is also consistent with the observations of Wang et al., who reported that Efg1 represses the expression of Ace2-regulated cell separation genes during hyphal development [39]. They found that in wild type strains, the Ace2-regulated expression of chitinase CHT3 occurs approximately 3 hours post-hyphal induction, a point at which multiple septa and daughter nuclei have formed within the hyphal filament. The 3-hour time point also corresponds to the time when we observed high levels of ACE2 expression. In EFG1 mutants, on the other hand, Wang et al. found that CHT3 is inappropriately expressed within the first hour of induction and is expressed at higher levels at 3 hours [39]. Our observations regarding the timing of Efg1 nuclear localization correlate well with these expression data in that Efg1 is present early when it suppresses Ace2-mediated CHT3 expression but is absent when CHT3 expression is induced. It is important to note that Efg1 has previously been proposed to function as both a transcriptional activator and repressor during hyphal morphogenesis [39], [41] and, taken together with the observations of Wang et al., our data are consistent with such a role.
At this point, further work will be required to understand the molecular mechanisms by which the RAM and PKA pathway interact. As noted above, ACE2 does possess a number of Efg1 consensus binding sites within its promoter. This suggests a possible feed-forward mechanism by which Efg1 activates the expression of ACE2, which, in turn, takes over transcription of co-regulated genes. However, chemical inhibition of the PKA pathway only modestly reduced expression of ACE2 during hyphal induction (Figure 7E). Similarly, efg1Δ/Δ mutants also exhibit very slight changes in ACE2 expression (data not shown). Although there may be an operative component of this feed-forward mechanism, it seems to be a relatively minor contributor to the crosstalk between these pathways.
In summary, we have shown that CHI-based genetic interaction screening is a useful approach for the analysis of complex phenotypes in C. albicans. The application of this approach to the RAM pathway has provided insights into the mechanisms by which the PKA and RAM signaling pathways function together during the transition from yeast to filamentous cells in C. albicans.
Materials and Methods
Strains, media, and growth conditions
All strains are derived from CAI4 (ura3Δ::imm434/ura3Δ::imm434). CAMM-292 (ura3Δ::imm434/ura3Δ::imm434/cbk1-Δ1::hisG/CBK1) [24] was used as the parental strain for transposon mutagenesis. A complete list of strains and genotypes is provided in Table S1. Yeast peptone dextrose supplemented with 80 mg/L uridine, synthetic dextrose medium lacking uracil, and SM were prepared using standard recipes [15], [52]. Induction of filamentation was carried out using SM plates (37°C, 3D) or liquid SM (37°C, 3 h). All phenotypes were confirmed on SM plates supplemented with uracil to control for possible positional effects of URA3 expression. Proportions of yeast, pseudohyphae and hyphae in liquid cultures were determined by light microscopy using morphological scoring criteria described by Sudbery et al. [53].
Transposon mutagenesis
C.albicans strain WO-1 pEMBLY23 genomic DNA library (NIH AIDS Research & Reference Reagent Program) was mutagenized (9 independent reactions) in vitro using the GPS3-Mutagenesis system from New England Biolabs (Beverly, MA) and a donor plasmid (pGPS3) containing the CaURA3-dpl200 cassette [16] inserted at the Spe I restriction site. Mutagenized genomic fragments were released by PvuII digestion and transformed into CAMM-292 using a lithium acetate-protocol with heat shock at 44°C for 20 min [54]. The library is available upon request from the Kumar laboratory (akumar@umich.edu).
Identification of transposon insertion sites
Transposon insertion sites were amplified by 3′ RACE (rapid amplification of cDNA ends) using primers complementary to the ends of the transposon construct, cloned into a TA vector, and sequenced. Insertion sites were then identified by BLASTN searches using the Candida Genome Database (www.candidagenome.org).
Construction of cbk1Δ/CBK1-derived double heterozygotes
Ten double heterozygotes that showed CHI were independently constructed from the Ura- parental strain cbk1Δ/CBK1 (CAMM292) using fusion PCR methods to generate URA3-based knockout cassettes [33]. The cassettes were used to transform CAMM292 to Ura prototrophy, and correct integration was confirmed by PCR. Two independent isolates were evaluated for all phenotypes.
qRT-PCR and Chromatin Immunoprecipitation assays
Total RNA was isolated using the RiboPure Yeast Kit (Ambion, Austin, TX) and reverse transcribed using the SuperScript III First Strand Synthesis Kit (Invitrogen, Carlsbad, CA). Changes in transcript levels of target genes were analyzed using the Platinum SYBR Green Mix (Invitrogen) and normalized to ACT1 levels using the 2−ΔΔCt method [55]. ChIP assays were performed as described previously [56] using Ura+ CAI4-dervatives containing ACE2-TAP and EFG1-MYC alleles.
