

Abstract
Esophageal adenocarcinoma is a major cause of cancer death in men in the developed world. Continuing poor outcomes with conventional therapies that predominantly target apoptosis pathways have lead to increasing interest in treatments that target the cell cycle. A large international effort has led to the development of a large number of inhibitors, which target cell cycle kinases, including cyclin-dependent kinases, Aurora kinases and polo-like kinase. Initial phase I/II trials in solid tumors have often demonstrated only modest clinical benefits of monotherapy. This may relate in part to a failure to identify the patient populations that will gain the most clinical benefit. Newer compounds lacking the side effect profile of first-generation compounds may show utility as adjunctive treatments targeted to an individual’s predicted response to treatment.
Keywords: Esophageal adenocarcinoma, Cell cycle, Cyclin-dependent kinase, Aurora kinases, Polo-like kinase
INTRODUCTION
Esophageal cancer is a major cause of cancer death worldwide[1]. It was the fourth most common cause of death from cancer in men in the United Kingdom between 2004 and 2006[2]. Although in the developed world the incidence and mortality of cancer in general has decreased with advances in diagnosis and treatment, the incidence and mortality of esophageal carcinoma have increased[1].
Esophageal cancer carries a poor prognosis with a 5-year survival rate of < 10%[3]. This probably reflects the fact that the majority of esophageal cancers present late with symptoms after invasion of the muscularis propria and lymph node metastasis have occurred[4]. Extensive disease means that few patients are suitable for definitive surgical therapy[4,5]. Poor outcomes from conventional therapies including surgery and radiochemotherapy have led to increasing interest in understanding the molecular mechanisms that underpin the development of esophageal cancer. This may assist in developing new diagnostic techniques and identifying potential therapeutic targets.
The mechanism by which cells reproduce has fascinated biologists since Virchow’s 1855 observation that cells could only arise from pre-existing cells. By the early 20th century, pathologists had recorded extensive descriptions of the cytological events of cell division, including division of the nucleus and partitioning of the cytoplasm to the formation of two daughter cells[6]. It has become increasingly clear since those early descriptions of the normal cell cycle that disorders in this process can lead to disease. It was not however until the 1970s, that molecular biology allowed a deeper understanding of the cell cycle and its role in health, disease and cancer development. The past three decades, in particular, have seen major advances in our understanding of the genetic and molecular mechanisms by which cells reproduce and how this process is regulated and controlled. It has also been aptly described that cell cycle deregulation, in the form of growth self-sufficiency and insensitivity to growth inhibitory signals, have become fundamental hallmarks of cancer development[7-9]. Targeting these pathways in cancer development for diagnostic and therapeutic use has become increasingly important. We assess in this review the potential for targeting the cell cycle to treat esophageal adenocarcinoma.
HALLMARKS OF CANCER
It is clear that cellular reproduction is carefully controlled and regulated to prevent uncontrolled proliferation of cells[10]. A number of alterations in cell physiology are required to lead to carcinogenesis[7]. First, a cell must become able to move from its dormant inactive state (known as quiescence) to enter the cell cycle without stimulation from external growth factors. Second, the cell must lose response to growth-inhibitory signals. Cells must evade senescence and programmed cell death to gain limitless replicative potential. Finally, it must be able to develop and maintain an adequate blood supply (angiogenesis), which allows the cancer cell to invade and metastasize throughout the organism[11].
Many genes responsible for the carcinogenesis have been identified. Broadly, they fall into two categories: oncogenes and tumor suppressor genes. Oncogenes are created by mutations in genes that cause them to become constitutively active, whereas in tumor suppressor genes, mutations reduce or inactive the gene product[12]. Oncogenes and tumor suppressor genes increase tumor cell number by stimulation of cell division or prevention of cell death.
CELL CYCLE
Embyronic cells can undergo DNA replication and nuclear division at rapid rates. A full cycle of embyronic cell division can last just 30 min[13]. Division of adult stem cells requires more complex control (Figure 1). Gaps or pauses are inserted between the phases of nuclear division (M phase) and DNA synthesis (S phase). These gaps are known as G1 (between M and S phases) and G2 (between S and M phases).
Figure 1.
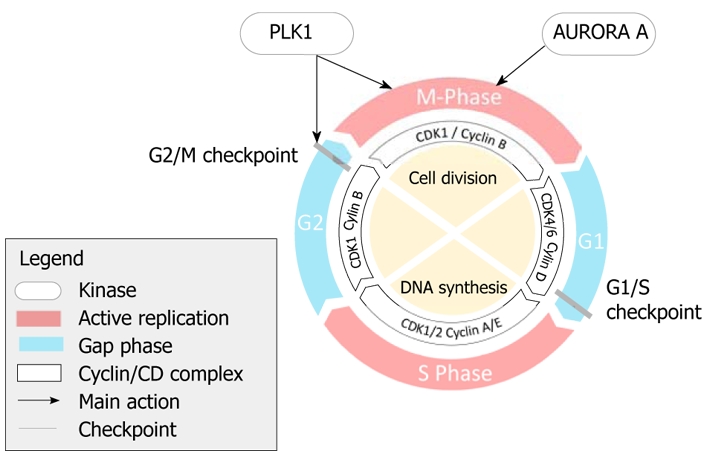
Cell cycle.
