

Abstract
Aggregates of charged amphiphilic molecules have been found to access a structure at elevated temperature that templates alignment of supramolecular fibrils over macroscopic scales. The thermal pathway leads to a lamellar plaque structure with fibrous texture that breaks upon cooling into large arrays of aligned nanoscale fibres and forms a strongly birefringent liquid. By manually dragging this liquid crystal from a pipette onto salty media, it is possible to extend this alignment over centimetres in noodle-shaped viscoelastic strings. Using this approach, the solution of supramolecular filaments can be mixed with cells at physiological temperatures to form monodomain gels of aligned cells and filaments. The nature of the self-assembly process and its biocompatibility would allow formation of cellular wires in situ that have any length and customized peptide compositions for use in biological applications.
Inspired largely by biological systems, molecular self-assembly continues to be a theme of great interest in science. The targets differ broadly, from accessing ordered materials and self-assembling devices1-3 to understanding how misfolded proteins self-assemble into stable fibres linked to human disease4,5. Long-range alignment of extracellular fibrils and cells in the heart6, brain, and spinal cord7 must involve highly complex self-assembly mechanisms that remain largely unknown. Access to similar three dimensional (3D) artificial systems of aligned fibrils and cells is therefore of scientific and biomedical interest8-10. Spontaneous long-range alignment of molecules is known to occur in liquid crystals11 but its fixation in the solid state normally requires chemical reactions12,13 that are not likely to be compatible with living cells. Electrospinning of polymers faces similar challenges because it requires the use of high mechanical and electrical energies14 that are not highly compatible with encapsulation of living cells. We report the discovery of a thermal pathway that leads highly designable peptide-based small molecules in water to form 2D plaques with filamentous texture that spontaneously template long-range alignment of bundled nanofibres upon cooling. This liquid crystal of supramolecular filaments can be mixed with cells at physiological temperatures and drawn by hand from a pipette into salt solutions to form monodomain gels of aligned filaments. Cells remain viable during the process and the monodomain gels can be drawn to arbitrary lengths and geometrical contours. We hypothesize that divalent ions and slow relaxation times of the long nanofibre bundles generated by this self-assembly pathway enable formation of the macroscopic monodomains.
We prepared 0.5–1.0 wt % aqueous solutions of peptide amphiphiles (PAs) known to self-assemble into high-aspect-ratio nanofibres15,16. One PA molecule investigated contains the peptide sequence V3A3E3(CO2H) and a C16 alkyl tail at the peptide's N-terminus, and its self-assembly into nanofibres is triggered by ions that screen the charged amino acid residues, resulting in the formation of gels. The diameter of these nanofibres, which contain β-sheets near their hydrophobic core, is roughly equivalent to the length of two PA molecules and lengths in excess of micrometres. We heated the aqueous solutions unscreened by added ions to 80 °C and kept them at this temperature for 30 minutes before cooling to 25 °C. After this heat treatment, the solution viscosity increased threefold from 5 cP to 15 cP. When calcium chloride was added to the heated and cooled PA solution, we observed the formation of a gel that was at least four-fold stiffer than one formed from an unheated solution (see Supplementary Information). Using polarized optical microscopy, we also found that gels or films formed from heated solutions contained large birefringent domains (tenths of millimetres) (Fig. 1), whereas those formed from unheated solutions appeared completely isotropic with no birefringence.
Figure 1. Strings and gels with long-range internal alignment.
