

Abstract
Gsα is a ubiquitously expressed G protein α-subunit that couples receptors to the generation of intracellular cyclic AMP. The Gsα gene GNAS is a complex gene that undergoes genomic imprinting, an epigenetic phenomenon that leads to differential expression from the two parental alleles. Gsα is imprinted in a tissue-specific manner, being expressed primarily from the maternal allele in a small number of tissues. Albright hereditary osteodystrophy is a monogenic obesity disorder caused by heterozygous Gsα mutations but only when the mutations are maternally inherited. Studies in mice indicate a similar parent-of-origin effect on energy and glucose metabolism, with maternal but not paternal mutations leading to obesity, reduced sympathetic nerve activity and energy expenditure, glucose intolerance and insulin resistance, with no primary effect on food intake. These effects result from Gsα imprinting leading to severe Gsα deficiency in one or more regions of the central nervous system, and are associated with a specific defect in melanocortins to stimulate sympathetic nerve activity and energy expenditure.
Index words: G protein, genomic imprinting, obesity, insulin resistance, sympathetic nervous system, melanocortin
1. Introduction
The heterotrimeric G proteins are a large family of membrane-associated proteins that couple with seven transmembrane receptors to transmit signals to the intracellular compartment. Each G protein is defined by its specific α subunit and is composed of α, β and γ subunits that are the product of separate genes. The G protein Gs contains Gsα, stimulatory α-subunit that couples cell surface receptors to adenylyl cyclase and mediates receptor-stimulated intracellular cAMP generation. Gsα is encoded by GNAS, a gene that is affected by genomic imprinting, and therefore heterozygous Gsα mutations in both humans and mice lead to effects on energy and glucose metabolism that are dependent on the parent-of-origin of the mutation (Weinstein et al., 2007). Recent studies show that this is due to Gsα imprinting in the central nervous system which leads to specific impairment on the actions of central melanocortins. In this paper we will review the evidence for Gsα imprinting effects on metabolic regulation and melanocortin action.
2. Gsα signaling and function
Gsα, a product of the imprinted GNAS gene, is a ubiquitously expressed G protein α-subunit which couples many seven-transmembrane receptors for hormones, neurotransmitters, and other extracellular stimuli to the stimulation of adenylyl cyclase and the generation of intracellular cyclic AMP (Weinstein et al., 2007). cAMP mediates its effects primarily by activation of protein kinase A, a serine/threonine protein kinase which was classically known to phosphorylate enzymes and other factors to stimulate release of glucose and free fatty acids into the circulation through increased gluconeogenesis, glycogenolysis, and lipolysis, and many other cellular substrates. Protein kinase A also chronically stimulates gene expression via phosphorylation of transcription factors such as cAMP response element binding protein (CREB) (Montminy, 1997). In addition, Gsα/cAMP mediates the effects of sympathetic nerve activity on peripheral tissues such as brown and white adipose tissue, liver, and muscle as this signaling pathway is stimulated by β adrenergic receptors (Bachman et al., 2002). cAMP also mediates some of its actions, particularly in neuroendocrine cells, by stimulating cAMP-regulated guanine nucleotide exchange factors leading to activation of ras-like proteins such as Rap1 (de Rooij et al., 1998). Gsα may also mediate its effects by stimulating other downstream effectors, such as Ca2+ channels (Mattera et al., 1989), and many interact with receptors outside of the seven transmembrane receptor family (Sun et al., 1997).
3. Organization and imprinting of the Gsα gene GNAS
The Gsα gene GNAS and its mouse ortholog Gnas have similar overall organizations and reside within syntenic regions at 20q13.2-13.3 and distal chromosome 2, respectively (Weinstein et al., 2007). GNAS and Gnas also undergo genomic imprinting and have similar overall imprinting patterns. Genomic imprinting is an epigenetic process in which a specific biochemical imprint ‘mark’ (e.g. DNA methylation) is erased in primordial germ cells and then reestablished during oogenesis or spermatogenesis (depending on the specific imprinted gene), resulting in suppression of gene expression from one parental allele (Reik and Walter, 2001). All imprinted genes have one or more regions in which the maternal and paternal alleles are differentially methylated. DNA methylation of a gene promoter region leads to silencing, but in other cases DNA methylation may occur on the transcriptionally active allele when it is outside the promoter region.
GNAS and Gnas generate multiple gene products in addition to Gsα through the use of alternative promoters and first exons that splice on to a set of common downstream exons (Fig. 1). The two most upstream promoters generate transcripts for NESP55 (neuroendocrine-specific protein of 55 kDa) (Hayward et al., 1998b; Kelsey et al., 1999; Peters et al., 1999) and the Gsα isoform XLαs (Hayward et al., 1998a; Kehlenbach et al., 1994; Kelsey et al., 1999; Peters et al., 1999), respectively. NESP55 and XLαs are oppositely imprinted, being expressed only from the maternal and paternal allele, respectively, due to methylation of the respective promoter regions on the opposite parental allele (Hayward et al., 1998b; Peters et al., 1999). NESP55 is a chromogranin-like protein expressed primarily in neuroendocrine cells that is unrelated to the G protein family (Ischia et al., 1997), and studies in both humans and mice suggest that it plays no significant role in metabolic regulation (Liu et al., 2000a; Plagge et al., 2005). XLαs is a Gsα isoform with a long amino-terminal extension encoded by its specific first exon that is expressed in the central nervous system a few other organs, and is capable of also mediating receptor-stimulated cAMP generation (Bastepe et al., 2002). XLαs knockout mice have elevated sympathetic nerve activity and are hypermetabolic with improved glucose metabolism, indicating that XLαs normally plays a role in downregulating sympathetic nerve activity in mice (Plagge et al., 2004; Xie et al., 2006). Alternatively, a truncated form of XLαs (XLN1) highly expressed in neurons (Pasolli et al., 2000) may be a dominant negative regulator of Gsα function. It is unclear whether XLαs plays an important role in metabolic control in humans, as GNAS mutations on the paternal allele that disrupt XLαs do not produce a similar phenotype.
Fig. 1.