Protein kinase A assay
Protein kinase A activity was measured in total cell lysates using the PepTag cAMP-dependent protein kinase assay kit (Promega, Madison WI) following a protocol previously developed for C. albicans [57]. Lysates were prepared from wild type and ace2Δ/Δ cells that had been exposed to SM for 3 h. Phosphorylation of the PepTag substrate was determined by agarose gel electrophoresis; the unphosphorylated substrate migrates in the opposite direction as the phosphorylated substrate. Images of the gel were captured on a gel-doc imaging system and processed using Adobe PhotoShop software. Identical contrast and levels were used for each image.
Microscopy
Light and fluorescence microscopy was performed using a Nikon ES80 epi-fluorescence microscope equipped with a CoolSnap CCD camera. Images were collected using NIS-Elements Software and processed in PhotoShop. Indirect immunofluorescence was performed as previously described using anti-Myc (Invitrogen) primary- and TexasRed-conjugated (Molecular Probes) secondary- antibodies [58]. DAPI and Calcofluor white staining was performed as described [52].
Supporting Information
Strains.
(DOC)
Table of GO terms, number of genes per GO category, p-values and example ORFs containing both Ace2 (MMCCASC) and Efg1 (CANNTG) binding sites within 1000 bp of the start codon. ORFs were identified by searching the CGD database (www.candidagenome.org) and analyzed using GO toolbox statistical software (http://genome.crg.es/GOToolBox/).
(DOC)
Acknowledgments
We thank Aaron Mitchell, William Fonzi, Joachim Ernst, Joe Bliss, Judy Berman, Carlos Vasquez, Alison Butler, and Melanie Wellington for strains, reagents, and/or helpful discussions.
Footnotes
The authors have declared that no competing interests exist.
This work was supported by National Institute of Allergy and Infectious Diseases grants 5R21AI084539 (to DJK and AK) and 5T32AI007464 (to YC-R), as well as by an American Cancer Society grant RSG-06-179-01-MBC (to AK). The funders had no role in study design, data collection and analysis, and decision to publish or preparation of the manuscript.
References
- 1.Vazquez JA, Sobel JD. Candidiasis. In: Dismukes WE, Pappas PG, Sobel JD, editors. Clinical Mycology. Oxford: Oxford University Press; 2003. pp. 143–188. [Google Scholar]
- 2.Lo HJ, Kohler JR, DiDomenico B, Loebenber D, Cacciapuoti A, et al. Nonfilamentous C. albicans mutants are avirulent. Cell. 1997;90:939–949. doi: 10.1016/s0092-8674(00)80358-x. [DOI] [PubMed] [Google Scholar]
- 3.Kumamoto CA, Vinces MD. Contributions of hyphae and hypha-co-regulated genes to Candida albicans virulence. Cell Microbiol. 2005;7:1546–1554. doi: 10.1111/j.1462-5822.2005.00616.x. [DOI] [PubMed] [Google Scholar]
- 4.Saville JP, Lazzell AL, Monteagudo C, Lopez-Ribot JL. Engineered control of cell morphology in vivo reveals distinct roles for yeast and filamentous forms of Candida albicans during infection. Eukaryot Cell. 2003;2:1053–1060. doi: 10.1128/EC.2.5.1053-1060.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noble SM, French S, Kohn LA, Chen V, Johnson AD. Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nat Genet. 2010;42:590–598. doi: 10.1038/ng.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whiteway M, Bachewich C. Morphogenesis in Candida albicans. Annu Rev Microbiol. 2007;61:529–553. doi: 10.1146/annurev.micro.61.080706.093341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hall RA, Cottier F, Muhlschlegel FA. Molecular networks in the fungal pathogen Candida albicans. Adv Appl Microbiol. 2009;67:191–212. doi: 10.1016/S0065-2164(08)01006-X. [DOI] [PubMed] [Google Scholar]
- 8.Tong AH, Boone C. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol Biol. 2006;313:171–192. doi: 10.1385/1-59259-958-3:171. [DOI] [PubMed] [Google Scholar]
- 9.Jin R, Dobry CJ, McGowan PJ, Kumar A. Large-scale analysis of yeast filamentous growth by systematic gene disruption and overexpression. Mol Biol Cell. 2008;19:284–296. doi: 10.1091/mbc.E07-05-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon SJ, Costanzo M, Baryshnikov A, Andrews B, Boone C. Systematic mapping of genetic interaction networks. Annu Rev Genet. 2009;43:601–625. doi: 10.1146/annurev.genet.39.073003.114751. [DOI] [PubMed] [Google Scholar]
