

Abstract
Synaptosomal expression of NCX1, NCX2, and NCX3, the three variants of the Na+-Ca2+ exchanger (NCX), was investigated in Alzheimer’s disease parietal cortex. Flow cytometry and immunoblotting techniques were used to analyze synaptosomes prepared from cryopreserved brain of cognitively normal aged controls and late stage Alzheimer’s disease patients. Major findings that emerged from this study are: (1) NCX1 was the most abundant NCX isoform in nerve terminals of cognitively normal patients; (2) NCX2 and NCX3 protein levels were modulated in parietal cortex of late stage Alzheimer’s disease: NCX2 positive terminals were increased in the Alzheimer’s disease cohort while counts of NCX3 positive terminals were reduced; (3) NCX1, NCX2 and NCX3 isoforms co-localized with amyloid-beta in synaptic terminals and all three variants are up-regulated in nerve terminals containing amyloid-beta. Taken together, these data indicate that NCX isoforms are selectively regulated in pathological terminals, suggesting different roles of each NCX isoform in Alzheimer’s disease terminals.
Keywords: Na+-Ca2+ exchanger (NCX), Alzheimer’s disease, calcium signaling, amyloid-beta, synapse
1. Introduction
Calcium (Ca2+) is one of the most important second messengers in the human brain: this ion is involved in the regulation of vital functions in neurons (i.e. release of neurotransmitters, neuronal excitability, gene expression, neuronal growth and programmed neuronal death). Calcium dyshomeostasis is associated with synaptic dysfunction in aging brain and neurodegeneration in late stages of Alzheimer’s disease (AD) [1–2]. Studies of synapses in AD patients and animal models suggest that synapses are the primary sites of Ca2+ dysregulation in AD [3–4].
Maintaining proper Ca2+ homeostasis is critical for the viability and functionality of neurons. The Na+-Ca2+ exchanger (NCX) is one of the major means of Ca2+ extrusion at the plasma membrane of many excitable and non-excitable cells. Depending on the intracellular levels of Na+ and Ca2+, NCX can operate in the forward mode, by extruding one Ca2+ against three entering Na+, using the Na+ gradient across the plasma membrane as a source of energy [5–6]. Alternatively, in the reverse mode, NCX can function as Na+ efflux-Ca2+ influx. Because of its high exchange capacity, NCX is well suited for rapid recovery from high intracellular Ca2+ concentrations ([Ca2+i]) and may play an important role in maintaining Ca2+ homeostasis and protecting cells from Ca2+ overload and eventual death [5–6].
Three genes have been cloned encoding the three different isoforms NCX1, NCX2, and NCX3 [7–10]. NCX1 is most abundant in the heart but is widely distributed in most cells [7, 10]. NCX2 expression is restricted to the brain and spinal cord [11]. NCX3 expression is restricted to the brain and skeletal muscle [12–13]. In the neocortex and hippocampus, NCX isoforms are widely distributed and are expressed in neurons, astrocytes, oligodendrocytes and capillary endothelial cells [7, 14–16].
All NCX isoforms display similar transport kinetics. The major up-regulation mechanism of NCX involves intracellular ATP [17]. NCX1 and NCX2 activity is affected by cellular depletion of ATP, whereas NCX3 activity is not [18]. Therefore, their ATP-dependent activity may underlie a specific role for each isoform under physiological and pathophysiological conditions (e.g. ischemia, oxidative stress).
Since all NCX isoforms are expressed in the same brain regions and, perhaps, in the same neurons, isolating their specific involvement in the maintenance of intracellular Ca2+ homeostasis is quite challenging. NCX1-, NCX2- and NCX3- specific knockout mice were generated over the past decade [11–12, 19]. These mouse models are useful tools for elucidating NCX1-3 specific function in physiological and pathophysiological processes in the central nervous system. NCX1-deficient mice are not viable. NCX1 null-mutation caused embryonic lethality, irregular heartbeats and apoptosis in the heart [19–20]. Recent studies indicated that cardiac-specific transgenic re-expression of NCX1 was not enough to rescue the lethal phenotype, suggesting an important non-cardiac role for NCX1 during embryogenesis (e.g. vascularization of yolk sac, placental development) [21–22]. Mice lacking NCX2 exhibit enhanced learning and memory [11]. Targeted disruption of NCX3 leads to defective neuromuscular transmission [12]. Under ischemic conditions, NCX3-deficient mice exhibit increased neuronal damage [13, 23]. Studies also showed that NCX plays a major role in restoring baseline Ca2+ levels following glutamate-induced depolarization in cortical and hippocampal neurons [11, 24]. These findings highlight NCX function in the regulation of Na+ and Ca2+ following synaptic activity.
Alzheimer’s disease is the most common form of dementia and is characterized by abnormal amyloid-beta (Aβ) metabolism. Accumulation of Aβ in the brain and synapse loss are pathological features of AD. The Ca2+ hypothesis of AD proposes that the amyloidogenic pathway contributes to the remodeling of Ca2+ signaling responsible for cognitive deficits [1, 25]. Recent in vivo Ca2+ imaging recordings performed on AD transgenic mice (APP/PS1) showed that severe alteration in Ca2+ signaling was detected within a short distance of Aβ plaques [4]. The resulting changes in intraneuronal Ca2+ concentration may afterwards result in localized synaptic dysfunction and synapse loss in AD brain [4, 26–27]. Degeneration of synapses is believed to occur early in the disease process and to markedly correlate with cognitive deficits [28]. The mechanism by which Aβ disrupts cellular Ca2+ homeostasis in nerve terminals is not yet fully understood. Very little is known about NCX expression and its function in AD.
Information about the pattern of NCX1-3 expression in brain of AD patients is not available. In this study, we used Western blot and flow cytometry analyses to quantify NCX1-3 levels in synaptosomal preparations of cryopreserved human AD parietal cortex. The goal of this investigation was to determine whether NCX1-3 synaptic expression is modulated in AD pathogenesis.
2. Materials and Methods
2.1. Human brain specimens
Human parietal cortices (A7, A39 and A40) were obtained at autopsy from the Alzheimer’s Disease Research Centers at the University of Southern California and the University of California at Los Angeles. Samples were obtained from 8 patients (6 females, 2 males, age 87.1 ± 2.6 yr, mean postmortem delay: 6.5 ± 0.7 h) diagnosed clinically and histopathologically with AD, and from 4 cognitively normal aged controls (3 females, 1 males, age 90.5 ± 5.1 yr; mean postmortem delay: 6.1 ± 1.1 h; Table 1).