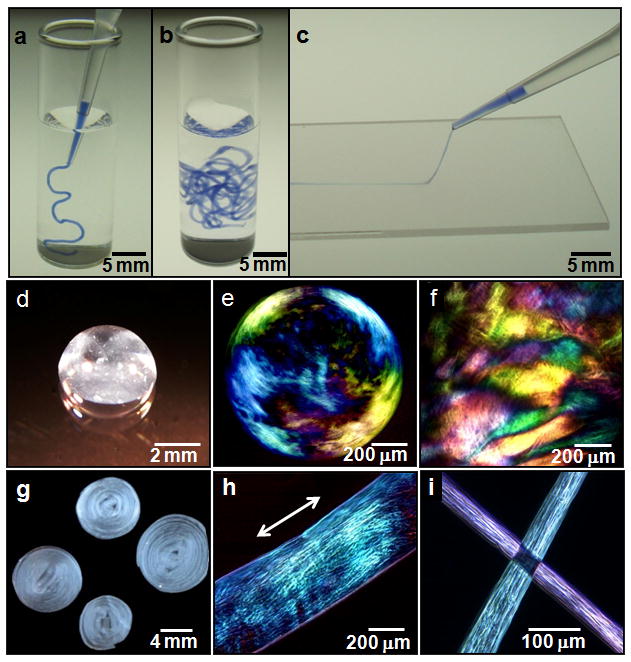
a, b, PA solution coloured with trypan blue injected into phosphate buffered saline after heat treatment. c, The same solution dragged through a thin layer of aqueous CaCl2 to form a noodle-like string. d, A knot made with PA string. e, Birefringence of bubble gel observed between cross polars suggesting the presence of macroscopically aligned domains. f, Similar domains in a gel film. g, PA noodle spirals prepared on a spin coater. h, Birefringence of a single string suggesting alignment along the string axis. i, Light extinction between cross polars at the crosspoint of two noodles demonstrating uniform alignment in each.
We observed that noodle-like strings of arbitrary length could be formed by manually drawing the aqueous PA solution into a salty medium from a pipette (Fig. 1a and b). When the solution was dragged on a surface covered by a thin layer of this medium (Fig. 1c), uniform birefringence was observed along the length of the string (Fig. 1h and i). This observation suggested that macroscopic alignment extending over centimetres was achieved. Using the same methods, the unheated solutions did not form mechanically stable string gels or show any birefringence. Scanning electron microscopy (SEM) indicated that strings formed from heated PA solutions contained extraordinarily long arrays of aligned nanofibre bundles (Fig. 2a and b). In great contrast, unheated PA solutions formed matrices of randomly entangled nanofibres (Fig. 2d and e). In order to verify this orientational order, we carried out small angle X-ray scattering (SAXS) experiments and found that only strings generated from the heated solutions revealed alignment (Fig. 2c and f). In contrast, simply dragging a PA solution that had not been heated does not lead to significant alignment. Similar strings can be made with other PA molecules, including those with bioactive epitopes, although their structural integrity depended on the amino acid sequence (Supplementary Information).
Figure 2. SEM evidence of massive alignment versus isotropy of nanofibre bundles.
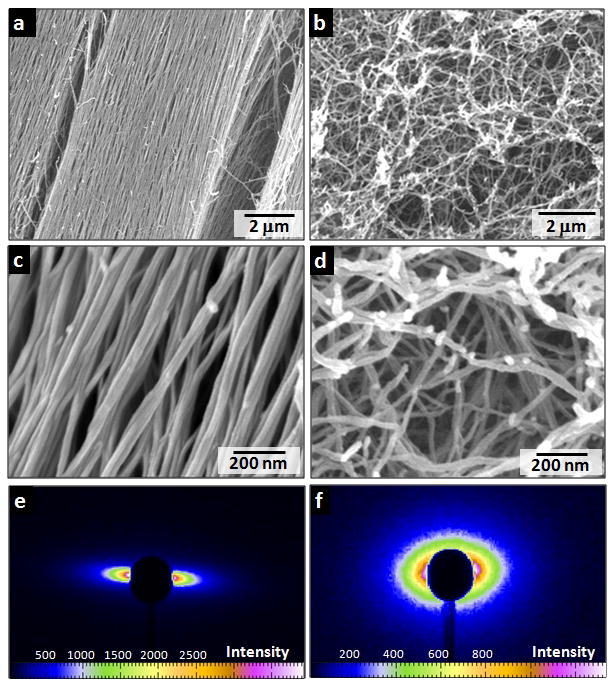
a, c, Aligned nanofibre bundles in macroscopic strings formed by dragging thermally treated amphiphile solutions onto a CaCl2 solution. b, d, Isotropic network of nanofibre bundles formed by adding CaCl2 to unheated amphiphile solutions. e, f, Small angle X-ray scattering of hydrogel strings prepared using PA solutions with and without heat treatment.