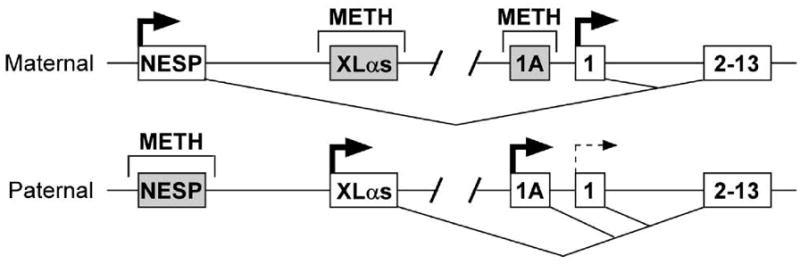
Organization and imprinting of the GNAS gene. The maternal and paternal alleles of the GNAS gene are depicted showing four alternative first exons for NESP55 (NESP), XLαs, 1A mRNA transcripts, and Gsα (exon 1) splicing onto common exons 2 through 13 (shown as a single box). Regions of differential methylation (METH) are shown above and splicing patterns are shown below each allele. Active promoters are shown in white with horizontal arrows while inactive promoters are shown in gray. The thin and dashed arrow for the paternal Gsα promoter indicates that this promoter is suppressed in a tissue-specific manner due to genomic imprinting.
The Gsα promoter is not methylated on either allele (Hayward et al., 1998a; Kozasa et al., 1988; Liu et al., 2000b; Peters et al., 1999). In spite of this, Gsα is imprinted in a tissue-specific manner being expressed primarily from the maternal allele in a number of tissues including pituitary somatotrophs, thyroid, renal proximal tubules, ovaries, and the paraventricular nucleus of the hypothalamus while being biallelically expressed in most other tissues (Campbell et al., 1994; Chen et al., 2009b; Davies and Hughes, 1993; Germain-Lee et al., 2002; Hayward et al., 2001; Liu et al., 2003; Mantovani et al., 2002; Weinstein et al., 2001; Yu et al., 1998).
Just upstream of the Gsα promoter region is an imprint control region (the exon 1A or exon A/B promoter region) (Fig. 1) that is methylated on the maternal allele (Ishikawa et al., 1990; Liu et al., 2000a; Liu et al., 2000b). In pseudohypoparathyroidism type 1B, a condition with renal parathyroid hormone resistance, the exon 1A methylation on the maternal allele is absent (Bastepe et al., 2001; Jan de Beur et al., 2003; Liu et al., 2000a; Liu et al., 2005b). Based upon this observation it has been suggested that this region has a negative regulatory cis-acting element that suppresses the paternal Gsα allele in a tissue-specific manner (Liu et al., 2005a; Williamson et al., 2004). For example, there may be a silencer that binds a tissue-specific repressor protein on the paternal allele, but this repressor fails to bind to the maternal allele due to methylation, allowing Gsα to always be expressed from the maternal allele. Consistent with this, mice with paternal deletion of the exon 1A region had reversal of Gsα imprinting with biallelic expression of Gsα in all tissues (Liu et al., 2005a; Williamson et al., 2004).
4. Parent-of-origin metabolic effects of Gsα mutation in humans and mice
4.1 Role of Gsα in energy balance in humans
Heterozygous Gsα loss-of-function mutations lead to Albright hereditary osteodystrophy, a congenital disorder characterized by the presence of short stature, brachydactyly (shortening of various long bones in the hands and feet), subcutaneous ossifications, and neurobehavioral abnormalities (Weinstein et al., 2006). When the mutation is on the maternal allele patients also develop multihormone resistance to parathyroid hormone, thyrotropin, growth hormone-releasing hormone, and gonadotropins, a condition known as pseudohypoparathyroidism type 1A. In contrast patients with mutations on the paternal allele only develop the features of Albright hereditary osteodystrophy without multihormone resistence, a condition known as pseudopseudohypoparathyroidism (Davies and Hughes, 1993; Weinstein et al., 2006). This is the direct result of tissue-specific Gsα imprinting, as mutation of the active maternal Gsα allele leads to severe Gsα deficiency and hormone resistance whereas mutation of the inactive paternal allele has little effect on Gsα expression or hormone sensitivity(Germain-Lee et al., 2002; Hayward et al., 2001; Liu et al., 2003; Mantovani et al., 2002; Yu et al., 1998).
Albright hereditary osteodystrophy is also a monogenic obesity disorder, with early onset obesity (within the first year) but only in patients with Gsα mutations on the maternal allele (pseudohypoparathyroidism type 1A but not pseudopseudohypoparathyroidism) (Long et al., 2007). Although adipocytes from pseudohypoparathyoidism type 1A patients have reduced lipolytic responses to epinephrine due to reduced Gsα levels (Carel et al., 1999), this is unlikely to be the cause of obesity as there is no evidence for Gsα imprinting in adipose tissue (Chen et al., 2005a; Germain-Lee et al., 2005; Mantovani et al., 2004). A more likely explanation for obesity in pseudohypoparathyroidism type 1A is a defect in the central nervous system leading to low sympathetic nerve activity and metabolic rate, as children with pseudohypoparathyroidism type 1A were shown to have extremely low serum norepinephrine levels when compared to controls or even similarly obese children without pseudohypoparathyroidism type 1A (Carel et al., 1999). In one recent case report, severe obesity developed in a pseudohypoparathyroidism type 1A patient during in the first year of life even in the absence of hyperphagia, also consistent with obesity in this disorder being primarily the result of low energy expenditure (Dekelbab et al., 2009). The incidence of insulin resistance and diabetes in pseudohypoparathyroidism type 1A has not been systematically examined, although severe insulin resistance in a young pseudohypoparathyroidism type 1A patient has been recently reported (Nwosu and Lee, 2009).
Other studies have shown an association of Gsα single nucleotide polymorphisms with obesity or weight loss. For example the silent T393C polymorphism was associated with increased obesity and insulin resistance in German women with polycystic ovarian syndrome, but not in a larger unselected population (Hahn et al., 2006). Another polymorphism within the Gsα promoter was shown to affect binding of the transcriptional factor upstream stimulatory factor 1, Gsα expression, lipolytic rates in adipocytes, and short-term weight loss, but was not associated with differences in baseline obesity (Frey et al., 2008a; Frey et al., 2008b).