Events in the cell cycle happen in a temporally organized sequence, with later events depending on successful completion of earlier events[14].
Control of the cell cycle is driven by the cyclin-dependent kinases (CDKs), a family of serine/threonine kinases. Cells cannot enter S phase, without CDK activation. In order to become catalytically active, CDKs need to bind to a cyclin subunit that acts as an activator. CDKs can also be modulated by inhibitors such as CDK inhibitor 1A (p21CIP1), CDK inhibitor 1B (p27KIP1) or CDK inhibitor 2B (p15INK4B)[10]. It has previously been thought that mammalian cells require the sequential activation of a number of the CDKs to complete the cell cycle successfully[15]. Recent evidence from mouse models has suggested that CDK1 alone is sufficient to complete the cell cycle, although other CDKs are required for normal development and cell type specialization[16]. Cell cycle defects can contribute to esophageal cancer development in a number of different ways (Figure 2).
Figure 2.

Compounds targeting the cell cycle.
Mitosis itself contains a series of phases that lead to chromosome separation and cell division. Mitosis is a vital step in the cell cycle, which involves carefully regulated interactions between multiple proteins. Abnormalities throughout the cell cycle can lead to genomic instability through unrestrained proliferation or defects in the transmission of genetic information to daughter cells. A number of established chemotherapy agents, including the vinca alkaloids and the taxanes work by targeting the mitotic phase of the cell cycle.
CELL CYCLE CHECKPOINTS
Cells need mechanisms that prevent progression of the cell cycle if there is significant genomic damage, until the damage is repaired or the cell undergoes apoptosis. These have become known as cell cycle checkpoints. There are two major checkpoints: the G1/S checkpoint and the G2/M checkpoint. Checkpoint kinases ATM and ATR mediate these checkpoints, through effector kinases such as CHK1 and CHK2, by preventing activation of CDKs and progression through the cell cycle[17]. Double-stranded DNA breaks activate preferentially ATM, whereas UV light activates ATR kinase. Defects in this DNA damage response can contribute to cancer formation by allowing tumor cell survival despite genome instability and enhanced mutation rates[18,19]. The DNA damage response is commonly activated in early neoplastic lesions[20,21].
G1/S checkpoint
The G1/S checkpoint occurs towards the end of the G1 phase, prior to entry into G2. During G1, the cell remains responsive to external mitogenic and anti-mitogenic stimuli. These can either cause the cell to become quiescent (entering the GO phase) or allow re-entry to the cell cycle. This decision is controlled by the pocket protein RB. Immediately after mitosis, RB is dephosphorylated by protein phosphatase type 1. Whilst in this dephosphorylated state, RB binds to a group of transcription factors called E2Fs and inhibits their activity. During G1, RB is hypophosphorylated by the complex of CDK4 and cyclin D. CDK2 and cyclin E complexes then act to hyperphosphorylate RB, which causes dissociation from E2Fs. Free E2Fs trigger increased transcription of CDK2 and cyclin E, which creates a positive feedback loop that drives the cell into DNA synthesis (S phase). CDC25 phosphatases act to regulate CDK and cyclin complexes by removing inhibitory phosphate groups thereby promoting cell cycle progression[13]. In genomic damage, CHK2 activates the p53 pathway, which stimulates production of p21CIP1 as well as phosphorylation of CDC25A. This prevents activation of the CDK/cyclin complexes[13].
G2/M Checkpoint
The G2/M checkpoint acts as a final check to prevent mitosis occurring if the genome is damaged. A complex of cyclin B and CDK1 regulates this transition. Throughout G2, the inhibitory kinases CHK1, WEE1 and MYT1 phosphorylate CDK1, which prevents its activation and progression to mitosis. PLK1 protein levels begin to accumulate during S phase and G2/M phases, having been relatively low during G1[22,23]. PLK1 transcription is most abundant in cells that are in G2/M phase[24]. In the absence of DNA damage, PLK1 is phosphorylated by Aurora A at its phosphorylation site at T210[25]. Phosphorylated PLK1 then activates CDC25 phosphatases that remove inhibitory phosphates from the ATP-binding site located at Thr14 and Tyr15 in human CDK1. This causes the activation of the CDK1/cyclin B complex and drives the cell into mitosis[26]. PLK1 also increases phosphorylation-dependent cyclin B import to the nucleus[27]. PLK1 phosphorylates WEE1 and MYT1, which leads to ubiquitination and degradation of WEE1 and inhibition of MYT1[28,29]. PLK1 is then inactivated and degraded during anaphase by ubiquitin-dependent degradation mediated by the anaphase promoting complex[30]. Cell cultures show severely impaired growth when PLK1 is either overexpressed or functionally depleted[31,32].