Table 1.
Clinical and neuropathological characteristics of patients and controls
| Patients | Sex | Age (y) | Ethnicity | PMI (h) | Parietal cortex atrophy | Neuritic plaques | Neuropil threads | Neuronal loss | Gliosis |
|---|---|---|---|---|---|---|---|---|---|
| Normal | |||||||||
| #1 | F | 89 | Caucasian | 8.4 | Moderate | None | None | None | None |
| #2 | F | 105 | Caucasian | 6.0 | Mild | None | None | None | None |
| #3 | M | 81 | Hispanic | 3.0 | Mild | None | None | None | None |
| #4 | F | 87 | Caucasian | 7.0 | None | None | None | None | None |
| AD (Braak & Braak score IV - V)+ Amyloid angiopathy | |||||||||
| #5 | F | 89 | Caucasian | 9.0 | Mild | Moderate | Sparse | None | None |
| #6 | F | 87 | Hispanic | 6.0 | Mild | Moderate | Sparse | None | Sparse |
| AD (Braak & Braak score V) | |||||||||
| #7 | M | 83 | Hispanic | 7.0 | Mild | Frequent | Moderate | None | None |
| #8 | F | 92 | Caucasian | 4.8 | Mild | Moderate | Sparse | None | None |
| #9 | F | 99 | Caucasian | 7.5 | Mild | Sparse | None | None | None |
| #10 | M | 76 | African-American | 5.0 | Moderate | Frequent | Moderate | Sparse | Sparse |
| AD (Braak & Braak score VI) | |||||||||
| #11 | F | 80 | ND | 11.0 | Moderate | Moderate | Frequent | None | None |
| #12 | M | 91 | ND | 7.0 | Moderate | Frequent | Frequent | None | None |
Neuropil threads and senile (neuritic) plaques are reported for parietal cortex and refer to assessment by silver (Gallyas) stain or immunohistochemistry. The plaque number includes plaques with and without amyloid cores; sparse (<5/field), mod (6 to 20/field), freq (21 to 30/field).
Abbreviations: F, female; M, male; ND, not documented; PMI, post-mortem interval.
2.2. Synaptosome preparation
Synaptosome-enriched fractions (SEF) were prepared from cryopreserved human brain tissue. Samples (1 to 3 g) were minced and slowly frozen on the day of autopsy in 10% dimethyl sulfoxide and 0.32 M sucrose and stored at −80°C until homogenization. The crude synaptosome fraction was prepared as previously described [29]. Briefly, the minced tissue was homogenized in 10 volumes of 0.32 M sucrose containing protease and phosphatase inhibitors. The homogenate was first centrifuged at 1,000 g for 10 min. The supernatant was centrifuged at 10,000 g for 20 min to obtain the crude synaptosomal pellet (P-2) which contains synaptic terminals that have resealed into functional spheres during homogenization in sucrose. Aliquots of P-2 are routinely cryopreserved in 0.32 M sucrose and stored at −80°C.
2.3. Western blotting analysis
Protein concentrations in synaptosomal preparations were determined using BCA assays (Thermo Scientific, Waltham, MA). Samples were boiled in Laemmli loading buffer (Invitrogen, Carlsbad, CA) and electrophoresed on 8% SDS-page gels (Expedeon, San Diego, CA). Membranes were blocked for 1 h at room temperature in 5% BSA, followed by incubation overnight at 4ºC with the primary antibodies in PBS containing 0.01% Tween 20 (PBS-T) and 1.5% (W/V) BSA and 0.05% sodium azide: NCX1 1:1000 (R3F1, Swant Switzerland); NCX2 1:30 (W1C3 monoclonal IgM antibody) [15] and NCX3 1:1000 (rabbit polyclonal IgG antibody) [15]. Membranes were then incubated for 1 h with the appropriate horseradish peroxidase (HRP)-conjugated (Jackson ImmunoResearch, West Grove, PA): anti-mouse IgG (1:10,000), anti-mouse IgM (1:20,000) and anti-rabbit IgG (1:30,000). Membranes were incubated with SuperSignal West Dura substrate (Thermo Scientific, Waltham, MA) and exposed to an OptiChemi HR Camera 600 (UVP Imaging, Upland, CA). Quantification of proteins was performed following optimal exposure time using the VisionWorksLS Image Acquisition and Analysis software (UVP Imaging, Upland, CA). To strip the immunoblots, membranes were incubated at room temperature in 0.1 M glycine at pH 2.5. At the end of each experiment, membranes were rinsed in large volumes of PBS-T and equal loading of protein was verified by staining PVDF membranes with Coomassie Blue. Control and AD samples were analyzed simultaneously within the same blot. All experiments were performed in duplicate.
2.4. Immunolabeling of synaptosomal fraction
P-2 aliquots were immunolabeled for flow cytometry analysis according to an intracellular antigen staining method [30]. Pellets were fixed in 0.25% buffered paraformaldehyde (PAF) and permeabilized in 0.2% Tween20/PBS (15 min., 37°C). NCX2 antibody was indirectly labeled with Dylight-488 IgM (1:500; Jackson ImmunoResearch, West Grove, PA). NCX1 and NCX3 antibodies were labeled directly with Zenon kit Alexa Fluor 488 or 647 reagents according to manufacturer instructions (Invitrogen, Carlsbad, CA). This mixture was added to P-2 aliquots and incubated at RT for 30 min. Pellets were washed 2 times with 1 ml 0.2% Tween20/PBS, then resuspended in PBS buffer for flow cytometry analysis. The synaptosomal pellet was dispersed for all washes and for incubations with fixative, detergent, antibodies and then collected by centrifugation (1310 × g at 4°C). Control and AD synaptosomes were immunolabeled in the same experiment.
2.5. Flow cytometry
For flow cytometry analysis, immunolabeled synaptosomes were transferred to a flow tube and analyzed as previously described [30]. Data was acquired using a BD-Calibur I analytic flow cytometer (Becton-Dickinson, San Jose, CA) equipped with argon 488 nm and 635 nm diode lasers. Ten thousand particles were collected and analyzed for each sample. Debris was excluded by establishing a size threshold set on forward light scatter. Alexa-488 and Dylight-488 were detected by the FL1 photomultiplier tube detector, whereas Alexa 647 was assessed by the FL4 photomultiplier tube detector. Analysis was performed using FCS Express software (DeNovo Software, Los Angeles, CA).