4.2 Parent-of-origin metabolic effects of Gsα mutations in mice
Similar to what is observed in pseudohypoparathyroidism type 1A and pseudopseudohypoparathyroidism patients (Long et al., 2007), mutation of the maternal but not the paternal Gsα allele (deletion of Gsα exon 1) leads to severe obesity which is associated with reduced sympathetic nerve activity and energy expenditure, with no primary abnormality in food intake (Chen et al., 2005a; Germain-Lee et al., 2005; Xie et al., 2008). In addition these mice also develop glucose intolerance, insulin resistance and hyperlipidemia, a phenotype reminiscent of the metabolic syndrome (Chen et al., 2005a). Other models with mutation of the maternal Gsα allele also develop obesity with reduced energy expenditure as well (Kelly et al., 2009; Yu et al., 2000).
The similar parent-of-origin effects of Gsα mutations on energy balance in humans and mice strongly suggests that obesity due to maternal Gsα mutations results from severe Gsα deficiency in one or more tissues due to mutation of the active maternal Gsα allele and suppressed Gsα expression from the inactive paternal allele due to tissue-specific imprinting. Consistent with this hypothesis, reversal of imprinting due to the presence of the 1A imprint control region deletion on the paternal allele led to complete reversal of the maternal Gsα metabolic phenotype (Xie et al., 2008).
5. Gsα imprinting in the central nervous system underlies the parent-of-origin metabolic effects of Gsα mutations
Although Gsα is expressed in liver, adipose tissue, pancreatic islets and muscle, these tissues are not involved in the parent-of-origins effects of Gsα mutations as Gsα expression is not affected by imprinting in these tissues (Germain-Lee et al., 2005; Mantovani et al., 2004; Weinstein et al., 2007; Yu et al., 2000; Yu et al., 1998) and Gsα knockouts in these tissues due not produce a phenotype similar to the germline maternal Gsα knockout (Chen et al., 2010; Chen et al., 2009a; Chen et al., 2005b; Xie et al., 2010; Xie et al., 2007). However studies of mice with disruption of either the maternal or paternal Gsα allele in the central nervous system (mBrGsKO and pBrGsKO mice, respectively) that were generated by reciprocal matings of Nestin-cre and Gsα-floxed mice indicate that Gsα imprinting in the central nervous system underlies the parent-of-origin effects of Gsα mutations on energy and glucose metabolism (Chen et al., 2009b). mBrGsKO mice develop severe obesity associated with lower sympathetic nerve activity and energy expenditure and greater metabolic efficiency (weight gain/calorie intake), but without hyperphagia. In addition, mBrGsKO mice became glucose intolerant and insulin resistant even before the development of obesity, indicating that Gsα deficiency in the central nervous system has a primary effect on peripheral glucose metabolism. In contrast, pBrGsKO maintain a normal metabolic phenotype.
Studies examining Gsα expression within the central nervous system in mice with maternal and paternal Gsα mutations show that Gsα undergoes imprinting in the paraventricular nucleus of the hypothalamus, but not in the nucleus of the solitary tract or hippocampus (Chen et al., 2009b). As the paraventricular nucleus is a major site of metabolic regulation it may at least account for some of the parent-of-origin effects of Gsα mutations on metabolism. In fact, mice with maternal Gsα mutation restricted to the paraventricular nucleus and a few other sites made using Single minded 1 (Sim1) promoter-cre recombinase mice also develop mild obesity, glucose intolerance, insulin resistance, and reduced energy expenditure, although the effects were more prominent in males than females and overall much milder than in mBrGsKO mice (M.C., L.S.W., unpublished results). These findings indicate that the paraventricular nucleus plays a role in the metabolic effects observed in mice with maternal Gsα mutations, but that other brain regions are likely to be involved as well. In contrast, mice with hetero- or homozygous Gsα mutation in the ventral medial nucleus of the hypothalamus (made using steroidogenic factor 1 (Sf1) promoter-cre mice) showed no abnormalities in energy or glucose metabolism (M.C., L.S.W., unpublished results). Overall, our findings in mBrGsKO and pBrGsKO mice show that the metabolic phenotype generated by germline maternal Gsα mutation is due to an effect of Gsα imprinting in the central nervous system.
6. Gsα deficiency in the central nervous system impairs the stimulation of energy expenditure by central melanocortins
The status of energy balance is communicated to the brain through various signals, including hormones (e.g. leptin, ghrelin, insulin), nutrients (e.g. glucose, fatty acids) and afferent neural inputs from the gut (Saper et al., 2002). The hypothalamus integrates these signals to regulate food intake and energy expenditure, and the brainstem also receives signals to primarily control hunger and satiety (Cone, 2005). Several hypothalamic nuclei are involved in control of energy balance, including the arcuate, paraventricular, and ventromedial nuclei and the lateral hypothalamic area. The arcuate nucleus, one of main targets of leptin and insulin, contains neurons expressing orexigenic polypeptides (neuropeptide Y and agouti-related protein), and others expressing anorexigenic polypeptides (proopiomelanocortin and cocaine- and amphetamine-regulated transcript).
Proopiomelanocortin neurons located in the arcuate nucleus are activated by leptin to inhibit food intake and stimulate sympathetic nerve activity and energy expenditure, leading to negative energy balance (Brito et al., 2007; Butler and Cone, 2002; Nogueiras et al., 2007). These neurons project to the paraventricular and ventral medial nuclei of the hypothalamus and other sites where they release α-melanocyte stimulating hormone, which activates melanocortin MC3 and MC4 receptors in downstream neurons (Bagnol et al., 1999; Cowley et al., 1999; Xu et al., 2003). Melanocortin MC4 and MC3 receptors are seven transmembrane receptors known to couple to Gsα. Most of the effects of central melanocortins on energy balance are mediated via melanocortin MC4 receptors (Chen et al., 2000b; Marsh et al., 1999). In addition to their locations in the hypothalamus, melanocortin MC4receptors are also expressed in other locations of the central nervous system involved in energy balance, including the hindbrain and the sympathetic preganglionic neurons in the intermediolateral nucleus of the spinal cord, the latter of which also receive neural projections from the paraventricular nucleus of the hypothalamus (Elias et al., 1998; Kishi et al., 2003; Saper et al., 1976; Swanson and Kuypers, 1980).
Melanocortin MC4 receptor mutations are the most common cause of monogenic obesity in humans (Farooqi et al., 2003; Krude et al., 1998; Vaisse et al., 1998; Yeo et al., 1998) and also lead to severe obesity in mice (Huszar et al., 1997; Marsh et al., 1999), in both cases being associated with both hyperphagia and reduced sympathetic nerve activity and energy expenditure. In contrast, melanocortin MC3 receptor mutation in mice results in more subtle changes in adiposity (Chen et al., 2000a). In addition to their effects on energy balance, melanocortin MC4 receptor mutations also lead to increased linear growth and primary effects on peripheral glucose metabolism (Fan et al., 2000; Nogueiras et al., 2007; Obici et al., 2001).