- 11.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braun BR, Johnson AD. TUP1, CPH1, and EFG1 make independent contributions to filamentation in Candida albicans. Genetics. 2000;155:57–67. doi: 10.1093/genetics/155.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noble SM, Johnson AD. Genetics of Candida albicans, a diploid human fungal pathogen. Annu Rev Genet. 2007;41:193–211. doi: 10.1146/annurev.genet.41.042007.170146. [DOI] [PubMed] [Google Scholar]
- 14.Bruno VM, Mitchell AP. Large-scale gene function analysis in Candida albicans. Trends Microbiol. 2004;12:157–161. doi: 10.1016/j.tim.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 15.Uhl MA, Biery M, Craig N, Johnson AD. Haploinsufficiency-based large scale forward genetic analysis of filamentous growth in the diploid fungal pathogen C. albicans. EMBO J. 2003;22:2668–2678. doi: 10.1093/emboj/cdg256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oh J, Fung E, Schlecht U, Davis RW, Giaver G, et al. Gene annotation and drug target discovery in Candida albicans with a tagged transposon mutant collection. PLoS Path. 2010;6:e1001140. doi: 10.1371/journal.ppat.1001140. doi: 10.1371/journal.ppat.1001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis DA, Bruno VM, Loza L, Filler SG, Mitchell AP. Candida albicans Mds3p, a conserved regulator of pH responses and virulence identified through insertional mutagenesis. Genetics. 2002;162:1753–1581. doi: 10.1093/genetics/162.4.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Epp E, Walther A, Lepine G, Leon Z, Mullick A, et al. Forward genetics in Candida albicans that reveals the Arp2/3 complex is required for hyphal formation but not endocytosis. Mol Microbiol. 2010;75:1182–1198. doi: 10.1111/j.1365-2958.2009.07038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Homann OR, Dea J, Noble SM, Johnson AD. A phenotypic profile of the Candida albicans regulatory network. PLoS Genet. 2009;5:e1000783. doi: 10.1371/journal.pgen.1000783. doi: 10.1371/journal.pgen.1000783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Becker JM, Kauffman SJ, Hauser M, Huang L, Lin M, et al. Pathway analysis of Candida albicans survival and virulence determinants in a murine infection model. Proc Nat Acad Sci USA. 2010;107:22044–22049. doi: 10.1073/pnas.1009845107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haarer B, Viggiano S, Hibbs MA, Troyanskaya OG, Amberg DC. Modeling complex genetic interactions in a simple eukaryotic genome: actin displays a rich spectrum of complex haploinsufficiencies. Genes Dev. 2007;21:148–159. doi: 10.1101/gad.1477507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stearns T, Botstein D. Unlinked noncomplementation: isolation of new conditional lethal mutantion in each of the tubulin genes of Saccharomyces cerevisiae. Genetics. 1988;119:249–260. doi: 10.1093/genetics/119.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vinh DB, Welch MD, Corsi AK, Wertman KF, Drubin DG. Genetic evidence for functional interactions between noncomplementing (Anc) gene products and actin cytoskeletal proteins in Saccharomyces cerevisiae. Genetics. 1993;135:275–286. doi: 10.1093/genetics/135.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McNemar MD, Fonzi WA. Conserved serine/threonine kinase encoded by CBK1 regulates expression of several hypha-associated transcripts and genes encoding cell wall proteins in Candida albicans. J Bacteriol. 2002;184:2058–2061. doi: 10.1128/JB.184.7.2058-2061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelly MT, MacCallum DM, Clancy SD, Odds FC, Brown AJ, et al. The Candida albicans CaACE2 gene affects morphogenesis, adherence, and virulence. Mol Microbiol. 2004;53:969–983. doi: 10.1111/j.1365-2958.2004.04185.x. [DOI] [PubMed] [Google Scholar]