2.6. Confocal microscopy
Synaptosome-enriched fractions were immunolabeled as described above and washed, then dispersed with a pipette and spread on slides. Slides were dried, coverslipped with Prolong Antifade (Molecular Probes, Eugene, OR), and stored at 4°C. NCX1-3 antibodies were coupled with Alexa 488 and 10G4, SNAP and GFAP antibodies were labeled with Alexa 568. Confocal fluorescence and differential interference contrast images of synaptosomes were taken using a 100X 1.4 Planapo objective lens on a Leica SP2 Confocal Inverted Microscope (Heidelberg, Germany) equipped with argon laser (488 nm excitation) and diode pump (561 nm excitation).
2.7. Data analysis and statistics
The mean ± SEM were calculated using SigmaPlot (Systat Software, San Jose, CA). A Student’s t test was used for simple comparisons of means.
3. Results
3.1. Western blot characterization of NCX1-3 in human cortex
After separation by SDS-page gel, NCX1-3 subtypes were characterized in human brain homogenate and synapse-enriched fraction of a cognitively normal aged control. Immunodetection with the NCX1-specific R3F1 antibody revealed a predominant band at approximately 120 kDa (Fig. 1A). A primary band of similar size was previously reported in rodent brain [15, 31]. Immunoblots labeled with the NCX2-specific W1C3 antibody revealed the full length protein (~ 100 kDa) and a smaller fragment around 60 kDa (Fig. 1B). This NCX2 immunolabeling pattern is consistent with previous studies performed with BHK cells overexpressing NCX2 and rodent brain homogenate [14–15]. Since NCX2 may be particularly prone to proteolytic cleavage, we compared NCX2 immunoreactivity in aged control human brain homogenate and synapse-enriched fractions, prepared within a postmortem interval of 6 h (Fig. 1B left panel), to NCX2 immunoreactivity in freshly isolated samples from C57/Bl6 wild-type mouse (Fig. 1B right panel). The 60 kDa fragment was found in human and murine samples indicating that the post-mortem delay does not significantly affect NCX2 proteolysis. The rabbit polyclonal NCX3 antibody labeled a major band close to 120 kDa in human brain homogenate and synapse-enriched fraction (Fig. 1C) [15, 31]. A band of similar size was previously reported in rodent brain [13, 15].
Fig. 1.

Characterization of the NCX1-3 variants by immunoblots using isoform-specific antibodies on synaptosomal preparation isolated from the parietal cortex of a cognitively normal aged control. Immunoblots for three exchanger subtype-specific antibodies used in the immunohistochemical studies. Brain homogenates (Homog) and synaptosome-enriched fractions (SEF) were separated by SDS-PAGE, blotted onto nitrocellulose and exposed to mouse monoclonal NCX1 antibody R3F1 (A), mouse monoclonal NCX2 antibody W1C3 (B) and rabbit polyclonal NCX3 antibody (C). Right arrows indicate NCX1-3 isoform specific bands. Left arrows indicate the position of the molecular weight ladder.
3.2. NCX2 co-localizes with SNAP-25 in human synaptosome-enriched fractions
Since previous fluorescent studies suggested that W1C3 recognized glial proteins, we examined NCX2 co-localization with GFAP, a glial marker and SNAP-25, a presynaptic marker in human synaptosomes-enriched fractions (SEF). NCX2 was extensively co-localized with SNAP-25 (Fig. 2A–C) compared to GFAP in our SEF (Fig. 2E–G). GFAP immunostaining revealed few glial processes (Fig. 2F) in washed SEF, and only rare NCX2 immunolabeled particles (Fig. 2E) were also positive for GFAP (Fig. 2G). Labeling with SNAP-25 demonstrated the presynaptic identity of the particles in washed SEF (Fig. 2B) and differential contrast images (Fig. 2D, and 2H) revealed the spherical structure and size of the synaptosome particles compared to polystyrene size standards (Fig. 2I–J).
Fig. 2.

Confocal analysis of NCX2 localization in human synaptosome-enriched fraction. A–C: Confocal analysis of washed SEF isolated from a 95 y/o AD case (parietal cortex) and dual labeled for NCX2 (A) and SNAP (B). The merge image and arrowheads show co-localization of NCX2 and SNAP-25 in a subset of synaptosomes (C). E–G, Confocal analysis of washed SEF dual labeled for NCX2 (E) and GFAP (F). The merge image and arrowhead indicate co-localization of NCX2 and GFAP (G). D and H, differential interference contrast images for each field; size standards are illustrated for 0.46 μm (I) and 1.53 μm beads (J).
3.3. Alteration of NCX1, NCX2 and NCX3 proteins in Alzheimer’s disease
NCX1, NCX2 and NCX3 expression was quantified following sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Immunoblotting analyses were performed with washed SEF isolated from normal and AD cases. Tissue was obtained from parietal brain regions A7, A39 and A40. Histopathological staging of AD was performed according to Braak’s classification [32]. Six late stage AD cases (Braak and Braak score IV–VI) were examined and compared to four cognitively normal aged cases. Normal cognitive status was documented in the control group: Mini-Mental Status Examination (MMSE) score was evaluated 2.0 ± 1.1 years before the patient demise and ranged from 27 to 30. Clinical and neuropathological characteristics of each patient are presented in Table 1. Representative NCX1, NCX2 and NCX3 immunoblots of two subjects within each group are shown in Fig. 3A, B and C respectively. Mean relative intensities were expressed as percentage of the control group, taken as 100%. NCX3 levels were reduced by 75% in the AD group (25.91 ± 7.21%, n = 6, p = 0.038) as compared to the control group (100 ± 37.76%, n = 4; Fig. 3C). In parietal cortex, NCX1 and NCX2 protein levels were unchanged in AD patients (Fig. 3A–B). NCX2 expression was determined by quantifying the full length protein (~ 100 KDa).
Fig. 3.