Mice with central nervous system-specific (mBrGsKO) or germline disruption of the maternal Gsα allele partially mimic melanocortin MC4 receptor null mice in that they develop severe obesity with reduced sympathetic nerve activity and energy expenditure as well as glucose intolerance and insulin resistance (Chen et al., 2005a; Chen et al., 2009b). These effects on sympathetic nerve activity and energy expenditure are likely due to impaired responsiveness of mBrGsKO mice to the effect of central melanocortins on energy expenditure, as mBrGsKO mice have a reduced increase in energy expenditure in response to a melanocortin agonist (Chen et al., 2009b). Consistent with this finding, mBrGsKO have evidence of reduced sympathetic nerve activity (reduced norepinephrine content in brown adipose tissue, heart rate, and diastolic blood pressure) and impaired diet-induced thermogenesis, a process known to be dependent on melanocortin MC4 receptor signaling (Butler et al., 2001; Voss-Andreae et al., 2007). This mechanism also likely underlies the obesity in pseudohypoparathyroidism type 1A patients, as these patients have low circulating norepinephrine levels (Carel et al., 1999).
However, mice with maternal Gsα mutations do not develop all the features seen with melanocortin MC4 receptor mutations, such as hyperphagia or increased linear growth (Chen et al., 2005a; Chen et al., 2009b). Consistent with the lack of hyperphagia, the ability of a melanocortin agonist to acutely reduce food intake was unaffected in mBrGsKO mice (Chen et al., 2009b). Early-onset obesity in the absence of hyperphagia has been well documented in at least one child with pseudohypoparathyroidism type 1A (Dekelbab et al., 2009). Sim1 is a transcription factor expressed almost exclusively in the paraventricular nucleus that is upregulated by melanocortin MC4 receptor signaling and mediates some of the effects of melanocortins (Kublaoui et al., 2006b). Sim1 mutations in humans and mice also lead to obesity, but in contrast to mBrGsKO mice, this effect is associated with hyperphagia and increased linear growth, with no primary effects on energy expenditure or glucose metabolism (Faivre et al., 2002; Holder et al., 2000; Holder et al., 2004; Kublaoui et al., 2008; Kublaoui et al., 2006a). Moreover, Sim1 haploinsufficient mice have the opposite pattern of melanocortin responsiveness to that seen in mBrGsKO (normal increase in energy expenditure and impaired ability to inhibit food intake) (Kublaoui et al., 2006a). Based upon these observations, we propose the possibility that melanocortins mediate their actions through independent signaling pathways downstream of melanocortin MC4 receptors, a Gsα-dependent pathway involved in regulating sympathetic nerve activity, energy expenditure, and glucose metabolism, and a Gsα-independent pathway working through Sim1 involved in food intake and linear growth. Further studies are required to determine whether is hypothesis is correct, and if so, whether other G proteins are involved in the melanocortin MC4 receptor/Sim1 pathway. We are also examining whether there is Gsα imprinting in other regions of the central nervous system.
7. Divergent metabolic effects of Gsα deficiency in different peripheral tissues
In addition to its effects in the central nervous system, Gsα plays important roles in peripheral tissues that also are involved in energy and glucose metabolism. We have generated several tissue-specific Gsα knockouts (in each case homozygous null) to examine these effects in other metabolically active tissues. Liver-specific Gsα knockout mice have increased insulin sensitivity and fasting hypoglycemia, associated with hepatic glucagon resistance and islet cell hyperplasia (Chen et al., 2005b). Skeletal-muscle Gsα specific have impaired glucose tolerance in the absence of insulin deficiency and resistance, most likely as the result of reduced skeletal muscle mass. In addition, there appears to be a switch of the muscle fiber type towards aerobic, slow twitch (red) fibers even though the muscles have metabolic characteristics more typical of anaerobic, fast-twitch (white) fibers (Chen et al., 2009a). Adipose tissue-specific Gsα knockout mice have markedly impaired adipogenesis (Chen et al., 2010). While these mice have reduced cold-induced thermogenesis due to resistance of brown adipose tissue to sympathetic stimulation, diet-induced thermogenesis is maintained indicating that these two forms of adaptive thermogenesis may occur in separate tissues. Finally, studies in pancreas-specific Gsα knockout models (Xie et al., 2010; Xie et al., 2007) indicate that Gsα signaling is required for normal β cell proliferation and maintanence of β cell mass and that Gsα may have opposite effects on proliferation of pancreatic α and β cells.