CELL CYCLE AS A TARGET FOR CANCER THERAPEUTICS
Many oncogenes and tumor suppressors have downstream effects on cellular functions involving cell cycle entry and exit. Healthy or normal cells have the ability to stop at predetermined checkpoints in the cell cycle in the presence of damage or unfavorable conditions. Cancer cells develop mechanisms that eliminate these checkpoints, which leads to uncontrolled proliferation. One example of this is the INK4 family member p16. This occurs as a result of epigenetic silencing by DNA hypermethylation at the p16 promoter, which leads to reduced transcription and loss of gene expression. p16 is a CDK inhibitor and loss of p16 function leads to unrestrained cellular proliferation. This has been demonstrated to occur with a number of different tumors[33]. Abnormalities of p16 function have been described in Barrett’s esophagus and esophageal adenocarcinoma[34]. DNA hypermethylation of the p16 promoter has also been shown to be a strong predictor of the progression to high-grade dysplasia and esophageal adenocarcinoma[35].
CDK inhibitors
Abnormal expression of CDKs and their partner cyclins has been noted in esophageal cancer[36-39]. Polymorphisms of CCND1, which encodes cyclin D1 has been shown to be associated with an increased risk of esophageal adenocarcinoma[40]. CCND1 amplification and nuclear staining of cyclin D1 have been shown to correlate negatively with survival[41,42]. Abnormal activity of the CDK/cyclin complexes in esophageal adenocarcinoma has been shown to be a marker of acquired chemoradioresistance[42,43]. The observation that inhibition of CDKs leads to cell cycle arrest and apoptosis has lead to the development of CDK inhibitors as antitumor drugs. There are a number of drugs that target these pathways. The pioneer compound for this group is flavopiridol, a semi-synthetic inhibitory flavonoid of CDKs. Flavopiridol prevents the phosphorylation and activation of CDK1, CDK2, CDK4 and CDK6, which leads to reduced expression of cyclin D1, cell cycle arrest, and induction of apoptosis[44].
In vitro, it has been demonstrated that even at nanomolar doses, flavopiridol can enhance the antitumor activity of cytotoxic drugs by increasing apoptosis[45]. Phase I and II studies have been undertaken with various combinations of chemotherapeutic agents with variable results. Most promising is the combination with irinotecan and cisplatin. A phase I trial of relapsed gastric and esophageal cancer patients showed that eight out 14 patients achieved a partial response[46]. Further clinical studies are awaited.
Aurora kinases inhibitors
The Aurora kinase family is an important family of serine/threonine kinases that are evolutionarily conserved and act as mitotic regulators throughout the cell cycle. There are three mammalian aurora kinases, Aurora A, Aurora B, and Aurora C, which have differing roles throughout mitosis[47]. Aurora A is required for centrosome maturation and spindle formation, in addition to its role at the G2/M checkpoint described above. Aurora B is required for chromosome segregation and cytokinesis. Small molecule inhibitors of Aurora B lead to premature mitotic exit without successful chromosome separation. Continued inhibition of Aurora B results in large multiploid cells that eventually undergo apoptosis[48]. This potentially has the advantage that Aurora B inhibitors could be combined with other agents that act during other phases of the cell cycle. Aurora C is abundant in the testes. Its global functions are unclear, however, it has recently been shown to have some overlap with the functions of Aurora B during mitosis[49]. Aurora kinases have been shown to be overexpressed in a number of different tumors. Aurora A has been shown to be overexpressed in Barrett’s esophagus and esophageal adenocarcinoma[50,51]. Cell line models suggest that Aurora A overexpression protects developing esophageal adenocarcinoma cells against drug-induced apoptosis[51]. In other forms of cancer, Aurora A expression has been shown to correlate with chromosomal instability[52]. A number of Aurora kinase inhibitors are undergoing phase I and II evaluation. Danusertib, a pan-Aurora kinase inhibitor has undergone phase I testing in patients with advanced solid tumors. Forty-six percent of patients treated with danuserib had stable disease following treatment and a number of prolonged objective responses were noted[53,54]. The major dose limiting effect of these drugs is neutropenia.
PLK1 inhibitors
PLKs form a group of prominent mitotic kinases. They were first described in mutants that failed to undergo a normal mitosis in Drosophilia melanogaster (polo)[55,56]. They are highly conserved from yeast to humans. There are four members of the polo family in mammals (PLK1-4)[57,58]. They are involved in multiple functions throughout the cell cycle in mitosis and meiosis. PLK1 is the best characterized of the four known PLKs[58].
PLK1 is a candidate for development as a therapeutic target because it contains two functionally relevant sites: a C-terminal regulatory region containing two polo box domains (PBDs) and an N-terminal catalytic kinase domain[59]. The highly conserved PBD has been identified as a phosphopeptide-binding motif[60]. The polo box motif is only observed in the PLK family and contains a characteristic sequence. Drugs that target the PBD are specific to the human family of PLKs.
PLK1 is overexpressed in a broad range of primary gastrointestinal tumors, including gastric, colorectal and pancreatic carcinoma[61-63]. In contrast, one study has noted downregulation of PLK1 within tumor cells[64]. There is now increasing evidence that PLK1 expression levels have prognostic significance within different cancers, including esophageal cancer[63]. Two separate reports of PLK1 overexpression in esophageal carcinoma primarily relate to squamous cell carcinoma (SCC) in the far east[63,65]. Given the high impact of environmental factors (e.g. aflatoxin) on SCC development in these populations, it is unclear whether the findings can be directly applied to western populations. There are no data on PLK1 expression in adenocarcinoma patients. Some reports of other cancers have suggested that PLK1 expression is a reliable marker of metastasis[66]. PLK1 has also been used in the context of larger arrays of genes as a prognostic marker to predict metastasis in breast cancers[67]. Current cancer staging systems and histological assessments often fail to predict individual outcomes reliably but correlation of PLK1 protein and mRNA expression levels with clinical stage has the potential to improve clinical decision making in a number of different tumors[68].
