

Abstract
Atherosclerosis and its clinical manifestations are widely prevalent throughout the world. Atherogenesis is highly complex and modulated by numerous genetic and environmental risk factors. A large body of basic scientific and clinical research supports the conclusion that inflammation plays a significant role in atherogenesis along the entire continuum of its progression. Inflammation adversely impacts intravascular lipid handling and metabolism, resulting in the development of macrophage foam cell, fatty streak, and atheromatous plaque formation. Given the enormous human and economic cost of myocardial infarction, ischemic stroke, peripheral arterial disease and amputation, and premature death and disability, considerable effort is being committed to refining our ability to correctly identify patients at heightened risk for atherosclerotic vascular disease and acute cardiovascular events so that they can be treated earlier and more aggressively. Serum markers of inflammation have emerged as an important component of risk factor burden. Lipoprotein-associated phospholipase A2 (Lp-PLA2) potentiates intravascular inflammation and atherosclerosis. A variety of epidemiologic studies support the utility of Lp-PLA2 measurements for estimating and further refining cardiovascular disease risk. Drug therapies to inhibit Lp-PLA2 are in development and show considerable promise, including darapladib, a specific molecular inhibitor of the enzyme. In addition to substantially inhibiting Lp-PLA2 activity, darapladib reduces progression of the necrotic core volume of human coronary artery atheromatous plaque. The growing body of evidence points to an important role and utility for Lp-PLA2 testing in preventive and personalized clinical medicine.
Keywords: Atherosclerosis, Inflammation, Low-density lipoprotein cholesterol, Lysophosphatidylcholine, Macrophage, Oxidized fatty acid, Phospholipid, Phospholipase
Key issues presented
Currently available methods for estimating cardiovascular risk often underestimate risk, especially in patients at intermediate risk (two or more risk factors or 10 yr Framingham risk of 10–20%).
Approaches for more accurately estimating cardiovascular risk are urgently needed to help identify patients who warrant more aggressive and comprehensive treatment of their cardiovascular risk burden in its entirety.
Atherosclerosis is a chronic inflammatory disease. Inflammation creates a toxic environment within the subendothelial space which stimulates atherosclerotic plaque development and plaque instability, leading to acute plaque rupture and coronary arterial luminal obstruction.
Markers of inflammation help to refine cardiovascular risk estimation. As serum levels of Lp-PLA2 rise, the risk for acute cardiovascular events increases in a continuous manner.
Key points for Lp-PLA2: (1) biomarker demonstrates low biovariability in serum; (2) enzyme plays a mechanistic role in atherogenesis; (3) serum levels reflect intravascular inflammation and the presence of unstable plaque; (4) increased expression of the enzyme within plaque associated with more complex and advanced lesions; (5) treatment with a specific molecular inhibitor beneficially impacts necrotic core volume of coronary plaque in humans; (6) increased serum levels associated with progressive elevation in risk for cardiovascular events.
Introduction
Despite enormous strides in the last five decades, acute cardiovascular events (myocardial infarction, stroke, and sudden death) remain the principal causes of morbidity and mortality in industrialized nations. A host of established and emerging risk factors for cardiovascular disease have been identified. Among the most important of these and best characterized epidemiologically are dyslipidemia, hypertension, age, smoking status, insulin resistance and diabetes mellitus, and family history for premature coronary artery disease (CAD). A variety of approaches to estimating risk for cardiovascular disease in the primary prevention setting have been developed [1]. Unfortunately, the use of quantitative risk estimation is severely underutilized and leaves many patients undertreated. In addition, although traditional risk factors for coronary heart disease (CHD) are well characterized, they do not fully account for risk. Simplifying risk estimation in primary prevention and improving the ability to identify patients with established atherosclerotic disease at heightened risk for either first time or recurrent events despite appropriate intervention are important clinical goals. The use of serum biomarkers holds considerable promise for helping to identify high risk patients in the contexts of both primary and secondary prevention [2–4]. Among the most commonly used serum biomarkers in cardiovascular medicine are those used to quantify the intensity of host inflammation.