8. Conclusion
One widely accepted hypothesis underlying the imprinting process is the parental conflict hypothesis which predicts that paternally transmitted alleles promote fetal growth as the father wants to maximize survival of his offspring while maternally transmitted alleles inhibit fetal growth so the mother can reserve resources for multiple litters (Moor, T 1991, Haig D 2004). Imprinted genes also appear to be involved in postnatal energy metabolism as several diseases caused by mutations in imprinted genes lead to obesity (e.g. Prader-Willi syndrome, pseudohypoparathyroidism type 1A) and population studies have identified a number of chromosomal regions associated with parent-of-origin effects on energy balance in humans (Dong et al., 2005; Gorlova et al., 2003; Lindsay et al., 2001; Rance et al., 2005). The effect of GNAS imprinting on energy balance is consistent with the parental conflict hypothesis, a loss of the paternally expressed XLαs leads to a lean phenotype with increased sympathetic nerve activity and energy expenditure while the oppositely imprinted GNAS gene product Gsα leads to obesity due to opposite effects on sympathetic nerve activity and energy expenditure.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, U. S. Department of Health and Human Services.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bachman ES, Dhillon H, Zhang CY, Cinti S, Bianco AC, Kobilka BK, Lowell BB. βAR signaling required for diet-induced thermogenesis and obesity resistance. Science. 2002;297:843–845. doi: 10.1126/science.1073160. [DOI] [PubMed] [Google Scholar]
- Bagnol D, Lu XY, Kaelin CB, Day HE, Ollmann M, Gantz I, Akil H, Barsh GS, Watson SJ. Anatomy of an endogenous antagonist: relationship between Agouti-related protein and proopiomelanocortin in brain. J Neurosci. 1999;19:RC26, 1–7. doi: 10.1523/JNEUROSCI.19-18-j0004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastepe M, Gunes Y, Perez-Villamil B, Hunzelman J, Weinstein LS, Jüppner H. Receptor-mediated adenylyl cyclase activation through XLαs, the extra-large variant of the stimulatory G protein α subunit. Mol Endocrinol. 2002;16:1912–1919. doi: 10.1210/me.2002-0054. [DOI] [PubMed] [Google Scholar]
- Bastepe M, Pincus JE, Sugimoto T, Tojo K, Kanatani M, Azuma Y, Kruse K, Rosenbloom AL, Koshiyama H, Jüppner H. Positional dissociation between the genetic mutation responsible for pseudohypoparathyroidism type Ib and the associated methylation defect at exon A/B: evidence for a long-range regulatory element within the imprinted GNAS1 locus. Hum Mol Genet. 2001;10:1231–1241. doi: 10.1093/hmg/10.12.1231. [DOI] [PubMed] [Google Scholar]
- Brito MN, Brito NA, Baro DJ, Song CK, Bartness TJ. Differential activation of the sympathetic innervation of adipose tissues by melanocortin receptor stimulation. Endocrinology. 2007;148:5339–5347. doi: 10.1210/en.2007-0621. [DOI] [PubMed] [Google Scholar]
- Butler AA, Cone RD. The melanocortin receptors: lessons from knockout models. Neuropeptides. 2002;36:77–84. doi: 10.1054/npep.2002.0890. [DOI] [PubMed] [Google Scholar]
- Butler AA, Marks DL, Fan W, Kuhn CM, Bartolome M, Cone RD. Melanocortin-4 receptor is required for acute homeostatic responses to increased dietary fat. Nat Neurosci. 2001;4:605–611. doi: 10.1038/88423. [DOI] [PubMed] [Google Scholar]
- Campbell R, Gosden CM, Bonthron DT. Parental origin of transcription from the human GNAS1 gene. J Med Genet. 1994;31:607–614. doi: 10.1136/jmg.31.8.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carel JC, Le Stunff C, Condamine L, Mallet E, Chaussain JL, Adnot P, Garabedian M, Bougneres P. Resistance to the lipolytic action of epinephrine : a new feature of protein Gs deficiency. J Clin Endocrinol Metab. 1999;84:4127–4131. doi: 10.1210/jcem.84.11.6145. [DOI] [PubMed] [Google Scholar]
- Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van der Ploeg LH. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet. 2000a;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- Chen AS, Metzger JM, Trumbauer ME, Guan XM, Yu H, Frazier EG, Marsh DJ, Forrest MJ, Gopal-Truter S, Fisher J, Camacho RE, Strack AM, Mellin TN, MacIntyre DE, Chen HY, Van der Ploeg LH. Role of the melanocortin-4 receptor in metabolic rate and food intake in mice. Transgenic Res. 2000b;9:145–154. doi: 10.1023/a:1008983615045. [DOI] [PubMed] [Google Scholar]
- Chen M, Chen H, Nguyen A, Gupta D, Wang J, Lai EW, Pacak K, Gavrilova O, Quon MJ, Weinstein LS. Gsα deficiency in adipose tissue leads to a lean phenotype with divergent effects on cold tolerance and diet-induced thermogenesis. Cell Metab. 2010;11:320–330. doi: 10.1016/j.cmet.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Feng HZ, Gupta D, Kelleher J, Dickerson KE, Wang J, Hunt D, Jou W, Gavrilova O, Jin JP, Weinstein LS. Gsα deficiency in skeletal muscle leads to reduced muscle mass, fiber-type switching, and glucose intolerance without insulin resistance or deficiency. Am J Physiol Cell Physiol. 2009a;296:C930–C940. doi: 10.1152/ajpcell.00443.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Gavrilova O, Liu J, Xie T, Deng C, Nguyen AT, Nackers LM, Lorenzo J, Shen L, Weinstein LS. Alternative Gnas gene products have opposite effects on glucose and lipid metabolism. Proc Natl Acad Sci USA. 2005a;102:7386–7391. doi: 10.1073/pnas.0408268102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Gavrilova O, Zhao WQ, Nguyen A, Lorenzo J, Shen L, Nackers L, Pack S, Jou W, Weinstein LS. Increased glucose tolerance and reduced adiposity in the absence of fasting hypoglycemia in mice with liver-specific Gsα deficiency. J Clin Invest. 2005b;115:3217–3227. doi: 10.1172/JCI24196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Wang J, Dickerson KE, Kelleher J, Xie T, Gupta D, Lai EW, Pacak K, Gavrilova O, Weinstein LS. Central nervous system imprinting of the G protein Gsα and its role in metabolic regulation. Cell Metab. 2009b;9:548–555. doi: 10.1016/j.cmet.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Pronchuk N, Fan W, Dinulescu DM, Colmers WF, Cone RD. Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron. 1999;24:155–163. doi: 10.1016/s0896-6273(00)80829-6. [DOI] [PubMed] [Google Scholar]