The unique PLD of PLK1 also makes it a good candidate for the development of alternative cancer therapies. Initial efforts have focused on specific phosphorothioate antisense oligonucleotides that are able to block protein translation[69]. Use of siRNAs, which cause depletion of PLK1, has also been considered. Whilst there are drawbacks of siRNAs, including off-target effects and nuclease sensitivity, these hold promise in cancers such as bladder cancer in which they can act locally[70]. There are now a number of small molecule inhibitors of PLK1, which act either in an ATP-competitive or non-ATP-competitive manner[68]. The multiple actions of PLK1 throughout the cell cycle mean that these new agents need to be carefully assessed for specificity and side effects. In particular, it is possible that anti-PLK1 agents have similar toxicity to other microtubule inhibitors. PLK1 inhibitors are now in early clinical testing (phase I and II). Early clinical experience suggests that neutropenia and thrombocytopenia are dose-related effects, although neuropathy has not been seen[71].
MPS1 inhibitors
Cell cycle translational research has focused on the development of inhibitors of the major kinases discussed above. There are additional mitotic kinases that may have relevance for inhibiting tumor growth. Inhibitors of MPS1, a kinetochore-associated kinase that is involved in the spindle assembly checkpoint, have been shown to arrest tumor cell proliferation in vitro[72,73]. This appears to be mediated at least in part by impaired Aurora B function at centromeres, which leads to impaired alignment of chromosomes[74]. Detailed information on MPS1 in esophageal cancer is lacking, however, MPS1 inhibition has been demonstrated as a chemotherapy sensitization strategy in vitro[75].
CONCLUSION
Established esophageal carcinoma chemotherapy regimes are relatively blunt tools that predominantly target apoptosis pathways and are often associated with significant side effects. This has led to a large international effort to develop targeted therapy.
Current therapies that target the cell cycle have largely disappointed with relatively modest effects seen in phase I/II trials (Table 1). This may be in part related to failure to identify the patient populations that will gain the most clinical benefit. Few treatments are targeted towards specific pathways or personalized to the individual tumor proteome or genomic signature.
Table 1.
Compounds targeting the cell cycle under active development
| Inhibitor | Main target | Sponsor | Clinical trials |
| BI2536 | PLK1 (partial inhibition of PLK2/3) | Boehringer Ingelheim | Phase II pancreatic cancer |
| Danusertib (Formerly PHA-739358) | Pan-aurora kinase inhibitor | Pfizer Italia | Phase II advanced solid tumors |
| MLN8237 | Aurora a inhibitor | Millennium | Phase I/II advanced solid tumours |
| BI6267 | PLK1 inhibitor | Boehringer Ingelheim | Phase II ovarian cancer/phase I advanced solid tumors |
| P276-00 | Small molecule cyclin inhibitor | Piramal Life Sciences | Phase I advanced malignancy/phase II head and neck malignancy |
| NMS-1286937 | PLK1 selective inhibitor | Nerviano Medical Sciences | Phase I advanced solid tumours |
| P1446A-05 | CDK selective inhibitor | Piramal Life Sciences | Phase I advanced malignancy |
| SCH727965 | CDK inhibitor | Schering–Plough | Phase I advanced malignancy |
| Seliciclib (Roscovitine) | CDK inhibitor | Cyclacel Pharmaceuticals | Phase I advanced malignancy |
CDK: Cyclin-dependent kinase; PLK1: Polo-like kinase 1.
Efforts are now being made to assess gene expression profiles from histological specimens from solid tumors such as breast cancer in an attempt to predict response to chemotherapy[76]. Initial steps in this direction have been taken by the UK Oesophageal Cancer Clinical and Molecular Stratification (OCCAMS) Study Group, which has demonstrated a four-gene signature associated with poor prognosis in esophageal adenocarcinoma, as well as a larger group of genes associated with lymph node metastasis[77]. Efforts have also been made to identify Barrett’s esophagus patients who are likely to progress to adenocarcinoma, however, little work has been undertaken on response to chemotherapy in the esophagus[78]. Careful studies are needed in esophageal adenocarcinoma to define patient populations that are likely to respond well to treatment with both established and novel chemotherapy regimes. Optimizing individual chemotherapy regimens for patients will assume greater significance as health economies demand most clinical benefit from limited resources. In this setting of personalized targeted therapy, new cell cycle treatments may hold promise as carefully selected adjuncts to existing chemotherapy regimes.
Patients with esophageal adenocarcinoma unfortunately often still present late with a large burden of disease. Given the large number of cells involved it is likely that some tumor cells will abrogate the inhibited pathways and escape from chemotherapy-induced apoptosis. Targeted cell cycle therapy in esophageal cancer presents an alternate strategy as cell cycle inhibitors affect multiple essential pathways involved in replication and DNA damage repair. They may provide a useful adjunct in patients with late presenting esophageal tumors who have failed standard chemotherapy regimens.