Atherosclerosis and acute coronary syndromes are now recognized as manifestations of vascular inflammation [5, 6]. Risk factors for CHD promote endothelial dysfunction. Dysfunctional endothelial cells express adhesion molecules which promote the binding and influx of inflammatory white blood cells (T-cells, monocytes, and mast cells) into the subendothelial space [7]. White blood cells produce interleukins, cytokines, and reactive oxygen species which create an inflammatory focus within the arterial wall. Atherogenic lipoproteins such as low-density lipoprotein access the subendothelial space where they undergo trapping within the network of intercellular matrix proteins, enzymatic oxidative modification, aggregation, and ultimately, uptake by macrophages leading to the development of foam cells [8, 9]. Atheromatous plaque progressively expands with formation of a lipid core. As inflammation worsens and the capacity of phagocytic cells to clear apoptotic and necrotic debris becomes compromised, a necrotic core forms [8]. A clinical consequence of steadily amplified inflammation within plaque is increased risk for cardiovascular events secondary to weakening and loss of architectural integrity. This raises the likelihood for sudden plaque rupture with subsequent formation of overlying thrombus and arterial luminal obstruction. Atherosclerosis can therefore be considered an inflammatory disease. There is mounting evidence that as serum levels of lipoprotein-associated phospholipase A2 (Lp-PLA2) rise, risk for myocardial infarction, stroke, and sudden death all increase significantly [10–13]. This review summarizes recent findings with this inflammatory biomarker, focusing on Lp-PLA2 mechanism of action, epidemiologic characterization, and clinical utility as a biomarker for risk prediction.
The inflammatory biomarker Lp-PLA2
A biomarker is defined as a substance used as an indicator of a biological state. Through various techniques it is objectively measured and used to assess normal or pathogenic biological processes, or of pharmacologic responses to a therapeutic intervention. By definition it is critical that this diagnostic tool be relied upon clinically to improve accuracy of diagnosis, delineate disease subtypes, monitor disease progression, or improve prognostication and risk assessment [14]. In addition, a biomarker may demonstrate treatment efficacy when used with disease modifying therapies, such as lipid lowering drugs.
Lipoprotein-associated phospholipase A2 (Lp-PLA2) is a novel biomarker of vascular-specific inflammation that provides information about atherosclerotic plaque inflammation and stability. Elevated levels of serum Lp-PLA2 are indicative of rupture prone plaque and a strong independent predictor of cardiovascular risk, including coronary artery disease, MI, and stroke [12, 13]. Lp-PLA2 is associated clinically with increased CHD risk, and there is a large body of published evidence from epidemiologic studies addressing the relationship of Lp-PLA2 and risk of cardiovascular disease [15–17].
Biologically, Lp-PLA2 is a vascular-specific proinflammatory enzyme that operates physiologically in the arterial intima (Fig. 1). Lp-PLA2 localizes to atherosclerotic plaque, particularly in plaques with a necrotic core and in ruptured plaques [18]. High levels of Lp-PLA2 are found in rupture prone plaques, and it appears Lp-PLA2 is released from these plaques into the circulation. Lp-PLA2 is primarily produced by macrophages and then bound to various lipoproteins, including the ApoB portion of low-density lipoprotein (LDL) and lipoprotein(a) [19]. Staining of coronary and carotid tissue demonstrates the presence of Lp-PLA2 in the thin fibrous cap of rupture-prone plaques, but not in the early-stage plaques [18, 20]. Coronary and carotid tissue concentrations of Lp-PLA2 are notably very high in the rupture-prone shoulder region of thin fibrous cap atheromas, and histopathologic stains reveal that Lp-PLA2 co-localizes with macrophages and oxidized LDL in atherosclerotic coronary and carotid plaques [21].
Fig. 1.