In order to gain a mechanistic understanding of the observed transformations, we examined the effects of heating on the PA solution structure by quick-freeze/deep-etch (QFDE) transmission electron microscopy (TEM)17. QFDE is a sample preparation technique that allows high-resolution imaging of hydrated structures while minimizing disruption of the sample due to fixation or processing. The freshly dissolved PA solution contained a variety of nanoscale, elongated objects less than a micron in length (Supplementary Information). Micrographs of the PA solution equilibrated at 80 °C for 30 minutes showed that the small aggregates largely disappeared; instead we observed thin “plaque-like” structures up to microns in both length and width (Fig. 3a). Some portions of these 2D plaques had a periodic surface texture with a characteristic spacing of about 7.5 nm, which corresponds to the expected diameter of a single canonical nanofibre formed by the PA molecules used15,18 (Fig. 3b). After cooling to room temperature, solutions were clearly composed of aligned filaments (Fig. 3d). The filaments did not have the diameter of canonical nanofibres (7–8 nm) but were instead tens of nanometres in diameter, and are therefore bundles of many cylindrical nanofibres. To further visualize the 3D structure of the plaque, we fixed the structure by adding calcium chloride at 80 °C and imaged the resulting structure by SEM. Although most of the sample cooled to ambient temperature was composed of large arrays of aligned nanofibres, some plaques were captured as well (Fig. 3e). These plaques measured about 40 nm in thickness and had lengths and widths comparable to those observed by QFDE-TEM. They often contained long parallel striations and, in some cases, appeared to be cracking into fibre bundles (Fig. 3f). These plaque structures were not observed without heating.
Figure 3. Morphological changes resulting from thermal treatment.

a, TEM obtained after a quick-freeze/deep-etch (QFDE) preparation of PA solution at 80 °C revealing a micron-sized, sheet-like plaque structure. b, Higher resolution QFDE-TEM of the sheet-like structures revealing surface pattern with a periodicity of about 7.5 nm. c, SAXS of PA solutions treated at different thermal conditions. d, QFDE-TEM of aligned nanofibre bundles templated by the sheet-like plaque after the PA solution was cooled to room temperature. e, SEM of plaques that were captured by adding CaCl2 at 80 °C. f, SEM of a plaque breaking into nanofibre bundles. g, Schematic representation of a plaque at high temperature and its rupture into fused bundles upon cooling (h). i, Schematic representation of the cross section of a plaque formed by fused fibers and of a fiber bundle (j).
We explored the effect of heating on structure in PA solutions using SAXS, circular dichroism (CD), differential scanning calorimetry (DSC), and Fourier-transform infrared spectroscopy (FT-IR). The X-ray scattering curve of PA solutions at 25 °C (Fig. 3c) shows a q−4 dependence within the low q range, indicative of the presence of large aggregates. Upon heating to 80 °C the −4 slope (log I versus log q) was retained, indicating that large aggregates are still present at high temperature19. This result is consistent with our observation of large plaque-like structures at 80 °C by QFDE-TEM. The scattering curves at 25 °C and 80 °C essentially superimpose at high q values, indicating that the local structure of supramolecular aggregates remains unchanged during heating. We also observe a broad scattering peak in the intermediate q range near 0.08 A−1. This q range corresponds to a spacing of approximately 8 nm, which is in the range of the diameter of the fibrous structures that exist in these solutions before heating. Upon heating this scattering maximum decreases in intensity, which is consistent with our observation that individual fibres are not observed at high temperature. The observed decrease in scattering at intermediate q values as solutions are heated is irreversible upon cooling of the samples. In fact, the fibrous structures observed after cooling by microscopy have diameters that are several times larger than the original ones and therefore their scattering contribution would be expected in the low rather than intermediate q range. Variable-temperature CD revealed the presence of β-sheet structure throughout the temperature range studied. Effectively no change was observed in the CD spectrum during heating and cooling cycles (Supplementary Information). Therefore, the heating and cooling of PA solutions does not significantly perturb the peptide secondary structure within the aggregates, which is also supported by X-ray diffraction studies (Supplementary Information). FT-IR studies of the solutions before and after heating show a major peak for parallel β-sheets (1625 cm−1) and a smaller peak at about 1660 cm−1 that can be attributed to anti-parallel β-sheets (Supplementary Information). DSC of a solution of the PA molecules did not show any peaks during heating and cooling cycles, indicating the absence of obvious phase transitions. We conclude from these experimental results that the reorganization of molecules from the 2D plaques to filaments upon cooling is a subtle one, which we believe reflects changes in the hydration of the aggregates, as explained below.