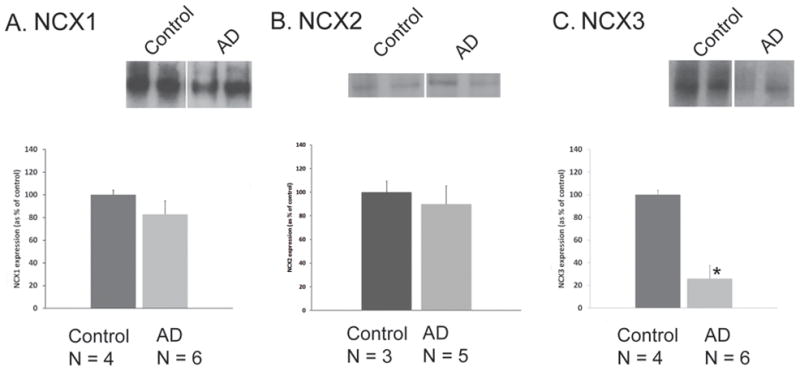
Immunoblot analysis of NCX1 (A), NCX2 (B), and NCX3 (C) protein expression in human parietal cortex isolated from control and AD patients. Representative Western blots of two individuals within each group are shown. Densitometric analyses of NCX1, NCX2, and NCX3 proteins are represented. Values represent means ± SEM. *P < 0.05, compared to controls.
Next, we analyzed NCX1-3 expression in intact synaptosomes using immunohistochemistry methods for flow cytometry. Synaptic terminals are not resolved by light microscopy, even at high magnification. Therefore, the present experiments used flow cytometry (FACS) analysis of synaptosomes, a method that quantifies multiple parameters on each nerve terminal in a sample [29], including fluorescence and forward scatter (FSC), which is proportional to the size of a particle. Gylys et al. have previously reported that non-synaptosomal elements are excluded by drawing a size-based gate that includes only particles that are ~ 0.75 to 1.5 microns [33–34]; thus ten thousand size-gated particles were acquired for each sample, with an acquisition gate based on size standard beads [33–34]. Based on background labeling in the presence of an isotype-specific control antibody, a gate was drawn to include only specific immunolabeling (Fig.4A); variation in protein level was then expressed as a change in the number of positive particles [35]. Background labeling is illustrated for a representative sample in Fig. 4A. Figure 4B illustrates the degree of synaptosomal purity (~ 99%) when synaptosomes were acquired based on size and analyzed as percentage of immunopositive particles for the synaptosomal-associated protein SNAP-25 [29].
Fig. 4.

Flow cytometry analysis of NCX1, NCX2 and NCX3 in control and AD synaptosomes. Human cortical synaptosomal preparation immunolabeled with (A) non-immune IgG and (B) SNAP-25. Representative density plots of normal (C, E and G) and AD (D, F and H) synaptosomes labeled for NCX1 (C–D), NCX2 (E–F) and NCX3 (G–H). The forward scatter parameter on the X-axis is proportional to size. The positive fraction for NCX1, NCX2 and NCX3 isotypes is shown for control vs. AD parietal cortex (I). Values represent means ± SEM. *P < 0.05, compared to controls.
Control and AD synaptosomes were immunolabeled with NCX isotype-specific antibodies and analyzed by flow cytometry analysis. Representative density plots of NCX1, NCX2 and NCX3 immunofluorescence in one normal and one AD patient are shown in Fig. 4C–H. The number of immunofluorescent particles included in the rectangular gate represent the synaptosomes specifically immunolabeled with isotype-specific antibodies for NCX1 (Fig. 4C–D), NCX2 (Fig. 4E–F) and NCX3 (Fig. 4G–H). In the control cohort (n = 4), the mean number of positive synaptosomes was 65.75% (± 2.43), 41.57% (± 10.03) and 45.96% (± 7.41) for NCX1, NCX2 and NCX3 respectively (Fig. 4I). Consistent with the immunoblotting analysis, NCX3 levels were significantly reduced (29.32 ± 4.05%, n = 8, p = 0.022) in the AD group compared to controls. NCX2 levels were significantly increased (64.57 ± 5.22%, n = 7, p = 0.049) in the AD cohort (Fig. 4I). This modest but significant increase in NCX2 expression was not observed by Western Blot. Flow cytometry analysis is a more sensitive and precise method of quantification than densitometric analysis of immunoblots: it quantifies individual immunolabeled synaptosomes in a population that is > 90% pure (Fig. 4B) as non-synaptosomal elements are excluded by size-gating [33–34, 36]. NCX1 expression was not significantly modified in AD (66.66 ± 2.83%, n = 8, p > 0.05).
3.4. NCX1-3 co-localized with Aβ in AD synaptosomes
Aβ presence in AD synaptosomes was previously reported [29]. In this study, we looked for Aβ and NCX1-3 co-localization within individual synaptic terminals of AD patients. Samples were dual labeled for Aβ (10G4 antibody) and NCX1-3 specific antibodies then analyzed by flow cytometry. Only size-gated particles were analyzed to ensure ~ 90% synaptosomal purity [33]. Ten thousand synaptosomes were acquired. The number of single-positive (Aβ−/NCX1-3+) and double-positive (Aβ+/NCX1-3+) were determined using quadrant analysis method. Positive immunofluorescent synaptosomes were identified by drawing a quadrant that excludes background labeling in the lower right, upper left and upper right quadrants (Fig. 5A). The degrees of Aβ/NCX1, Aβ/NCX2 and Aβ/NCX3 co-localization in a representative sample from AD parietal cortex are illustrated in Fig. 5B, 5C and 5D respectively. Particles positive for NCX1-3 only (Aβ−/NCX1-3+) are in the lower right quadrant. Particles positive for Aβ only (Aβ+/NCX1-3−) are in the upper left quadrant and dual positives (Aβ+/NCX1-3+) are in the upper right quadrant. In AD parietal cortex, a significantly higher fraction of double-positive synaptosomes was observed for all NCX isotypes related to the number of single-positive (Fig. 5E). The percentage of Aβ+/NCX1+ was 37.30 ± 2.58% (n = 7, p = 0.014) as compared to 26.11 ± 2.93% (n = 4) of Aβ−/NCX1+. The percentage of Aβ+/NCX2+ was 49.66 ± 4.43% (n = 8, p < 0.01) compared to 12.63 ± 1.77 % (n = 4) of Aβ−/NCX2+. The percentage of Aβ+/NCX3+ was 18.93 ± 2.91% (n = 8, p < 0.01) compared to 8.26 ± 1.43% (n = 4) of Aβ−/NCX3+.
Fig. 5.