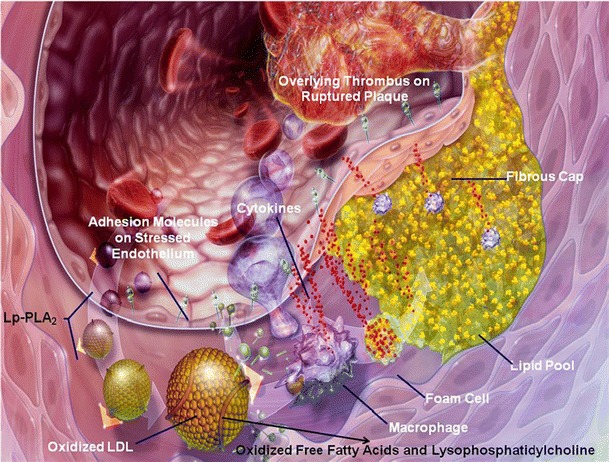
Lipoprotein-associated phospholipase A2 and atherogenesis. Dysfunctional endothelial cells express a variety of adhesion molecules that promote the binding, rolling, and stable arrest of inflammatory white blood cells, such as T-cells, monocytes, and mast cells. These inflammatory white cells express a large number of interleukins and cytokines which help to create an inflammatory nidus within the vessel wall. Monocytes alter their three-dimensional actin cytoskeleton and follow a gradient of monocyte chemoattractant protein-1 down into the subendothelial space by diapadesing between endothelial cells. Monocytes can transform into resident tissue macrophages. Low-density lipoprotein particles carry both lipid and Lp-PLA2 into the arterial wall. Macrophages also produce Lp-PLA2 in situ within plaque. The lipid in LDL particles undergoes oxidation mediated by myeloperoxidase, 5′-lipoxygenase, and other agents. Oxidized LDL stimulates increased expression of scavenging receptors on the surface of macrophages. As lipid is taken up into macrophages, they are converted into foam cells which can coalesce to form fatty streaks, which then evolve into atherosclerotic plaques. Lp-PLA2 specifically hydrolyzes phosphatidylcholine into oxidized free fatty acid and lysophosphatidylcholine. These lipids potentiate inflammation and plaque progression. The cap region of a plaque can become architecturally weakened as matrix metalloproteinases are produced within plaque. The plaque can rupture with overlying thrombus formation, resulting in acute myocardial ischemia and an acute coronary syndrome
Lp-PLA2 hydrolyzes phospholipids on oxidized LDL particles in the subendothelial space (Fig. 2). Lp-PLA2 hydrolyzes the center (sn-2) ester bond of phospholipids which yields oxidized fatty acids and lysophosphatidylcholine (lysoPC), a molecule with a range of potentially atherogenic effects, including chemoattraction of monocytes, increased expression of adhesion molecules, and inhibition of endothelial nitric oxide production [22, 23]. In this manner, a vicious cycle is set up that leads to recruitment of monocytes to the intima, where they differentiate to become macrophages, and, ultimately, foam cells, while at the same time locally producing more Lp-PLA2. Furthermore, lysoPC has been found to be cytotoxic to vascular smooth muscle cells and can induce local production of matrix metalloproteinases (MMP’s), which can thin the fibrous cap and destabilize the architectural integrity of an atheromatous plaque through destruction of the collagen matrix, increasing its propensity to rupture [24].
Fig. 2.