Canonical PA nanofibres are known to be internally hydrated20 and their charged surfaces should also contain electrostricted water molecules around charged amino acid residues. It is reasonable to assume that as the system is heated, bound water molecules associated with the supramolecular aggregates become bulk water molecules driven by entropy. This dehydration process could allow the fibrous aggregates to interact more closely, facilitating their fusion as depicted in Fig. 3g. We believe the observed flat and textured plaques are the result of these fused, dehydrated fibres. During cooling, water molecules should simply rehydrate the supramolecular structures, but this process actually generates filaments with diameters that are several times greater than those of canonical nanofibres that exist prior to the thermal treatment (see schematic in Fig. 3h). Based on microscopy the plaque appears to break into nanofibre bundles with diameters of approximately 40 nm (Fig. 2c) rather than individual canonical fibres with diameters of about 8 nm. The diameter of the bundles corresponds to the plaque's thickness observed by SEM (Fig. 3e).
The possibility of a transition from the plaque to a fused nanofibre bundle can be understood by computing the contributions to the difference in free energy per thermal energy kBT per amphiphile for a cylindrical fibre Ff and a lamella Fl,
| (1) |
where and are the electrostatic free energies of a fibre and a lamella, respectively, and and are the cohesive free energies of molecules in these two different morphologies. The fraction of ions condensed on the surface of PA fibres and plaques is estimated from the modified Poisson–Boltzmann equation21, which shows that the charges are neutralized by counterions for both the plaque and the fibre (see Supplementary Information). Therefore, the transition should be dominated by the difference in cohesive energies of lamellae and fibres, , which is given by,
| (2) |
where ΔHPA is the enthalpy difference per PA molecule between lamellar and fibre aggregates, and ΔSPA and ΔSwater are the entropy differences of the PA and water molecules, respectively. Since the β-sheet signature in the CD spectrum does not change significantly during cooling, we assumed that the internal energy of the β-sheet is similar in the fibre and the plaque. Therefore, the enthalpy difference between lamellae and fibres must originate from the coupling of interactions among peptide segments and hydrophobic tails. This coupling is supported by our previous spectroscopic experiments that showed order can exist in the hydrophobic core of PA nanofibres and is enhanced by β-sheet orientation along the fibre axis22. The fibre architecture could also optimize interactions among peptide segments and alkyl segments. We estimate ΔHPA to be dominated by van der Waals forces, which are of the order of thermal energy. However, the increase in entropy of the PA and water molecules in the plaque state can offset the enthalpy difference at elevated temperature. Specifically, the higher entropy in the plaque can originate in greater translation of water molecules (ΔSwater), since there is less water interface per PA molecule than in the fibre structure, and therefore fewer restricted water molecules per amphiphile. This would reasonably predict a transition temperature from fibrous to planar assemblies of the peptide amphiphiles (see Supplementary Information).
The observed rupture of the plaque at lower temperatures into bundles of fibres that give rise to an aqueous lyotropic liquid crystal at an unusually low concentration suggests a novel mechanism of membrane rupture. The plaque observed by QFDE-TEM at 80°C reveals a periodic surface texture with a characteristic spacing of about 7.5 nm, which corresponds to the expected diameter of a single canonical nanofibre15. This strongly suggests the plaque results from the fusion of nanofibres as the dehydration occurs at elevated temperature. The microscopy also revealed the existence of ripples in the plaque of larger dimension than the fibres (Figure 3a,b). We propose that fluctuations of the anisotropic plaque structure with 1D fibrous texture are crucial for its metamorphosis into arrays of highly aligned nanofibres. It is known that typically only membranes in curved geometries such as cylinders break by Rayleigh instabilities23; flat membranes generally rupture by creating holes24. Therefore, the possible breaking mechanism of a plaque into bundled fibres that gives rise to a lyotropic liquid crystal is unique given its underlying anisotropic 