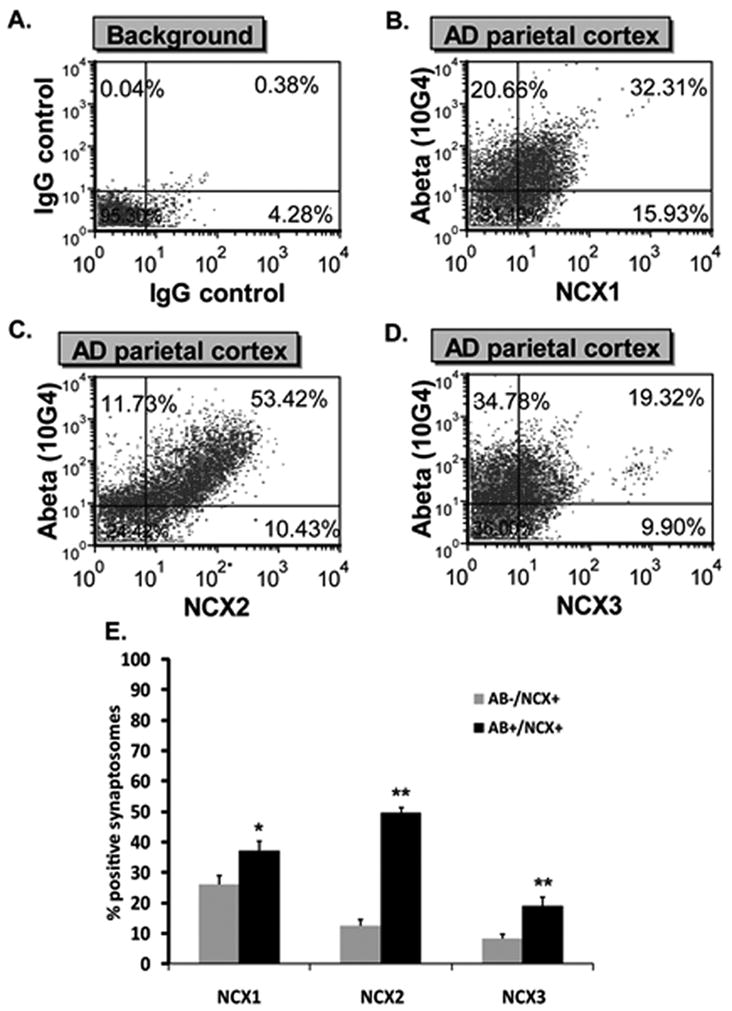
Co-localization of NCX1-3 and Aβ. A. Quadrant analysis of human cortical synaptosomal preparation immunolabeled with non-immune IgG. B–D: Quadrant analysis for a representative sample of AD parietal cortex double labeled for NCX1 and Aβ (B), NCX2 and Aβ (C) and NCX3 and Aβ (D). Particles positive for Aβ only are in the upper left quadrant and particles positive for both Aβ and NCXs are in the upper right quadrant. Numbers represent the percentage of synaptosomes in each quadrant. (E) NCX1-3 variation in single-labeled (Aβ−/NCX+ only, grey bars) and dual-labeled (Aβ+/NCX+, black bars) fractions. Values represent means ± SEM. *P < 0.05 and **P < 0.01, single-labeled compared to dual-labeled fractions.
Co-localization of NCX1-3 and Aβ within spherical particles (0.5–1.5 μm) was confirmed by confocal analysis of SEF isolated from AD superior parietal cortex (Fig. 6). Synaptosomal preparations were dual labeled for Aβ (Fig. 6B, 6F and 6J) and NCX1 (Fig. 6A), NCX2 (Fig. 6E) and NCX3 (Fig. 6I) respectively. Arrowheads showed co-localization of Aβ and NCX1-3 in a subset of synaptosomes; differential contrast images (Fig. 6D, 6H and 6L) revealed the spherical structure of the synaptosome particles.
Fig. 6.

Confocal analysis of NCX1-3 and Aβ co-localization. Washed SEF isolated from a 95 y/o AD case (parietal cortex) were dual labeled for Aβ (B, F and J) and NCX1 (A), NCX2 (E), NCX3 (I) respectively. Overlay images with yellow color indicating co-localization (arrowheads) of NCX1-3 and Aβ in a subset of synaptosomes (C, G and K). D, H and L: differential interference contrast images for each field.
4. Discussion
The results of this study demonstrated for the first time that selective changes occur in the pattern of NCX1-3 protein expression in AD synaptosomes. Major findings can be summarized as follows: (i) NCX1-3 are widely expressed in human synaptosomes isolated from parietal cortex of AD and control patients; (ii) NCX2 expression was modestly but significantly increased and NCX3 levels were significantly reduced in AD terminals compared to controls and (iii) all NCX isotypes co-localized with Aβ in AD parietal cortex.
Previous in situ studies in rat neocortex reported that NCX1-3 isoforms are expressed at the plasma membrane of neurons of adult rat brain [14, 37], but only minor presynaptic expression was observed for NCX1-3 isotypes [14]. However, our flow cytometry analyses support a high density of NCX1-3 molecules in presynaptic terminals. This apparent conflict may result from different fixation protocols; labeling methods for flow cytometry use light fixation (0.25% PAF) compared to electron microscopy (EM) immunohistochemistry (4% PAF or 1% glutaraldehyde). The heavy fixation required for EM analysis is more likely to mask NCX1-3 epitopes. The possibility that NCX1-3 antibodies cross-react with nonspecific synaptosomal antigens in our flow experiments is unlikely: our immunoblots on human synaptosomal preparations revealed size specific NCX1-3 bands as described in rodent neocortex and BHK cell lines overexpressing NCX1-3 [14–15]. Furthermore, NCX activity has been measured in brain synaptosomes [38–39], providing functional support to our findings, and the present results are also supported by work showing NCX expression and function in presynaptic boutons of cultured hippocampal neurons [37, 40].