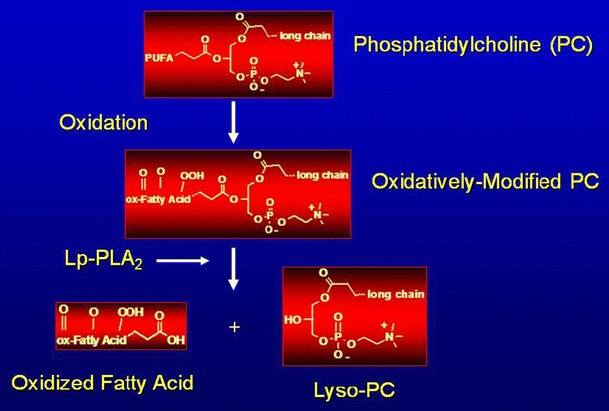
Lp-PLA2 hydrolyzes oxidized LDL to release proinflammatory lipids. Oxidative enzymes can oxidize phospholipids in LDL particles. Oxidized phosphatidylcholine is hydrolyzed by Lp-PLA2 to release oxidized fatty acid and lysophosphatidylcholine
In terms of its utility as a circulating biomarker, Lp-PLA2 produced by activated macrophages and foam cells reenters the bloodstream and can be quantitatively measured. As reported by Lavi et al. [19], Lp-PLA2 blood concentrations sampled simultaneously in the human coronary sinus demonstrated a net increase in Lp-PLA2 levels as blood traverses the coronary vascular bed from individuals with significant atherosclerotic plaque. However, when no coronary plaque is present, a decrease in Lp-PLA2 levels is found. This study also showed that the lysoPC produced by Lp-PLA2-mediated hydrolysis of oxidized LDL is highly associated with coronary artery endothelial dysfunction.
An important unmet clinical need is satisfied by measuring a circulating biomarker which signals the presence of plaque prone to rupture, since more than two-thirds of MIs occur in persons with less than 50% stenosis on coronary angiography, and it was found by Kolodgie et al. [18] that over 75% of sudden coronary deaths at necropsy were attributable to plaque rupture and thrombosis. Current well-accepted diagnostic tools available to physicians for assessing cardiovascular risk include traditional risk factor counting, a variety of risk estimation procedures (e.g., Framingham and Reynolds risk scoring), lipid and lipoprotein measurement, carotid ultrasound imaging, stress tests, echocardiography, nuclear imaging, coronary angiography and coronary intravascular ultrasonography. However, none of these is able to assess whether a patient has vulnerable, rupture-prone plaques. It is often discussed that current risk assessment approaches do not include noninvasive, inexpensive and reliable means of identifying the potential of plaque rupture; and even though emerging technologies, such as virtual histology intravascular ultrasound (IVUS), intravascular ultrasound palpography and thermography, optical coherence tomography (OCT), or carotid magnetic resonance imaging (CMRI), may help assess plaque composition and morphologic characteristics, these approaches are either invasive or very expensive for widespread utilization [16, 25].
Lp-PLA2 is a marker of vascular-specific inflammation and reflects the presence of rupture prone plaque. It is an independent predictor of cardiovascular risk, over and above traditional risk factors. However, tests for other robust biomarkers may be clinically useful in an additive manner. For example, it has been shown that while these markers are independent predictors of risk, Lp-PLA2 added to hs-CRP provides significantly more risk assessment information over hs-CRP alone. With regard to hs-CRP as a standalone test, the measured values are quite variable, requiring several independent measurements over the course of time to confirm the level, since general inflammation, infection and adiposity could be driving the value, while Lp-PLA2 has high specificity and low biovariability.
Lp-PLA2 as an independent predictor of CVD: the epidemiologic evidence
Numerous epidemiological studies consistently demonstrate that an elevated plasma level of Lp-PLA2 is independently associated with risk of coronary heart disease (CHD) and ischemic stroke [13, 14, 26–29]. As can be seen from the studies listed in Table 1 and on Fig. 3, Lp-PLA2 is a robust independent predictor of risk for the development of future heart disease and stroke as well as a strong prognostic indicator of cardiovascular risk in both men and women with established cardiovascular disease. While both the enzyme’s mass and activity have been associated with cardiovascular risk in human clinical research, at this time only the Lp-PLA2 mass assay is cleared by the U.S. Food and Drug Administration for clinical use and carries the CE Mark. Therefore, this review will tend to use “levels” referring to circulating mass of the Lp-PLA2 enzyme as detected by immunoassay.
Table 1.