Footnotes
Supported by UK National Institute of Health Research/Cancer Research Network and Research and Development Department of Wrightington Wigan and Leigh NHS Foundation Trust (to Ang YS); Wrightington Wigan and Leigh NHS Foundation Trust Cancer Therapy Fund (to Dibb M)
Peer reviewer: Luis Grande, Professor, Department of Surgery, Hospital del Mar, Passeig Marítim 25-29, Barcelona 08003, Spain
S- Editor Tian L L- Editor Kerr C E- Editor Ma WH
References
- 1.Umar SB, Fleischer DE. Esophageal cancer: epidemiology, pathogenesis and prevention. Nat Clin Pract Gastroenterol Hepatol. 2008;5:517–526. doi: 10.1038/ncpgasthep1223. [DOI] [PubMed] [Google Scholar]
- 2. Available from: http://www.statistics.gov.uk/StatBase/Product.asp?vlnk=7720.
- 3.Coleman MP, Gatta G, Verdecchia A, Estève J, Sant M, Storm H, Allemani C, Ciccolallo L, Santaquilani M, Berrino F. EUROCARE-3 summary: cancer survival in Europe at the end of the 20th century. Ann Oncol. 2003;14 Suppl 5:v128–v149. doi: 10.1093/annonc/mdg756. [DOI] [PubMed] [Google Scholar]
- 4.Bird-Lieberman EL, Fitzgerald RC. Early diagnosis of oesophageal cancer. Br J Cancer. 2009;101:1–67. doi: 10.1038/sj.bjc.6605126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perlmutter AE, Karimi KM, McFadden DW, Nigam A. Management of esophageal carcinoma. W V Med J. 2005;101:60–63. [PubMed] [Google Scholar]
- 6.Nurse P, Masui Y, Hartwell L. Understanding the cell cycle. Nat Med. 1998;4:1103–1106. doi: 10.1038/2594. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 8.Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 9.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 10.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 11.Morales CP, Souza RF, Spechler SJ. Hallmarks of cancer progression in Barrett’s oesophagus. Lancet. 2002;360:1587–1589. doi: 10.1016/S0140-6736(02)11569-8. [DOI] [PubMed] [Google Scholar]
- 12.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 13.Massagué J. G1 cell-cycle control and cancer. Nature. 2004;432:298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- 14.Nurse P. A long twentieth century of the cell cycle and beyond. Cell. 2000;100:71–78. doi: 10.1016/s0092-8674(00)81684-0. [DOI] [PubMed] [Google Scholar]
- 15.Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene. 2009;28:2925–2939. doi: 10.1038/onc.2009.170. [DOI] [PubMed] [Google Scholar]
- 16.Santamaría D, Barrière C, Cerqueira A, Hunt S, Tardy C, Newton K, Cáceres JF, Dubus P, Malumbres M, Barbacid M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature. 2007;448:811–815. doi: 10.1038/nature06046. [DOI] [PubMed] [Google Scholar]
- 17.Takaki T, Trenz K, Costanzo V, Petronczki M. Polo-like kinase 1 reaches beyond mitosis--cytokinesis, DNA damage response, and development. Curr Opin Cell Biol. 2008;20:650–660. doi: 10.1016/j.ceb.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 20.Bartkova J, Horejsí Z, Koed K, Krämer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 21.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr, Kastrinakis NG, Levy B, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 22.Golsteyn RM, Mundt KE, Fry AM, Nigg EA. Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J Cell Biol. 1995;129:1617–1628. doi: 10.1083/jcb.129.6.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamanaka R, Smith MR, O’Connor PM, Maloid S, Mihalic K, Spivak JL, Longo DL, Ferris DK. Polo-like kinase is a cell cycle-regulated kinase activated during mitosis. J Biol Chem. 1995;270:21086–21091. doi: 10.1074/jbc.270.36.21086. [DOI] [PubMed] [Google Scholar]
- 24.Uchiumi T, Longo DL, Ferris DK. Cell cycle regulation of the human polo-like kinase (PLK) promoter. J Biol Chem. 1997;272:9166–9174. doi: 10.1074/jbc.272.14.9166. [DOI] [PubMed] [Google Scholar]
- 25.Macůrek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008;455:119–123. doi: 10.1038/nature07185. [DOI] [PubMed] [Google Scholar]
- 26. Pelengaris S, Khan M. The Molecular biology of Cancer. 2006 [Google Scholar]
- 27.Toyoshima-Morimoto F, Taniguchi E, Shinya N, Iwamatsu A, Nishida E. Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature. 2001;410:215–220. doi: 10.1038/35065617. [DOI] [PubMed] [Google Scholar]