4.1. NCX1-3 in AD terminals
Recent avenues of research have provided evidence in support of the “Ca2+ hypothesis of AD”. Where does NCX function in this process? Because of its large capacity for intracellular Ca2+, NCX is likely to play an important role in nerve terminal physiology, particularly in situations like AD, where elevated [Ca2+i] leads to cell demise. A possible role for NCX in AD was reported in the early 1990’s prior to the cloning of all NCX isoforms [41]. Colvin and colleagues showed that NCX activity was increased in AD plasma membrane vesicles but were unable to determine whether the increase in Ca2+ transport was due to changes in NCX protein levels or in plasmalemmal lipid composition [41]. They suggested that increased NCX activity may result from the regulation of NCX-specific isoforms. However, they were not able to investigate their hypothesis because of the lack of NCX isotype-specific antibodies. Since then, NCX1, NCX2 and NCX3 proteins have been identified. The generation of NCX1-3 specific antibodies has allowed us to study their specific expression in terminals isolated from AD and cognitively normal individuals.
In the present study, two different methods (flow cytometry and immunoblotting) were used to quantify NCX1-, NCX2- and NCX3-specific immunoreactivity in intact synaptosomes isolated from brain tissue of cognitively normal and AD patients. Our flow cytometry data revealed that NCX1 is 1.5 times more abundant than NCX2 and NCX3 in the parietal cortex of cognitively normal patients. Quantitative flow cytometry also showed that NCX2 levels were increased and NCX3 levels reduced in the parietal cortex of AD patients. NCX2 upregulation in AD terminals may be the result of a compensatory mechanism to balance the loss of NCX3 expression. Accumulation of NCX2 might also result from a blockade of retrograde axonal transport in AD neurons, a possible downstream effect of Aβ accumulation. The decrease of NCX3 immunoreactivity in nerve terminals may be associated with cholinergic and glutamatergic synapse loss, which are pathologic features of late stage AD [42–43], or might result from decreased transcriptional activity in AD neurons.
A net increase of NCX proteins may result in increased NCX activity in AD brains [41]. Such an increase would protect nerve terminals that are unable to regulate increases in [Ca2+i] caused by Aβ-induced activation of voltage-dependent calcium channels (VDCC), formation of Ca2+-permeable pores in the plasma membrane, or through ER Ca2+-depletion signaling pathways in AD [44].
The present experiments demonstrated co-localization of NCX1, NCX2 and NCX3 with Aβ, and all three NCX isoforms were up-regulated in pathological terminals that contained Aβ. In terms of peptide species, soluble Aβ oligomers are thought to be the key peptides associated with cognitive decline in AD [45]. In the present experiments, the most likely oligomeric species associated with NCX1-3 include a 56 kDa assembly and small oligomers < 20 kDa, which are prominent in synaptic terminals [29, 46]. Increased levels of NCX1, NCX2 and NCX3 in Aβ-positive terminals are likely to follow oligomeric Aβ–induced Ca2+ imbalance and may be an indication of the participation of NCX1-3 in Ca2+ homeostasis in surviving synapses affected by the intraterminal toxicity of Aβ oligomers [47]. Recent studies have demonstrated that NCX1 and NCX3 are up-regulated in response to the activation of the pro-survival kinase, Akt [48] Since Akt is overactive in AD neurons [49], it is attractive to speculate that NCX1 and NCX3 up-regulation in Aβ-positive terminals may reflect a synaptic response to Aβ buildup and increased [Ca2+i] [49–50]. Thus, NCX1 and NCX3 may contribute to the survival of nerve terminals with Aβ-induced [Ca2+ elevations, consistent with previous observations of increased Na+-dependent Ca2+i] uptake in AD terminals [41].
Previous anatomical studies showed that all isoforms are widely expressed in neuronal structures of rat cerebral cortex and hippocampus [14, 37]. However, additional studies on their localization in chemically defined neuronal population (i.e. glutamatergic, cholinergic) and regional distribution in human brain are still needed to understand the NCX isoform-specific role in AD pathophysiology. Moreover, pharmacological studies are not sufficient to study the contribution of different NCX subtypes to Ca2+ homeostasis in AD terminals due to the lack of selective NCX subtype inhibitors. Further studies using NCX1 conditional knockout mice, NCX2- and NCX3- null-mutant mice will contribute to elucidating NCX1-3 specific function in Aβ-induced neurodegeneration and synapse loss.
4.2. Conclusion
In summary, our results demonstrate a selective regulation of NCX1, NCX2 and NCX3 isoforms in AD cortex, specifically in terminals containing Aβ. Further studies will concentrate on in vitro and in vivo examination of NCX-related Ca2+ fluctuations contributing to the progression of AD pathology.
Acknowledgments
This work was supported by grants from the Alzheimer’s Association (NIRG-03-6103) and the NIH (5R01AG027465) (to K.H.G) and the NIH (CA 16042 and AI 28697) (to the Jonsson Cancer Center at the University of California Los Angeles). Tissue was obtained from the Alzheimer’s Disease Research Center Neuropathology Cores of the University of Southern California (NIA 050 AG05142) and the University of California Los Angeles (NIA P50 AG 16570). H.V.V. supported in part by the Daljit S. and Elaine Sarkaria Chair in Diagnostic Medicine. The authors wish to thank Drs. E. Porzig, D. Nicoll and K.D. Phillipson for generously providing NCX2 and NCX3 antibodies. We acknowledge Dr. P. Kehoe for her useful discussion during the writing of this manuscript. The authors wish to thank the families who generously donated the brain samples for the present research program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Khachaturian ZS. Hypothesis on the regulation of cytosol calcium concentration and the aging brain. Neurobiol Aging. 1987;8:345–346. doi: 10.1016/0197-4580(87)90073-x. [DOI] [PubMed] [Google Scholar]