- Davies SJ, Hughes HE. Imprinting in Albright’s hereditary osteodystrophy. J Med Genet. 1993;30:101–103. doi: 10.1136/jmg.30.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- Dekelbab BH, Aughton DJ, Levine MA. Pseudohypoparathyroidism type 1A and morbid obesity in infancy. Endocr Pract. 2009;15:249–253. doi: 10.4158/EP.15.3.249. [DOI] [PubMed] [Google Scholar]
- Dong C, Li WD, Geller F, Lei L, Li D, Gorlova OY, Hebebrand J, Amos CI, Nicholls RD, Price RA. Possible genomic imprinting of three human obesity-related genetic loci. Am J Hum Genet. 2005;76:427–437. doi: 10.1086/428438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias CF, Lee C, Kelly J, Aschkenasi C, Ahima RS, Couceyro PR, Kuhar MJ, Saper CB, Elmquist JK. Leptin activates hypothalamic CART neurons projecting to the spinal cord. Neuron. 1998;21:1375–1385. doi: 10.1016/s0896-6273(00)80656-x. [DOI] [PubMed] [Google Scholar]
- Faivre L, Cormier-Daire V, Lapierre JM, Colleaux L, Jacquemont S, Genevieve D, Saunier P, Munnich A, Turleau C, Romana S, Prieur M, De Blois MC, Vekemans M. Deletion of the SIM1 gene (6q16.2) in a patient with a Prader-Willi-like phenotype. J Med Genet. 2002;39:594–596. doi: 10.1136/jmg.39.8.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Dinulescu DM, Butler AA, Zhou J, Marks DL, Cone RD. The central melanocortin system can directly regulate serum insulin levels. Endocrinology. 2000;141:3072–3079. doi: 10.1210/endo.141.9.7665. [DOI] [PubMed] [Google Scholar]
- Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–1095. doi: 10.1056/NEJMoa022050. [DOI] [PubMed] [Google Scholar]
- Frey UH, Hauner H, Jockel KH, Manthey I, Brockmeyer N, Siffert W. A novel promoter polymorphism in the human gene GNAS affects binding of transcription factor upstream stimulatory factor 1, Gαs protein expression and body weight regulation. Pharmacogenet Genomics. 2008a;18:141–151. doi: 10.1097/FPC.0b013e3282f49964. [DOI] [PubMed] [Google Scholar]
- Frey UH, Michalsen A, Merse S, Dobos GJ, Siffert W. A functional GNAS promoter polymorphism is associated with altered weight loss during short -term fasting. Eur J Med Res. 2008b;13:576–578. [PubMed] [Google Scholar]
- Germain-Lee EL, Ding C, Deng Z, Crane JL, Saji M, Ringel MD, Levine MA. Paternal imprinting of Gαs in the human thyroid as the basis of TSH resistance in pseudohypoparathyroidism type 1a. Biochem Biophys Res Commun. 2002;296:67–72. doi: 10.1016/s0006-291x(02)00833-1. [DOI] [PubMed] [Google Scholar]
- Germain-Lee EL, Schwindinger W, Crane JL, Zewdu R, Zweifel LS, Wand G, Huso DL, Saji M, Ringel MD, Levine MA. A mouse model of Albright hereditary osteodystrophy generated by targeted disruption of exon 1 of the Gnas gene. Endocrinology. 2005;146:4697–4709. doi: 10.1210/en.2005-0681. [DOI] [PubMed] [Google Scholar]
- Gorlova OY, Amos CI, Wang NW, Shete S, Turner ST, Boerwinkle E. Genetic linkage and imprinting effects on body mass index in children and young adults. Eur J Hum Genet. 2003;11:425–432. doi: 10.1038/sj.ejhg.5200979. [DOI] [PubMed] [Google Scholar]
- Hahn S, Frey UH, Siffert W, Tan S, Mann K, Janssen OE. The CC genotype of the GNAS T393C polymorphism is associated with obesity and insulin resistance in women with polycystic ovary syndrome. Eur J Endocrinol. 2006;155:763–770. doi: 10.1530/eje.1.02275. [DOI] [PubMed] [Google Scholar]
- Hayward BE, Barlier A, Korbonits M, Grossman AB, Jacquet P, Enjalbert A, Bonthron DT. Imprinting of the Gsα gene GNAS1 in the pathogenesis of acromegaly. J Clin Invest. 2001;107:R31–R36. doi: 10.1172/JCI11887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward BE, Kamiya M, Strain L, Moran V, Campbell R, Hayashizaki Y, Bonthron DT. The human GNAS1 gene is imprinted and encodes distinct paternally and biallelically expressed G proteins. Proc Natl Acad Sci USA. 1998a;95:10038–10043. doi: 10.1073/pnas.95.17.10038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward BE, Moran V, Strain L, Bonthron DT. Bidirectional imprinting of a single gene: GNAS1 encodes maternally, paternally, and biallelically derived proteins. Proc Natl Acad Sci USA. 1998b;95:15475–15480. doi: 10.1073/pnas.95.26.15475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder JL, Jr, Butte NF, Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000;9:101–108. doi: 10.1093/hmg/9.1.101. [DOI] [PubMed] [Google Scholar]
- Holder JL, Jr, Zhang L, Kublaoui BM, DiLeone RJ, Oz OK, Bair CH, Lee YH, Zinn AR. Sim1 gene dosage modulates the homeostatic feeding response to increased dietary fat in mice. Am J Physiol Endocrinol Metab. 2004;287:E105–113. doi: 10.1152/ajpendo.00446.2003. [DOI] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Ischia R, Lovisetti-Scamihorn P, Hogue-Angeletti R, Wolkersdorfer M, Winkler H, Fischer-Colbrie R. Molecular cloning and characterization of NESP55, a novel chromogranin-like precursor of a peptide with 5-HT1B receptor antagonist activity. J Biol Chem. 1997;272:11657–11662. doi: 10.1074/jbc.272.17.11657. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Bianchi C, Nadal-Ginard B, Homcy CJ. Alternative promoter and 5′ exon generate a novel Gsα mRNA. J Biol Chem. 1990;265:8458–8462. [PubMed] [Google Scholar]
- Jan de Beur S, Ding C, Germain-Lee EL, Cho J, Maret A, Levine MA. Discordance between genetic and epigenetic defects in pseudohypoparathyroidism type 1b revealed by inconsistent loss of maternal imprinting at GNAS1. Am J Hum Genet. 2003;73:314–322. doi: 10.1086/377136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehlenbach RH, Matthey J, Huttner WB. XLαs is a new type of G protein. Nature. 1994;372:804–809. doi: 10.1038/372804a0. [DOI] [PubMed] [Google Scholar]