- 28.Nakajima H, Toyoshima-Morimoto F, Taniguchi E, Nishida E. Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J Biol Chem. 2003;278:25277–25280. doi: 10.1074/jbc.C300126200. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, Osada H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci USA. 2004;101:4419–4424. doi: 10.1073/pnas.0307700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsvetkov L. Polo-like kinases and Chk2 at the interface of DNA damage checkpoint pathways and mitotic regulation. IUBMB Life. 2004;56:449–456. doi: 10.1080/15216540400010826. [DOI] [PubMed] [Google Scholar]
- 31.Mundt KE, Golsteyn RM, Lane HA, Nigg EA. On the regulation and function of human polo-like kinase 1 (PLK1): effects of overexpression on cell cycle progression. Biochem Biophys Res Commun. 1997;239:377–385. doi: 10.1006/bbrc.1997.7378. [DOI] [PubMed] [Google Scholar]
- 32.Smith MR, Wilson ML, Hamanaka R, Chase D, Kung H, Longo DL, Ferris DK. Malignant transformation of mammalian cells initiated by constitutive expression of the polo-like kinase. Biochem Biophys Res Commun. 1997;234:397–405. doi: 10.1006/bbrc.1997.6633. [DOI] [PubMed] [Google Scholar]
- 33.Wajed SA, Laird PW, DeMeester TR. DNA methylation: an alternative pathway to cancer. Ann Surg. 2001;234:10–20. doi: 10.1097/00000658-200107000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barrett MT, Sanchez CA, Galipeau PC, Neshat K, Emond M, Reid BJ. Allelic loss of 9p21 and mutation of the CDKN2/p16 gene develop as early lesions during neoplastic progression in Barrett’s esophagus. Oncogene. 1996;13:1867–1873. [PubMed] [Google Scholar]
- 35.Wang JS, Guo M, Montgomery EA, Thompson RE, Cosby H, Hicks L, Wang S, Herman JG, Canto MI. DNA promoter hypermethylation of p16 and APC predicts neoplastic progression in Barrett’s esophagus. Am J Gastroenterol. 2009;104:2153–2160. doi: 10.1038/ajg.2009.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawakubo H, Ozawa S, Ando N, Kitagawa Y, Mukai M, Ueda M, Kitajima M. Alterations of p53, cyclin D1 and pRB expression in the carcinogenesis of esophageal squamous cell carcinoma. Oncol Rep. 2005;14:1453–1459. doi: 10.3892/or.14.6.1453. [DOI] [PubMed] [Google Scholar]
- 37.Arber N, Gammon MD, Hibshoosh H, Britton JA, Zhang Y, Schonberg JB, Roterdam H, Fabian I, Holt PR, Weinstein IB. Overexpression of cyclin D1 occurs in both squamous carcinomas and adenocarcinomas of the esophagus and in adenocarcinomas of the stomach. Hum Pathol. 1999;30:1087–1092. doi: 10.1016/s0046-8177(99)90227-7. [DOI] [PubMed] [Google Scholar]
- 38.Hirai T, Kuwahara M, Yoshida K, Osaki A, Toge T. The prognostic significance of p53, p21 (Waf1/Cip1), and cyclin D1 protein expression in esophageal cancer patients. Anticancer Res. 1999;19:4587–4591. [PubMed] [Google Scholar]
- 39.Morgan RJ, Newcomb PV, Hardwick RH, Alderson D. Amplification of cyclin D1 and MDM-2 in oesophageal carcinoma. Eur J Surg Oncol. 1999;25:364–367. doi: 10.1053/ejso.1999.0658. [DOI] [PubMed] [Google Scholar]
- 40.Casson AG, Zheng Z, Evans SC, Geldenhuys L, van Zanten SV, Veugelers PJ, Porter GA, Guernsey DL. Cyclin D1 polymorphism (G870A) and risk for esophageal adenocarcinoma. Cancer. 2005;104:730–739. doi: 10.1002/cncr.21229. [DOI] [PubMed] [Google Scholar]
- 41.Miller CT, Moy JR, Lin L, Schipper M, Normolle D, Brenner DE, Iannettoni MD, Orringer MB, Beer DG. Gene amplification in esophageal adenocarcinomas and Barrett’s with high-grade dysplasia. Clin Cancer Res. 2003;9:4819–4825. [PubMed] [Google Scholar]
- 42.Bani-Hani K, Martin IG, Hardie LJ, Mapstone N, Briggs JA, Forman D, Wild CP. Prospective study of cyclin D1 overexpression in Barrett’s esophagus: association with increased risk of adenocarcinoma. J Natl Cancer Inst. 2000;92:1316–1321. doi: 10.1093/jnci/92.16.1316. [DOI] [PubMed] [Google Scholar]
- 43.Milas L, Akimoto T, Hunter NR, Mason KA, Buchmiller L, Yamakawa M, Muramatsu H, Ang KK. Relationship between cyclin D1 expression and poor radioresponse of murine carcinomas. Int J Radiat Oncol Biol Phys. 2002;52:514–521. doi: 10.1016/s0360-3016(01)02693-1. [DOI] [PubMed] [Google Scholar]
- 44.Syrigos KN, Zalonis A, Kotteas E, Saif MW. Targeted therapy for oesophageal cancer: an overview. Cancer Metastasis Rev. 2008;27:273–288. doi: 10.1007/s10555-008-9117-z. [DOI] [PubMed] [Google Scholar]
- 45.Motwani M, Rizzo C, Sirotnak F, She Y, Schwartz GK. Flavopiridol enhances the effect of docetaxel in vitro and in vivo in human gastric cancer cells. Mol Cancer Ther. 2003;2:549–555. [PubMed] [Google Scholar]