1D sub-structure imparted by the nanofibres composed of β-sheets. Long-range forces have been proposed to cause the rupture of surfactant membranes via concentration fluctuations25-27. The fibrous texture on the surface, however, generates an anisotropic surface tension, which may lead to the formation of waves of fluctuating composition on the surface similar to binary immiscible lipid membranes28. The waves due to the membrane tension appear as surface ripples when the β-sheets align, and this may generate the concentration of fluctuations required for rupture. However, the size of the successful composition fluctuation has to be large enough (larger than the membrane thickness D) in order to generate a critical size of a neck for rupture; otherwise the strain generated by the composition fluctuations in the internal structure (the interpenetrated bilayers) opposes the growth of the fluctuation29, and the lateral composition fluctuations are restored. One can assume that overall the breaking of the surface is due to a decrease in the overall surface tension γ, which is the change of free energy (F) as the interface area (A) increases, ∂F/∂A. That is, the bundled fibre surface tension γf is lower than that of the lamellar γl due to the increase of hydration and order of PA molecules within the bundle. Unfortunately, the linear theory that assumes γ is constant under a deformation of the interface is not appropriate to describe the breaking of a lamella. To a first order approximation one can assume that the interfacial energy of the fluctuation that leads to the rupture of the plaque is of the order of the γf. Consider fluctuations perpendicular to the lamellar surface plane (x,y) described by a function h(x,y), which induce a decrease in γl to γf that may result in the rupture of the plaque. In order to induce rupture, the resulting free energy change ΔF =Ff − F, is negative, or ΔF < 0, where Ff is the free energy of the fluctuating plaque given by
| (3) |
where hx = ∂h/∂x and hy = ∂h/∂y, and F the flat plaque free energy is given by,
| (4) |
If we assume a one-dimensional fluctuation along x of maximum amplitude h0, which is half the thickness of the lamella D (h0 = D/2) and wavelength λ, then
| (5) |
Therefore, by approximating (1 + hx2 + hy2)1/2 as (1 + hx2 / 2) and assuming γf is a constant in the integrant of Ff, we find that after the integration of both Ff and F breaking occurs if ΔF/A ≤ 0, or
| (6) |
where Δγ = γf - γl < 0 and A is the total area of the plaque of volume V=AD. This inequality gives a bound for the characteristic size λc above which fluctuations lead to rupture, given by (2π h0/λc)2 = -2Δγ /γf or λc = h0 π (2γf /(γl-γf))1/2; that is, all λ ≥ λc lead to rupture and all λ < λc would lead to stable plaques. Since h0 = D/2 at rupture and there is no long range diffusion in the rupture process, λc is the most probable size for rupture, and, given that γf/(γl-γf) is of the order of one (see Supplementary Information), λc is of the order of the membrane thickness D, in agreement with the expected size of the breaking of a cylinder through a Rayleigh instability.
It has been shown previously that liquid crystals can be aligned with elongational flow30. Theoretical models suggest that during uniaxial stretching of polymeric nematics, if the product of strain rate ε̇ and the conformational relaxation time λ is greater than unity, one can expect high degrees of uniaxial orientation31. In the context of this model, the product (known as Weissenberg number) would be greater than unity for thermally treated PA gel strings and less than unity for gel strings formed with unheated samples. Since we used the same longitudinal strain rate for both samples (Fig. 2c and d), the aggregates in thermally treated solutions must have longer relaxation times. Thus we hypothesize the formation of larger diameter supramolecular bundles derived from the rupture of the plaque enables the fixing of macroscopic orientation over arbitrary length in hand-drawn string gels. It is also interesting to note that the low strain rate used here (1–4 s-1) is ideal for supramolecular aggregates, since their noncovalent interactions may not be stable in some cases at the large strain rates commonly used in electrospinning of polymers (104–105 s-1)32. The mechanism proposed here is fundamentally different from that observed in gel spinning of polymers33 and the polymerization of monomers in heated liquid-crystalline solution34. While these strings were produced by hand drawing, this methodology could be extended to automated printing technology.