- Kelly ML, Moir L, Jones L, Whitehill E, Anstee QM, Goldin RD, Hough A, Cheeseman M, Jansson JO, Peters J, Cox RD. A missense mutation in the non-neural G-protein α-subunit isoforms modulates susceptibility to obesity. Int J Obes (Lond) 2009;33:507–518. doi: 10.1038/ijo.2009.30. [DOI] [PubMed] [Google Scholar]
- Kelsey G, Bodle D, Miller HJ, Beechey CV, Coombes C, Peters J, Williamson CM. Identification of imprinted loci by methylation-sensitive representational difference analysis: application to mouse distal chromosome 2. Genomics. 1999;62:129–138. doi: 10.1006/geno.1999.6022. [DOI] [PubMed] [Google Scholar]
- Kishi T, Aschkenasi CJ, Lee CE, Mountjoy KG, Saper CB, Elmquist JK. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J Comp Neurol. 2003;457:213–235. doi: 10.1002/cne.10454. [DOI] [PubMed] [Google Scholar]
- Kozasa T, Itoh H, Tsukamoto T, Kaziro Y. Isolation and characterization of the human Gsα gene. Proc Natl Acad Sci USA. 1988;85:2081–2085. doi: 10.1073/pnas.85.7.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. 1998;19:155–157. doi: 10.1038/509. [DOI] [PubMed] [Google Scholar]
- Kublaoui BM, Gemelli T, Tolson KP, Wang Y, Zinn AR. Oxytocin deficiency mediates hyperphagic obesity of Sim1 haploinsufficient mice. Mol Endocrinol. 2008;22:1723–1734. doi: 10.1210/me.2008-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kublaoui BM, Holder JL, Jr, Gemelli T, Zinn AR. Sim1 haploinsufficiency impairs melanocortin-mediated anorexia and activation of paraventricular nucleus neurons. Mol Endocrinol. 2006a;20:2483–2492. doi: 10.1210/me.2005-0483. [DOI] [PubMed] [Google Scholar]
- Kublaoui BM, Holder JL, Jr, Tolson KP, Gemelli T, Zinn AR. SIM1 overexpression partially rescues agouti yellow and diet-induced obesity by normalizing food intake. Endocrinology. 2006b;147:4542–4549. doi: 10.1210/en.2006-0453. [DOI] [PubMed] [Google Scholar]
- Lindsay RS, Kobes S, Knowler WC, Bennett PH, Hanson RL. Genome-wide linkage analysis assessing parent-of-origin effects in the inheritance of type 2 diabetes and BMI in Pima Indians. Diabetes. 2001;50:2850–2857. doi: 10.2337/diabetes.50.12.2850. [DOI] [PubMed] [Google Scholar]
- Liu J, Chen M, Deng C, Bourc’his D, Nealon JG, Erlichman B, Bestor TH, Weinstein LS. Identification of the control region for tissue-specific imprinting of the stimulatory G protein α-subunit. Proc Natl Acad Sci USA. 2005a;102:5513–5518. doi: 10.1073/pnas.0408262102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Erlichman B, Weinstein LS. The stimulatory G protein α-subunit Gsα is imprinted in human thyroid glands: implications for thyroid function in pseudohypoparathyroidism types 1A and 1B. J Clin Endocrinol Metab. 2003;88:4336–4341. doi: 10.1210/jc.2003-030393. [DOI] [PubMed] [Google Scholar]
- Liu J, Litman D, Rosenberg MJ, Yu S, Biesecker LG, Weinstein LS. A GNAS1 imprinting defect in pseudohypoparathyroidism type IB. J Clin Invest. 2000a;106:1167–1174. doi: 10.1172/JCI10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Nealon JG, Weinstein LS. Distinct patterns of abnormal GNAS imprinting in familial and sporadic pseudohypoparathyroidism type IB. Hum Mol Genet. 2005b;14:95–102. doi: 10.1093/hmg/ddi009. [DOI] [PubMed] [Google Scholar]
- Liu J, Yu S, Litman D, Chen W, Weinstein LS. Identification of a methylation imprint mark within the mouse Gnas locus. Mol Cell Biol. 2000b;20:5808–5817. doi: 10.1128/mcb.20.16.5808-5817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DN, McGuire S, Levine MA, Weinstein LS, Germain-Lee EL. Body mass index differences in pseudohypoparathyroidism type 1a versus pseudopseudohypoparathyroidism implicate paternal imprinting of Gαs in the development of human obesity. J Clin Endocrinol Metab. 2007;92:1073–1079. doi: 10.1210/jc.2006-1497. [DOI] [PubMed] [Google Scholar]
- Mantovani G, Ballare E, Giammona E, Beck-Peccoz P, Spada A. The Gs gene: predominant maternal origin of transcription in human thyroid gland and gonads. J Clin Endocrinol Metab. 2002;87:4736–4740. doi: 10.1210/jc.2002-020183. [DOI] [PubMed] [Google Scholar]
- Mantovani G, Bondioni S, Locatelli M, Pedroni C, Lania AG, Ferrante E, Filopanti M, Beck-Peccoz P, Spada A. Biallelic expression of the Gsα gene in human bone and adipose tissue. J Clin Endocrinol Metab. 2004;89:6316–6319. doi: 10.1210/jc.2004-0558. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Hollopeter G, Huszar D, Laufer R, Yagaloff KA, Fisher SL, Burn P, Palmiter RD. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat Genet. 1999;21:119–122. doi: 10.1038/5070. [DOI] [PubMed] [Google Scholar]
- Mattera R, Graziano MP, Yatani A, Zhou Z, Graf R, Codina J, Birnbaumer L, Gilman AG, Brown AM. Splice variants of the alpha subunit of the G protein Gs activate both adenylyl cyclase and calcium channels. Science. 1989;243:804–807. doi: 10.1126/science.2536957. [DOI] [PubMed] [Google Scholar]
- Montminy M. Transcriptional regulation by cyclic AMP. Annu Rev Biochem. 1997;66:807–822. doi: 10.1146/annurev.biochem.66.1.807. [DOI] [PubMed] [Google Scholar]
- Nogueiras R, Wiedmer P, Perez-Tilve D, Veyrat-Durebex C, Keogh JM, Sutton GM, Pfluger PT, Castanada TR, Neschen S, Hofmann SM, Howles PN, Morgan DA, Benoit SC, Szanto I, Schrott B, Schurmann A, Joost HG, Hammond C, Hui DY, Woods SC, Rahmouni K, Butler AA, Farooqi IS, O’Rahilly S, Rohner-Jeanrenaud F, Tschop MH. The central melanocortin system directly controls peripheral lipid metabolism. J Clin Invest. 2007;117:3475–3488. doi: 10.1172/JCI31743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwosu BU, Lee MM. Pseudohypoparathyroidism type 1a and insulin resistance in a child. Nat Rev Endocrinol. 2009;5:345–350. doi: 10.1038/nrendo.2009.81. [DOI] [PubMed] [Google Scholar]