- 46.Shah MA, Kortmansky J, Motwani M, Drobnjak M, Gonen M, Yi S, Weyerbacher A, Cordon-Cardo C, Lefkowitz R, Brenner B, et al. A phase I clinical trial of the sequential combination of irinotecan followed by flavopiridol. Clin Cancer Res. 2005;11:3836–3845. doi: 10.1158/1078-0432.CCR-04-2651. [DOI] [PubMed] [Google Scholar]
- 47.Kimura M, Uchida C, Takano Y, Kitagawa M, Okano Y. Cell cycle-dependent regulation of the human aurora B promoter. Biochem Biophys Res Commun. 2004;316:930–936. doi: 10.1016/j.bbrc.2004.01.178. [DOI] [PubMed] [Google Scholar]
- 48.Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Shapiro GI. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res. 2006;66:7668–7677. doi: 10.1158/0008-5472.CAN-05-3353. [DOI] [PubMed] [Google Scholar]
- 49.Slattery SD, Mancini MA, Brinkley BR, Hall RM. Aurora-C kinase supports mitotic progression in the absence of Aurora-B. Cell Cycle. 2009;8:2984–2994. [PubMed] [Google Scholar]
- 50.Agnese V, Cabibi D, Calcara D, Terrasi M, Pantuso G, Fiorentino E, Intrivici C, Colucci G, Aragona F, Gebbia N, et al. Aurora-A overexpression as an early marker of reflux-related columnar mucosa and Barrett’s oesophagus. Ann Oncol. 2007;18 Suppl 6:vi110–vi115. doi: 10.1093/annonc/mdm237. [DOI] [PubMed] [Google Scholar]
- 51.Dar AA, Zaika A, Piazuelo MB, Correa P, Koyama T, Belkhiri A, Washington K, Castells A, Pera M, El-Rifai W. Frequent overexpression of Aurora Kinase A in upper gastrointestinal adenocarcinomas correlates with potent antiapoptotic functions. Cancer. 2008;112:1688–1698. doi: 10.1002/cncr.23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miyoshi Y, Iwao K, Egawa C, Noguchi S. Association of centrosomal kinase STK15/BTAK mRNA expression with chromosomal instability in human breast cancers. Int J Cancer. 2001;92:370–373. doi: 10.1002/ijc.1200. [DOI] [PubMed] [Google Scholar]
- 53.Cohen RB, Jones SF, Aggarwal C, von Mehren M, Cheng J, Spigel DR, Greco FA, Mariani M, Rocchetti M, Ceruti R, et al. A phase I dose-escalation study of danusertib (PHA-739358) administered as a 24-hour infusion with and without granulocyte colony-stimulating factor in a 14-day cycle in patients with advanced solid tumors. Clin Cancer Res. 2009;15:6694–6701. doi: 10.1158/1078-0432.CCR-09-1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steeghs N, Eskens FA, Gelderblom H, Verweij J, Nortier JW, Ouwerkerk J, van Noort C, Mariani M, Spinelli R, Carpinelli P, et al. Phase I pharmacokinetic and pharmacodynamic study of the aurora kinase inhibitor danusertib in patients with advanced or metastatic solid tumors. J Clin Oncol. 2009;27:5094–5101. doi: 10.1200/JCO.2008.21.6655. [DOI] [PubMed] [Google Scholar]
- 55.Sunkel CE, Glover DM. polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J Cell Sci. 1988;89(Pt 1):25–38. doi: 10.1242/jcs.89.1.25. [DOI] [PubMed] [Google Scholar]
- 56.Llamazares S, Moreira A, Tavares A, Girdham C, Spruce BA, Gonzalez C, Karess RE, Glover DM, Sunkel CE. polo encodes a protein kinase homolog required for mitosis in Drosophila. Genes Dev. 1991;5:2153–2165. doi: 10.1101/gad.5.12a.2153. [DOI] [PubMed] [Google Scholar]
- 57.Nigg EA. Polo-like kinases: positive regulators of cell division from start to finish. Curr Opin Cell Biol. 1998;10:776–783. doi: 10.1016/s0955-0674(98)80121-x. [DOI] [PubMed] [Google Scholar]
- 58.Martin BT, Strebhardt K. Polo-like kinase 1: target and regulator of transcriptional control. Cell Cycle. 2006;5:2881–2885. doi: 10.4161/cc.5.24.3538. [DOI] [PubMed] [Google Scholar]
- 59.Leung GC, Hudson JW, Kozarova A, Davidson A, Dennis JW, Sicheri F. The Sak polo-box comprises a structural domain sufficient for mitotic subcellular localization. Nat Struct Biol. 2002;9:719–724. doi: 10.1038/nsb848. [DOI] [PubMed] [Google Scholar]
- 60.Elia AE, Cantley LC, Yaffe MB. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science. 2003;299:1228–1231. doi: 10.1126/science.1079079. [DOI] [PubMed] [Google Scholar]
- 61.Weichert W, Kristiansen G, Schmidt M, Gekeler V, Noske A, Niesporek S, Dietel M, Denkert C. Polo-like kinase 1 expression is a prognostic factor in human colon cancer. World J Gastroenterol. 2005;11:5644–5650. doi: 10.3748/wjg.v11.i36.5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Macmillan JC, Hudson JW, Bull S, Dennis JW, Swallow CJ. Comparative expression of the mitotic regulators SAK and PLK in colorectal cancer. Ann Surg Oncol. 2001;8:729–740. doi: 10.1007/s10434-001-0729-6. [DOI] [PubMed] [Google Scholar]