- 2.Stutzmann GE. The pathogenesis of Alzheimers disease Is It a lifelong “Calciumopathy”? Neuroscientist. 2007;13:546–559. doi: 10.1177/1073858407299730. [DOI] [PubMed] [Google Scholar]
- 3.Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium. 2003;34:385–97. doi: 10.1016/s0143-4160(03)00128-3. [DOI] [PubMed] [Google Scholar]
- 4.Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–25. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- 6.Annunziato L, Pignataro G, Di Renzo GF. Pharmacology of brain Na+/Ca2+ exchanger: From molecular biology to therapeutic perspectives. Pharmacol Rev. 2004;56:633–654. doi: 10.1124/pr.56.4.5. [DOI] [PubMed] [Google Scholar]
- 7.Quednau BD, Nicoll DA, Philipson KD. Tissue specificity and alternative splicing of the Na+/Ca2+ exchanger isoforms NCX1, NCX2, and NCX3 in rat. Am J Physiol. 1997;272:C1250–61. doi: 10.1152/ajpcell.1997.272.4.C1250. [DOI] [PubMed] [Google Scholar]
- 8.Nicoll DA, Quednau BD, Qui Z, Xia YR, Lusis AJ, Philipson KD. Cloning of a third mammalian Na+-Ca2+ exchanger, NCX3. J Biol Chem. 1996;271:24914–21. doi: 10.1074/jbc.271.40.24914. [DOI] [PubMed] [Google Scholar]
- 9.Li Z, Matsuoka S, Hryshko LV, Nicoll DA, Bersohn MM, Burke EP, Lifton RP, Philipson KD. Cloning of the NCX2 isoform of the plasma membrane Na(+)-Ca2+ exchanger. J Biol Chem. 1994;269:17434–9. [PubMed] [Google Scholar]
- 10.Nicoll DA, Longoni S, Philipson KD. Molecular cloning and functional expression of the cardiac sarcolemmal Na(+)-Ca2+ exchanger. Science. 1990;250:562–5. doi: 10.1126/science.1700476. [DOI] [PubMed] [Google Scholar]
- 11.Jeon D, Yang Y-M, Jeong M-J, Philipson KD, Rhim H, Shin H-S. Enhanced learning and memory in mice lacking Na+/Ca2+ exchanger 2. Neuron. 2003;38:965–976. doi: 10.1016/s0896-6273(03)00334-9. [DOI] [PubMed] [Google Scholar]
- 12.Sokolow S, Manto M, Gailly P, Molgo J, Vandebrouck C, Vanderwinden JM, Herchuelz A, Schurmans S. Impaired neuromuscular transmission and skeletal muscle fiber necrosis in mice lacking Na/Ca exchanger 3. J Clin Invest. 2004;113:265–73. doi: 10.1172/JCI18688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Molinaro P, Cuomo O, Pignataro G, Boscia F, Sirabella R, Pannaccione A, Secondo A, Scorziello A, Adornetto A, Gala R, Viggiano D, Sokolow S, Herchuelz A, Schurmans S, Di Renzo G, Annunziato L. Targeted disruption of Na+/Ca2+ exchanger 3 (NCX3) gene leads to a worsening of ischemic brain damage. J Neurosci. 2008;28:1179–84. doi: 10.1523/JNEUROSCI.4671-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minelli A, Castaldo P, Gobbi P, Salucci S, Magi S, Amoroso S. Cellular and subcellular localization of Na+-Ca2+ exchanger protein isoforms, NCX1, NCX2, and NCX3 in cerebral cortex and hippocampus of adult rat. Cell Calcium. 2007;41:221–34. doi: 10.1016/j.ceca.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Thurneysen T, Nicoll DA, Philipson KD, Porzig H. Sodium/calcium exchanger subtypes NCX1, NCX2 and NCX3 show cell-specific expression in rat hippocampus cultures. Brain Res Mol Brain Res. 2002;107:145–56. doi: 10.1016/s0169-328x(02)00461-8. [DOI] [PubMed] [Google Scholar]
- 16.Papa M, Canitano A, Boscia F, Castaldo P, Sellitti S, Porzig H, Taglialatela M, Annunziato L. Differential expression of the Na+-Ca2+ exchanger transcripts and proteins in rat brain regions. J Comp Neurol. 2003;461:31–48. doi: 10.1002/cne.10665. [DOI] [PubMed] [Google Scholar]
- 17.DiPolo R, Beauge L. Differential up-regulation of Na+-Ca2+ exchange by phosphoarginine and ATP in dialysed squid axons. J Physiol. 1998;507(Pt 3):737–47. doi: 10.1111/j.1469-7793.1998.737bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linck B, Qiu Z, He Z, Tong Q, Hilgemann DW, Philipson KD. Functional comparison of the three isoforms of the Na+/Ca2+ exchanger (NCX1, NCX2, NCX3) Am J Physiol. 1998;274:C415–23. doi: 10.1152/ajpcell.1998.274.2.C415. [DOI] [PubMed] [Google Scholar]
- 19.Wakimoto K, Fujimura H, Iwamoto T, Oka T, Kobayashi K, Kita S, Kudoh S, Kuro-o M, Nabeshima Y-i, Shigekawa M, Imai Y, Komuro I. Na+/Ca2+ exchanger-deficient mice have disorganized myofibrils and swollen mitochondria in cardiomyocytes. Comp Biochem and Physiol B: Biochem and Mol Biol. 2003;135:9–15. doi: 10.1016/s1096-4959(03)00057-5. [DOI] [PubMed] [Google Scholar]
- 20.Koushik SV, Wang J, Rogers R, Moskophidis D, Lambert NA, Creazzo TL, Conway SJ. Targeted inactivation of the sodium-calcium exchanger (Ncx1) results in the lack of a heartbeat and abnormal myofibrillar organization. FASEB J. 2001;15:1209–11. doi: 10.1096/fj.00-0696fje. [DOI] [PubMed] [Google Scholar]
- 21.Cho CH, Lee SY, Shin HS, Philipson KD, Lee CO. Partial rescue of the Na+-Ca2+ exchanger (NCX1) knock-out mouse by transgenic expression of NCX1. Exp Mol Med. 2003;35:125–35. doi: 10.1038/emm.2003.18. [DOI] [PubMed] [Google Scholar]
- 22.Conway SJ, Kruzynska-Frejtag A, Wang J, Rogers R, Kneer PL, Chen H, Creazzo T, Menick DR, Koushik SV. Role of sodium-calcium exchanger (Ncx1) in embryonic heart development: a transgenic rescue? Ann N Y Acad Sci. 2002;976:268–81. doi: 10.1111/j.1749-6632.2002.tb04749.x. [DOI] [PubMed] [Google Scholar]
- 23.Cross JL, Meloni BP, Bakker A, Sokolow S, Herchuelz A, Schurmans S, Knuckey NW. Neuronal injury in NCX3 knockout mice following permanent focal cerebral ischemia and in NCX3 knockout cortical neuronal cultures following oxygen-glucose deprivation and glutamate exposure. J Exp Stroke & Transl Med. 2009;2 [Google Scholar]
- 24.Ranciat-McComb NS, Bland KS, Huschenbett J, Ramonda L, Bechtel M, Zaidi A, Michaelis ML. Antisense oligonucleotide suppression of Na+/Ca2+ exchanger activity in primary neurons from rat brain. Neurosci Lett. 2000;294:13–16. doi: 10.1016/s0304-3940(00)01524-x. [DOI] [PubMed] [Google Scholar]