- Obici S, Feng Z, Tan J, Liu L, Karkanias G, Rossetti L. Central melanocortin receptors regulate insulin action. J Clin Invest. 2001;108:1079–1085. doi: 10.1172/JCI12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasolli HA, Klemke M, Kehlenbach RH, Wang Y, Huttner WB. Characterization of the extra-large G protein α-subunit XLαs. I. Tissue distribution and subcellular localization. J Biol Chem. 2000;275:33622–33632. doi: 10.1074/jbc.M001335200. [DOI] [PubMed] [Google Scholar]
- Peters J, Wroe SF, Wells CA, Miller HJ, Bodle D, Beechey CV, Williamson CM, Kelsey G. A cluster of oppositely imprinted transcripts at the Gnas locus in the distal imprinting region of mouse chromosome 2. Proc Natl Acad Sci USA. 1999;96:3830–3835. doi: 10.1073/pnas.96.7.3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagge A, Gordon E, Dean W, Boiani R, Cinti S, Peters J, Kelsey G. The imprinted signaling protein XLαs is required for postnatal adaptation to feeding. Nat Genet. 2004;36:818–826. doi: 10.1038/ng1397. [DOI] [PubMed] [Google Scholar]
- Plagge A, Isles AR, Gordon E, Humby T, Dean W, Gritsch S, Fischer-Colbrie R, Wilkinson LS, Kelsey G. Imprinted Nesp55 influences behavioral reactivity to novel environments. Mol Cell Biol. 2005;25:3019–3026. doi: 10.1128/MCB.25.8.3019-3026.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rance KA, Fustin JM, Dalgleish G, Hambly C, Bunger L, Speakman JR. A paternally imprinted QTL for mature body mass on mouse chromosome 8. Mamm Genome. 2005;16:567–577. doi: 10.1007/s00335-005-0012-4. [DOI] [PubMed] [Google Scholar]
- Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- Saper CB, Chou TC, Elmquist JK. The need to feed: homeostatic and hedonic control of eating. Neuron. 2002;36:199–211. doi: 10.1016/s0896-6273(02)00969-8. [DOI] [PubMed] [Google Scholar]
- Saper CB, Loewy AD, Swanson LW, Cowan WM. Direct hypothalamo-autonomic connections. Brain Res. 1976;117:305–312. doi: 10.1016/0006-8993(76)90738-1. [DOI] [PubMed] [Google Scholar]
- Sun H, Chen Z, Poppleton H, Scholich K, Mullenix J, Weipz GJ, Fulgham DL, Bertics PJ, Patel TB. The juxtamembrane, cytosolic region of the epidermal growth factor receptor is involved in association with α-subunit of Gs. J Biol Chem. 1997;272:5413–5420. [PubMed] [Google Scholar]
- Swanson LW, Kuypers HG. The paraventricular nucleus of the hypothalamus: cytoarchitectonic subdivisions and organization of projections to the pituitary, dorsal vagal complex, and spinal cord as demonstrated by retrograde fluorescence double-labeling methods. J Comp Neurol. 1980;194:555–570. doi: 10.1002/cne.901940306. [DOI] [PubMed] [Google Scholar]
- Vaisse C, Clement K, Guy-Grand B, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. 1998;20:113–114. doi: 10.1038/2407. [DOI] [PubMed] [Google Scholar]
- Voss-Andreae A, Murphy JG, Ellacott KL, Stuart RC, Nillni EA, Cone RD, Fan W. Role of the central melanocortin circuitry in adaptive thermogenesis of brown adipose tissue. Endocrinology. 2007;148:1550–1560. doi: 10.1210/en.2006-1389. [DOI] [PubMed] [Google Scholar]
- Weinstein LS, Chen M, Xie T, Liu J. Genetic diseases associated with heterotrimeric G proteins. Trends Pharmacol Sci. 2006;27:260–266. doi: 10.1016/j.tips.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Weinstein LS, Xie T, Zhang QH, Chen M. Studies of the regulation and function of the Gsα gene Gnas using gene targeting technology. Pharmacol Ther. 2007;115:271–291. doi: 10.1016/j.pharmthera.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein LS, Yu S, Warner DR, Liu J. Endocrine manifestations of stimulatory G protein α-subunit mutations and the role of genomic imprinting. Endocr Rev. 2001;22:675–705. doi: 10.1210/edrv.22.5.0439. [DOI] [PubMed] [Google Scholar]
- Williamson CM, Ball ST, Nottingham WT, Skinner JA, Plagge A, Turner MD, Powles N, Hough T, Papworth D, Fraser WD, Maconochie M, Peters J. A cis-acting control region is required exclusively for the tissue-specific imprinting of Gnas. Nat Genet. 2004;36:894–899. doi: 10.1038/ng1398. [DOI] [PubMed] [Google Scholar]
- Xie T, Chen M, Gavrilova O, Lai EW, Liu J, Weinstein LS. Severe obesity and insulin resistance due to deletion of the maternal Gsα allele is reversed by paternal deletion of the Gsα imprint control region. Endocrinology. 2008;149:2443–2450. doi: 10.1210/en.2007-1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Chen M, Weinstein LS. Pancreas-specific Gsα deficiency has divergent effects on pancreatic α and β cell proliferation. J Endocrinol. 2010 doi: 10.1677/JOE-10-0030. published online June 11, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Chen M, Zhang QH, Ma Z, Weinstein LS. β cell-specific deficiency of the stimulatory G protein α-subunit Gsα leads to reduced β cell mass and insulin-deficient diabetes. Proc Natl Acad Sci USA. 2007;104:19601–19606. doi: 10.1073/pnas.0704796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Plagge A, Gavrilova O, Pack S, Jou W, Lai EW, Frontera M, Kelsey G, Weinstein LS. The alternative stimulatory G protein α-subunit XLαs is a critical regulator of energy and glucose metabolism and sympathetic nerve activity in adult mice. J Biol Chem. 2006;281:18989–18999. doi: 10.1074/jbc.M511752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, Tecott LH, Reichardt LF. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O’Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20:111–112. doi: 10.1038/2404. [DOI] [PubMed] [Google Scholar]
- Yu S, Gavrilova O, Chen H, Lee R, Liu J, Pacak K, Parlow AF, Quon MJ, Reitman ML, Weinstein LS. Paternal versus maternal transmission of a stimulatory G protein α subunit knockout produces opposite effects on energy metabolism. J Clin Invest. 2000;105:615–623. doi: 10.1172/JCI8437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Yu D, Lee E, Eckhaus ME, Lee R, Corria Z, Accili D, Westphal H, Weinstein LS. Variable and tissue-specific hormone resistance in heterotrimeric Gs protein α-subunit (Gsα) knockout mice is due to tissue-specific imprinting of the Gsα gene. Proc Natl Acad Sci USA. 1998;95:8715–8720. doi: 10.1073/pnas.95.15.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]