We used the strings of aligned PA nanofibres to direct the orientation of cells in 3D environments. After dispersing human mesenchymal stem cells (hMSCs) in heated and cooled PA solutions, we manually dragged these solutions onto salty media (160 mM NaCl and 5–10 mM CaCl2) to form noodle-shaped strings with encapsulated stem cells. These cells remain viable during the process of string formation and start to elongate along the string director axis within 12 hours. Optical, fluorescence, and electron microscopy (Fig. 4a to c) demonstrated that both cell bodies and filopodia were aligned with PA nanofibre bundles in the extracellular space. These effects on hMSC orientation are hypothesized to result from contact guidance along the preferentially oriented matrix35. A “cellular wire” based on the noodle-like string could also serve as a bridge to direct cells spatially for function or migration from one site to another; current methods to generate long-range 3D alignment of a synthetic, fibrous matrix over arbitrary lengths in the presence of living cells are limited. To demonstrate further biological applications of the noodle construct, we formed a string in the presence of HL-1 cardiomyocytes, a cell line with spontaneous electrical activity that requires extensive cell-cell contacts to propagate signals, and then observed the cellular behaviour over time.36 The cells survived and proliferated extensively to fill the entire structure. By fluorescently visualizing intracellular calcium concentration, we observed pockets of spontaneous electrical activity by day 6 and action potentials that were able to propagate throughout the entire macroscopic structure by day 10 (Fig. 4e). Plots of the calcium fluorescence over time show these propagating signals, demonstrating the presence of a continuous functional cardiac syncytium (Fig. 4f). This finding suggests potential for these ‘cellular wires’ in regenerative medicine applications in which restoring electrical communication in tissues is of critical importance e.g., in the treatment of cardiac arrhythmia or spinal cord injury. In addition to this ability to propagate electric signals, it is clear that the strong cell alignment of the constructs, as found in muscle and nervous tissue, will also be helpful in developing regenerative strategies. In another example we created a monodomain PA string gel containing dispersed carbon nanotubes that retained the same degree of alignment as in pure PA strings (Fig. 4d). These black strings had electrical conductivities on the order of 1–10 S cm−1 in a dry state, depending on the concentration of carbon nanotubes. The incorporation of carbon nanotubes into the structure without disrupting the final construct suggests the use of the methodology to align 1D nanostructures within biocompatible matrices.
Figure 4. Cell alignment in strings of aligned filaments.
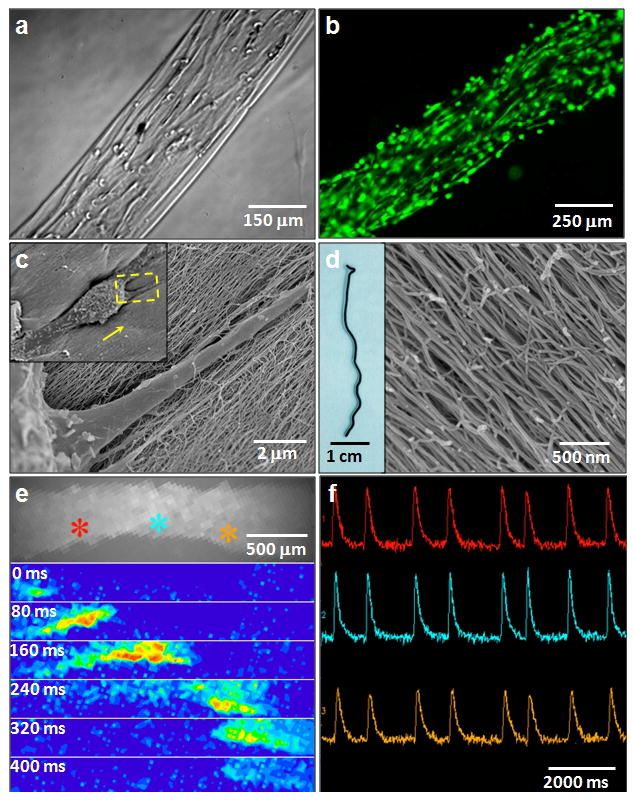
a, Preferential alignment of encapsulated human mesenchymal stem cells (hMSCs) along the string axis. b, Calcein-labeled aligned cells cultured in string. c, SEM images at different magnifications of a single cell in a string (inset is the zoom out view with arrow indicates alignment direction). d, A conductive black string formed by dispersing carbon nanotubes in PA solutions before heating. The SEM micrograph on the right shows aligned nanofibre bundles in the black string. e, (Top) Calcium fluorescence image of HL-1 cardiomyocytes encapsulated in a noodle-like string. (Below) Successive spatial maps of calcium fluorescence intensity travelling at 80 ms intervals, showing the propagation of an electrical signal throughout the entire string and demonstrating a functional cardiac syncytium f, Calcium fluorescence intensity signal in time at three points in the string marked by colour in e), showing repeated propagating action potentials.