- 26.Mulhern SM, Logue ME, Butler G. Candida albicans transcription factor Ace2 regulates metabolism and is required for filamentation in hypoxic conditions. Eukaryot Cell. 2006;5:2001–2013. doi: 10.1128/EC.00155-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song Y, Cheon SA, Lee KE, Lee S-Y, Lee B-K, et al. Role of the RAM network in cell polarity and hyphal morphogenesis in Candida albicans. Mol Biol Cell. 2008;19:5456–5477. doi: 10.1091/mbc.E08-03-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelson B, Kurischko C, Horecka J, Mody M, Nair L, et al. RAM: a conserved signaling network that regulates Ace2p transcriptional activity and polarized morphogenesis. Mol Biol Cell. 2003;14:3782–3803. doi: 10.1091/mbc.E03-01-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biery MC, Stewart FJ, Stellwagen AE, Ralidgh EA, Craig NL. A simple in vitro Tn7-based transposition system with low target site selectivity for genome and gene analysis. Nucleic Acids Res. 2000;28:1067–1077. doi: 10.1093/nar/28.5.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar A, Seringhaus M, Biery MC, Sarnovsky RJ, Umansky L, et al. Large-scale mutagenesis of yeast genome using a Tn7-derived multipurpose transposon. Genome Res. 2004;14:1975–1986. doi: 10.1101/gr.2875304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seringhaus M, Kumar A, Hartigan J, Snyder M, Gerstein M. Genomic analysis of insertion behavior and target specificity of mini-Tn7 and Tn3 transposons in Saccharomyces cerevisiae. Nuclei Acid Res. 2006;34:e57. doi: 10.1093/nar/gkl184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson RB, Davis D, Enloe BM, Mitchell AP. A recyclable Candida albicans URA3 cassette for PCR product-directed gene disruptions. Yeast. 2000;16:65–70. doi: 10.1002/(SICI)1097-0061(20000115)16:1<65::AID-YEA508>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 33.Noble SM, Johnson AD. Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot Cell. 2005;4:298–309. doi: 10.1128/EC.4.2.298-309.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jansen JM, Wanless AG, Seidel CW, Weiss EL. Cbk1 regulation of the RNA-binding protein Ssd1 integrates cell fate with translational control. Curr Biol. 2009;19:2114–2120. doi: 10.1016/j.cub.2009.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jorgensen P, Nelson B, Robinson MD, Chen Y, Andrews B, et al. High-resolution genetic mapping with ordered arrays of Saccharomyces cerevisiae deletion mutants. Genetics. 2002;162:1091–1099. doi: 10.1093/genetics/162.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ross KE, Cohen-Fix O. The role of Cdh1 in maintaining genomic stability in budding yeast. Genetics. 2003;165:489–503. doi: 10.1093/genetics/165.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Havens KA, Gardner MK, Kamieniecki RJ, Dresser ME, Dawson DS. Slk19 regulates spindle dynamics through two independent mechanisms. Genetics. 2010;186:1247–1260. doi: 10.1534/genetics.110.123257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parnell EJ, Stillman DJ. Betting a transcription factor to only one nucleus following mitosis. PLoS Biol. 2008;6:e229. doi: 10.1371/journal.pbio.0060229. doi: 10.1371/journal.pbio.0060229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang A, Raniga PP, Lane S, Lu Y, Liu H. Hyphal chain formation in Candida albicans: Cdc28-Hgc1 phosphorylation of Efg1 represses cell separation genes. Mol Cell Biol. 2009;29:4406–4416. doi: 10.1128/MCB.01502-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dihlmann MA, Herth W, Hurt EC. NSP1 depletion in yeast affects nuclear pore formation and nuclear import. Eur J Cell Biol. 1992;59:280–295. [PubMed] [Google Scholar]
- 41.Doedt T, Krishnamurthy S, Bockmuhl DP, Tebarth B, Stempel C, et al. APES proteins regulate morphogenesis and metabolism in Candida albicans. Mol Biol Cell. 2004;15:3167–3180. doi: 10.1091/10.1091/mbc.E03-11-0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willis IM, Moir RD. Integration of nutritional and stress signaling pathways by Maf1. Trends Biochem Sci. 2006;32:51–53. doi: 10.1016/j.tibs.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 43.Robertson LS, Fink GR. Three yeast A kinases have specific signaling functions in pseudohyphal growth. Proc Nat Acad Sci USA. 1998;95:13783–13787. doi: 10.1073/pnas.95.23.13783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolyniak MJ, Sundstrom P. Role of actin cytoskeletal dynamics in activation of the cyclic AMP pathway and HWP1 gene expression in Candida albicans. Eukaryot Cell. 2007;6:1824–1840. doi: 10.1128/EC.00188-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zou H, Fong H-M, Zhu Y, Wang Y. Candida albicans Cyr1, Cap1 and G-actin form a sensor/effector apparatus for activating cAMP synthesis in hyphal growth. Mol Microbiol. 2010;75:579–591. doi: 10.1111/j.1365-2958.2009.06980.x. [DOI] [PubMed] [Google Scholar]