- 25.Mattson MP, Barger SW, Cheng B, Lieberburg I, Smith-Swintosky VL, Rydel RE. beta-Amyloid precursor protein metabolites and loss of neuronal Ca2+ homeostasis in Alzheimer’s disease. Trends Neurosci. 1993;16:409–14. doi: 10.1016/0166-2236(93)90009-b. [DOI] [PubMed] [Google Scholar]
- 26.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–9. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bezprozvanny I. Amyloid Goes Global. Sci Signal. 2009;2:pe16. doi: 10.1126/scisignal.263pe16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration. 1996;5:417–21. doi: 10.1006/neur.1996.0056. [DOI] [PubMed] [Google Scholar]
- 29.Fein JA, Sokolow S, Miller CA, Vinters HV, Yang F, Cole GM, Gylys KH. Co-localization of amyloid beta and tau pathology in Alzheimer’s disease synaptosomes. Am J Pathol. 2008;172:1683–92. doi: 10.2353/ajpath.2008.070829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gylys KH, Fein JA, Cole GM. Quantitative characterization of crude synaptosomal fraction (P-2) components by flow cytometry. J Neurosci Res. 2000;61:186–92. doi: 10.1002/1097-4547(20000715)61:2<186::AID-JNR9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 31.Gobbi P, Castaldo P, Minelli A, Salucci S, Magi S, Corcione E, Amoroso S. Mitochondrial localization of Na+/Ca2+ exchangers NCX1-3 in neurons and astrocytes of adult rat brain in situ. Pharmacol Res. 2007;56:556–65. doi: 10.1016/j.phrs.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 32.Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction. Eur Neurol. 1993;33:403–8. doi: 10.1159/000116984. [DOI] [PubMed] [Google Scholar]
- 33.Gylys KH, Fein JA, Yang F, Cole GM. Enrichment of presynaptic and postsynaptic markers by size-based gating analysis of synaptosome preparations from rat and human cortex. Cytometry A. 2004;60:90–6. doi: 10.1002/cyto.a.20031. [DOI] [PubMed] [Google Scholar]
- 34.Gylys KH, Fein JA, Yang F, Miller CA, Cole GM. Increased cholesterol in Abeta-positive nerve terminals from Alzheimer’s disease cortex. Neurobiol Aging. 2007;28:8–17. doi: 10.1016/j.neurobiolaging.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 35.Lopez JR, Lyckman A, Oddo S, Laferla FM, Querfurth HW, Shtifman A. Increased intraneuronal resting [Ca2+] in adult Alzheimer’s disease mice. J Neurochem. 2008;105:262–71. doi: 10.1111/j.1471-4159.2007.05135.x. [DOI] [PubMed] [Google Scholar]
- 36.Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer’s disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165:1809–17. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Canitano A, Papa M, Boscia F, Castaldo P, Sellitti S, Taglialatela M, Annunziato L. Brain distribution of the Na+/Ca2+ exchanger-encoding genes NCX1, NCX2, and NCX3 and their related proteins in the central nervous system. Ann N Y Acad Sci. 2002;976:394–404. doi: 10.1111/j.1749-6632.2002.tb04766.x. [DOI] [PubMed] [Google Scholar]
- 38.Bartschat DK, Blaustein MP. Calcium-activated potassium channels in isolated presynaptic nerve terminals from rat brain. J Physiol. 1985;361:441–57. doi: 10.1113/jphysiol.1985.sp015654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fontana G, Rogowski RS, Blaustein MP. Kinetic properties of the sodium-calcium exchanger in rat brain synaptosomes. J Physiol. 1995;485(Pt 2):349–64. doi: 10.1113/jphysiol.1995.sp020734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reuter H, Porzig H. Localization and functional significance of the Na+/Ca2+exchanger in presynaptic boutons of hippocampal cells in culture. Neuron. 1995;15:1077–1084. doi: 10.1016/0896-6273(95)90096-9. [DOI] [PubMed] [Google Scholar]
- 41.Colvin RA, Davis N, Wu A, Murphy CA, Levengood J. Studies of the mechanism underlying increased Na+/Ca2+ exchange activity in Alzheimer’s disease brain. Brain Res. 1994;665:192–200. doi: 10.1016/0006-8993(94)91338-2. [DOI] [PubMed] [Google Scholar]
- 42.Kashani A, Lepicard E, Poirel O, Videau C, David JP, Fallet-Bianco C, Simon A, Delacourte A, Giros B, Epelbaum J, Betancur C, El Mestikawy S. Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol Aging. 2008;29:1619–30. doi: 10.1016/j.neurobiolaging.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 43.Tiraboschi P, Hansen LA, Alford M, Masliah E, Thal LJ, Corey-Bloom J. The decline in synapses and cholinergic activity is asynchronous in Alzheimer’s disease. Neurology. 2000;55:1278–83. doi: 10.1212/wnl.55.9.1278. [DOI] [PubMed] [Google Scholar]
- 44.Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008;31:454–63. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–7. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 46.Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-[beta] protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 47.Green KN, LaFerla FM. Linking calcium to Abeta and Alzheimer’s disease. Neuron. 2008;59:190–4. doi: 10.1016/j.neuron.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 48.Formisano L, Saggese M, Secondo A, Sirabella R, Vito P, Valsecchi V, Molinaro P, Di Renzo G, Annunziato L. The Two Isoforms of the Na+/Ca2+ Exchanger, NCX1 and NCX3, Constitute Novel Additional Targets for the Prosurvival Action of Akt/Protein Kinase B Pathway. Mol Pharmacol. 2008;73:727–737. doi: 10.1124/mol.107.042549. [DOI] [PubMed] [Google Scholar]
- 49.Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O’Connor R, O’Neill C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J Neurochem. 2005;93:105–17. doi: 10.1111/j.1471-4159.2004.02949.x. [DOI] [PubMed] [Google Scholar]
- 50.Martín D, Salinas M, López-Valdaliso R, Serrano E, Recuero M, Cuadrado A. Effect of the Alzheimer amyloid fragment Aβ (25–35) on Akt/PKB kinase and survival of PC12 cells. J Neurochem. 2001;78:1000–1008. doi: 10.1046/j.1471-4159.2001.00472.x. [DOI] [PubMed] [Google Scholar]