We found in this work that supramolecular architectures of self-assembling molecules can drastically change in a pathway dependent manner. We specifically discovered a thermal pathway that converts isotropic solutions of peptide-containing molecules to liquid crystals in which molecules group into long filaments of bundled nanofibres. This transformation allows the formation of monodomain fibrous gels, which may contain viable cells as well. These systems offer potential to study phenomena in macroscopically aligned arrays of cells and could also lead to the development of therapies that require directed cell migration, directed cell growth, or the spatial cell interconnections in tissues such as heart, brain, and spinal cord.
Supplementary Material
Acknowledgments
This work was supported by the U.S. Department of Energy-Basic Energy Sciences (DE-FG02-00ER45810, DE-FG02-08ER46539), National Institutes of Health (5-R01-EB003806, 5-R01-DE015920, 5-P50-NS054287), National Science Foundation (DMR-0605427), Department of Homeland Security Fellowship (M.A.G.), Non-Equilibrium Energy Research Center (NERC), an Energy Frontier Research Center funded by DOE-BES (award number DE-SC0000989 for L.C.P), Northwestern University's NIH Biotechnology Training Program (pre-doctoral fellowship to J.R.M.), Ben Gurion University of Negev, Israel (post-doctoral fellowship for R.B.), and Generalitat de Catalunya (visiting scholar sponsorship for C.A.). Experiments made use of the following facilities at Northwestern University: J. B. Cohen X-ray Diffraction Facility, IMSERC, EPIC Facilities of the NUANCE Center, Keck Biophysics Facility, Biological Imaging Facility, and the Institute for BioNanotechnology in Medicine and its Cleanroom Core Facility. The NUANCE Center is supported by the NSF-NSEC, NSF-MRSEC, Keck Foundation, the State of Illinois, and Northwestern University. We acknowledge facilities support by the Materials Research Center through NSF-MRSEC grant DMR-0520513. X-ray measurements were carried out at the DuPont–Northwestern–Dow Collaborative Access Team (DND-CAT) Synchrotron Research Center located at Sector 5 of the Advanced Photon Source. DND-CAT is supported by the E.I. DuPont de Nemours & Co., The Dow Chemical Company, the US National Science Foundation through Grant DMR-9304725 and the State of Illinois through the Department of Commerce and the Board of Higher Education Grant IBHE HECA NWU 96. Use of the Advanced Photon Source was supported by the US Department of Energy–Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38 and DE-AC02-06CH11357. Use of the BioCARS Sector 14 was supported by the National Institutes of Health, National Center for Research Resources, under grant number RR007707. We also thank Greg Darnell and Steven Weigand for X-ray assistance and Dr. William Claycomb for the generous gift of the HL-1 cells.
Footnotes
Author Contributions: S. Z., M. A. G., A. M., L. C. P., R. B., C. A., and J. R. M. performed experiments. M. A. G. and M. O. generated numerical data. M. A. G., M. O., and S.I.S developed a theoretical model. S. Z., M. A. G., L. C. P., R. B., J. R. M., M. O. and S. I. S. wrote the paper.
Additional Information: Supplementary Information accompanies the paper on www.nature.com/naturematerials.
Reprints and permissions information is available at npg.nature.com/reprintsandpermissions.
References
- 1.Whitesides GM, Mathias JP, Seto CT. Science. 1991;254(5036):1312–1319. doi: 10.1126/science.1962191. [DOI] [PubMed] [Google Scholar]
- 2.Reches M, Gazit E. Science. 2003;300(5619):625–627. doi: 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- 3.Kim SO, et al. Nature. 2003;424(6947):411–414. doi: 10.1038/nature01775. [DOI] [PubMed] [Google Scholar]