- 46.Bockmuhl DP, Krishnamurthy S, Gerads M, Sonneborn A, Ernst JF. Distinct and redundant roles of the two protein kinase A isoforms Tpk1p and Tpk2p in moprhogenesis and growth of C. albicans. Mol Microbiol. 2001;42:1243–1257. doi: 10.1046/j.1365-2958.2001.02688.x. [DOI] [PubMed] [Google Scholar]
- 47.Carlisle PL, Banerjee M, Lazzell A, Monteagudo C, Lopez-Ribot JL, et al. Expression levels of a filament-specific transcriptional regulator are sufficient to determine Candida albicans morphology and virulence. Proc Natl Acad Sci USA. 2009;106:599–604. doi: 10.1073/pnas.0804061106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tebarth B, Doedt T, Krishnamurthy S, Weide M, Monterola F, et al. Adaptation of the morphogenetic pathway in Candida albicans by negative autoregulation and PKA-dependent repression of the EGF1 gene. J Mol Biol. 2003;329:949–962. doi: 10.1016/s0022-2836(03)00505-9. [DOI] [PubMed] [Google Scholar]
- 49.Cloutier M, Castilla R, Bolduc N, Zelada A, Martineau P, et al. The two isoforms of cAMP-dependent protein kinase catalytic subunit are involved in the control of dimorphism in the human fungal pathogen Candida albicans. Fungal Genet Biol. 2003;38:133–141. doi: 10.1016/s1087-1845(02)00520-0. [DOI] [PubMed] [Google Scholar]
- 50.Ernst JF, Tielker D. Response to hypoxia in fungal pathogens. Cell Microbiol. 2009;11:183–190. doi: 10.1111/j.1462-5822.2008.01259.x. [DOI] [PubMed] [Google Scholar]
- 51.Schneper L, Krauss A, Miyamoto, Fang S, Broach JR. The ras/protein kinase A pathway acts in parallel with the Mob2/Cbk1 pathway to effect cell cycle progression and proper bud site selection. Eukaryot Cell. 2004;3:108–120. doi: 10.1128/EC.3.1.108-120.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burke DJ, Dawson D, Stearns T. Methods in Yeast Genetics. Woodville NY: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- 53.Sudbery P, Gow N, Berman J. The distinct morphogenic states of Candida albicans. Trends Microbiol. 2004;12:317–324. doi: 10.1016/j.tim.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 54.Walther A, Wendland J. An improved transformation protocol for the human fungal pathogen Candida albicans. Curr Genet. 2003;42:339–343. doi: 10.1007/s00294-002-0349-0. [DOI] [PubMed] [Google Scholar]
- 55.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C T method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 56.Xu T, Shively CA, Jin R, Eckwahl MJ, Dobry CJ, et al. A profile of differentially abundant proteins at the yeast cell periphery during pseudohyphal growth. J Biol Chem. 2010;285:15476–15488. doi: 10.1074/jbc.M110.114926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hnisz D, Majer O, Frohner IE, Komnenovic V, Kuchler K. The Set3/Hos2 histone deacetylase complex attenuates cAMP/PKA signaling to regulate morphogenesis and virulence of Candida albicans. PLoS Pathog. 2010;6:e1000889. doi: 10.1371/journal.ppat.1000889. doi: 10.1371/journal.ppat.1000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kumar A, Agarwal S, Heyman JA, Matson S, Heidtman M, et al. Subcellular localization of the yeast proteome. Genes Dev. 2002;16:707–719. doi: 10.1101/gad.970902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Strains.
(DOC)
Table of GO terms, number of genes per GO category, p-values and example ORFs containing both Ace2 (MMCCASC) and Efg1 (CANNTG) binding sites within 1000 bp of the start codon. ORFs were identified by searching the CGD database (www.candidagenome.org) and analyzed using GO toolbox statistical software (http://genome.crg.es/GOToolBox/).
(DOC)