- 63.Tokumitsu Y, Mori M, Tanaka S, Akazawa K, Nakano S, Niho Y. Prognostic significance of polo-like kinase expression in esophageal carcinoma. Int J Oncol. 1999;15:687–692. doi: 10.3892/ijo.15.4.687. [DOI] [PubMed] [Google Scholar]
- 64.Simizu S, Osada H. Mutations in the Plk gene lead to instability of Plk protein in human tumour cell lines. Nat Cell Biol. 2000;2:852–854. doi: 10.1038/35041102. [DOI] [PubMed] [Google Scholar]
- 65.Feng YB, Lin DC, Shi ZZ, Wang XC, Shen XM, Zhang Y, Du XL, Luo ML, Xu X, Han YL, et al. Overexpression of PLK1 is associated with poor survival by inhibiting apoptosis via enhancement of survivin level in esophageal squamous cell carcinoma. Int J Cancer. 2009;124:578–588. doi: 10.1002/ijc.23990. [DOI] [PubMed] [Google Scholar]
- 66.Kneisel L, Strebhardt K, Bernd A, Wolter M, Binder A, Kaufmann R. Expression of polo-like kinase (PLK1) in thin melanomas: a novel marker of metastatic disease. J Cutan Pathol. 2002;29:354–358. doi: 10.1034/j.1600-0560.2002.290605.x. [DOI] [PubMed] [Google Scholar]
- 67.Ahr A, Karn T, Solbach C, Seiter T, Strebhardt K, Holtrich U, Kaufmann M. Identification of high risk breast-cancer patients by gene expression profiling. Lancet. 2002;359:131–132. doi: 10.1016/S0140-6736(02)07337-3. [DOI] [PubMed] [Google Scholar]
- 68.Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006;6:321–330. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 69.Spänkuch B, Steinhauser I, Wartlick H, Kurunci-Csacsko E, Strebhardt KI, Langer K. Downregulation of Plk1 expression by receptor-mediated uptake of antisense oligonucleotide-loaded nanoparticles. Neoplasia. 2008;10:223–234. doi: 10.1593/neo.07916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nogawa M, Yuasa T, Kimura S, Tanaka M, Kuroda J, Sato K, Yokota A, Segawa H, Toda Y, Kageyama S, et al. Intravesical administration of small interfering RNA targeting PLK-1 successfully prevents the growth of bladder cancer. J Clin Invest. 2005;115:978–985. doi: 10.1172/JCI23043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jackson JR, Patrick DR, Dar MM, Huang PS. Targeted anti-mitotic therapies: can we improve on tubulin agents? Nat Rev Cancer. 2007;7:107–117. doi: 10.1038/nrc2049. [DOI] [PubMed] [Google Scholar]
- 72.Dorer RK, Zhong S, Tallarico JA, Wong WH, Mitchison TJ, Murray AW. A small-molecule inhibitor of Mps1 blocks the spindle-checkpoint response to a lack of tension on mitotic chromosomes. Curr Biol. 2005;15:1070–1076. doi: 10.1016/j.cub.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 73.Schmidt M, Budirahardja Y, Klompmaker R, Medema RH. Ablation of the spindle assembly checkpoint by a compound targeting Mps1. EMBO Rep. 2005;6:866–872. doi: 10.1038/sj.embor.7400483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jelluma N, Brenkman AB, van den Broek NJ, Cruijsen CW, van Osch MH, Lens SM, Medema RH, Kops GJ. Mps1 phosphorylates Borealin to control Aurora B activity and chromosome alignment. Cell. 2008;132:233–246. doi: 10.1016/j.cell.2007.11.046. [DOI] [PubMed] [Google Scholar]
- 75.Janssen A, Kops GJ, Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc Natl Acad Sci USA. 2009;106:19108–19113. doi: 10.1073/pnas.0904343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gianni L, Zambetti M, Clark K, Baker J, Cronin M, Wu J, Mariani G, Rodriguez J, Carcangiu M, Watson D, et al. Gene expression profiles in paraffin-embedded core biopsy tissue predict response to chemotherapy in women with locally advanced breast cancer. J Clin Oncol. 2005;23:7265–7277. doi: 10.1200/JCO.2005.02.0818. [DOI] [PubMed] [Google Scholar]
- 77.Peters CJ, Rees JR, Hardwick RH, Hardwick JS, Vowler SL, Ong CA, Zhang C, Save V, O’Donovan M, Rassl D, et al. A 4-gene signature predicts survival of patients with resected adenocarcinoma of the esophagus, junction, and gastric cardia. Gastroenterology. 2010;139:1995–2004.e15. doi: 10.1053/j.gastro.2010.05.080. [DOI] [PubMed] [Google Scholar]
- 78.Lao-Sirieix P, Boussioutas A, Kadri SR, O’Donovan M, Debiram I, Das M, Harihar L, Fitzgerald RC. Non-endoscopic screening biomarkers for Barrett’s oesophagus: from microarray analysis to the clinic. Gut. 2009;58:1451–1459. doi: 10.1136/gut.2009.180281. [DOI] [PubMed] [Google Scholar]