- 4.Nelson R, et al. Nature. 2005;435(7043):773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobson CM. Nature. 2003;426(6968):884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 6.Chung CY, Bien H, Entcheva E. J of Cardiovasc Electr. 2007;18(12):1323–1329. doi: 10.1111/j.1540-8167.2007.00959.x. [DOI] [PubMed] [Google Scholar]
- 7.Davies SJA, Goucher DR, Doller C, Silver J. J Neurosci. 1999;19(14):5810–5822. doi: 10.1523/JNEUROSCI.19-14-05810.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feinberg AW, et al. Science. 2007;317(5843):1366–1370. doi: 10.1126/science.1146885. [DOI] [PubMed] [Google Scholar]
- 9.Merzlyak A, Indrakanti S, Lee SW. Nano Lett. 2009;9(2):846–852. doi: 10.1021/nl8036728. [DOI] [PubMed] [Google Scholar]
- 10.Bettinger CJ, Langer R, Borenstein JT. Angew Chem Int Ed. 2009;48(30):5406–5415. doi: 10.1002/anie.200805179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kato T, Mizoshita N, Kishimoto K. Angew Chem Int Ed. 2006;45(1):38–68. doi: 10.1002/anie.200501384. [DOI] [PubMed] [Google Scholar]
- 12.Gin DL, Gu WQ, Pindzola BA, Zhou WJ. Acc Chem Res. 2001;34(12):973–980. doi: 10.1021/ar000140d. [DOI] [PubMed] [Google Scholar]
- 13.Stupp SI, Osenar P. Polymerization in Organized Media. In: Schlüter AD, editor. Synthesis of Polymers. Wiley-VCH; Germany: 1999. [Google Scholar]
- 14.Greiner A, Wendorff JH. Angew Chem Int Ed. 2007;46(30):5670–5703. doi: 10.1002/anie.200604646. [DOI] [PubMed] [Google Scholar]
- 15.Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294(5547):1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 16.Behanna HA, Donners JJJM, Gordon AC, Stupp SI. J Am Chem Soc. 2005;127(4):1193–1200. doi: 10.1021/ja044863u. [DOI] [PubMed] [Google Scholar]
- 17.Ruberti JW, et al. Invest Ophth Vis Sci. 2003;44(4):1753–1759. doi: 10.1167/iovs.02-0496. [DOI] [PubMed] [Google Scholar]
- 18.Bull SR, et al. Bioconjugate Chem. 2005;16(6):1343–1348. doi: 10.1021/bc050153h. [DOI] [PubMed] [Google Scholar]
- 19.Glatter O, Kratky O, editors. Small Angle X-ray Scattering. Academic Press; London: 1982. [Google Scholar]
- 20.Tovar JD, Claussen RC, Stupp SI. J Am Chem Soc. 2005;127(20):7337–7345. doi: 10.1021/ja043764d. [DOI] [PubMed] [Google Scholar]
- 21.Cheng H, Zhang K, Libera JA, de la Cruz MO, Bedzyk MJ. Biophys J. 2006;90:1164–1174. doi: 10.1529/biophysj.105.070649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang HZ, Guler MO, Stupp SI. Soft Matter. 2007;3(4):454–462. doi: 10.1039/b614426h. [DOI] [PubMed] [Google Scholar]
- 23.Lenz P, Nelson DR. Phys Rev E. 2003;67(3):031502. doi: 10.1103/PhysRevE.67.031502. [DOI] [PubMed] [Google Scholar]
- 24.Sandre O, Moreaux L, Brochard-Wyart F. Proc Natl Acad Sci USA. 1999;96(19):10591–10596. doi: 10.1073/pnas.96.19.10591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Wit A, Gallez D, Christov CI. Phys Fluids. 1994;6(10):3256–3266. [Google Scholar]
- 26.Oron A, Davis SH, Bankoff SG. Rev Mod Phys. 1997;69(3):931. [Google Scholar]
- 27.Liu YS, Li MH, Bansil R, Steinhart M. Macromolecules. 2007;40(26):9482–9490. [Google Scholar]
- 28.Solis FJ, Funkhouser CM, Thornton K. Europhys Lett. 2008;(3):38001. [Google Scholar]
- 29.Mayes AM, de la Cruz MO. Acta Metall. 1989;37(2):615–620. [Google Scholar]
- 30.Idem Y, Ophir Z. Polym Eng Sci. 1983;23(5):261–265. [Google Scholar]
- 31.Larson RG, Mead DW. Liq Cryst. 1993;15(2):151–169. [Google Scholar]
- 32.Reneker DH, Yarin AL, Fong H, Koombhongse S. J Appl Phys. 2000;87(9):4531–4547. [Google Scholar]
- 33.Barham PJ, Keller A. J Mat Sci. 1985;20:2281–2302. [Google Scholar]
- 34.Fujikake H, Murashige T, Sato H, Kawakita M, Kikuchi H. Appl Phys Lett. 2003;82(10):1622–1624. [Google Scholar]
- 35.Tranquillo RT. Biochem Soc Symp. 1999;65:27–42. [PubMed] [Google Scholar]
- 36.Claycomb WC, et al. Proc Natl Acad Sci USA. 1998;95(6):2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.