Summary of key Lp-PLA2 primary and secondary observational studies
| Studies | Population examined | Endpoints evaluated | Major findings |
|---|---|---|---|
| Primary prevention | |||
| Atherosclerosis risk in communities [12] | Apparently healthy adults 45–64 years | CHD | Adjusting for traditional risk factors, 2-fold increase risk among persons with LDL <130 mg/dL and highest Lp-PLA2 levels |
| Atherosclerosis risk in communities [13] | Apparently healthy adults 45–64 years | Ischemic stroke | 2-fold increased risk after adjusting for smoking, systolic hypertension, lipid levels and diabetes status. Persons with the highest levels of Lp-PLA2 and hs-CRP were at the highest risk |
| Women’s health initiative [26] | Apparently healthy postmenopausal women | Ischemic stroke | Fully adjusted 1.6 fold increased risk was seen among non-users of hormone replacement therapy with the highest levels of Lp-PLA2. With high hs-CRP and high Lp-PLA2 these same women had more than twice the risk of stroke (odds ratio: 2.26) |
| MONItoring of trends and determinants in CArdiovascular disease (MONICA) Study [54] | Apparently healthy men 45–64 years | CHD | Each 1-standard deviation rise in Lp-PLA2 was associated with a 37% increase in risk for CHD related events |
| Secondary prevention | |||
| PEACE [27] | Stable CHD predominately men | CVD events | Fully adjusted 1.4 fold increase risk of composite endpoint (cardiovascular death, MI, coronary revascularization, unstable angina or stroke) |
| Northern Manhattan stroke study [28] | Patients with previous ischemic stroke | Recurrent ischemic stroke | 2-fold increased risk after accounting for factors such as age, atrial fibrillation, smoking, hypertension, hs-CRP |
| Mayo [29] | Patients undergoing coronary angiography | Major adverse cardiovascular events | Lp-PLA2 levels were associated with a higher incidence of major adverse events during follow-up, independent of traditional risk factors and CRP |
Fig. 3.
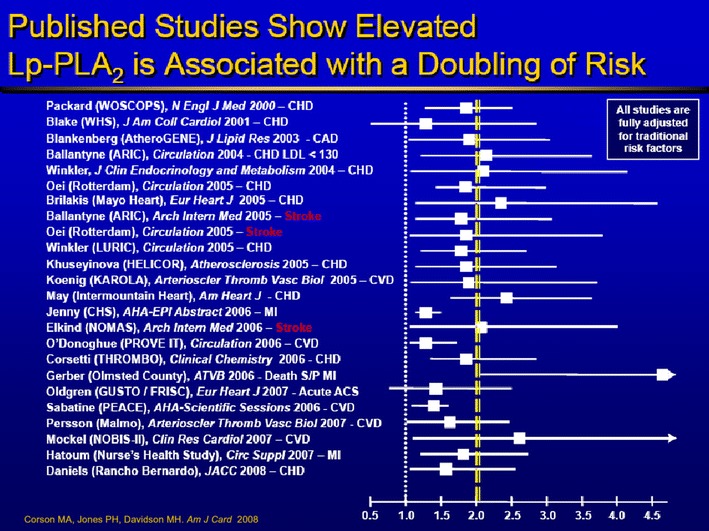
Epidemiologic evidence demonstrates the clinical utility of Lp-PLA2. More than two dozen clinical studies demonstrate the utility of Lp-PLA2 and are peer-reviewed and published [16]
A large meta-analysis of 32 prospective clinical studies on Lp-PLA2 was recently published in Lancet [30]. The Lp-PLA2 Studies Collaboration (LSC) investigated the associations of Lp-PLA2 mass and activity with cardiovascular disease (CVD) risk in 79,036 individuals, with over 17,000 outcomes, including risk of coronary heart disease (CHD), stroke and mortality in various clinical populations, comprising 474,976 person-years at risk. The LSC analyzed approximately 36,000 individuals with no history of vascular disease, about 35,000 patients with history of stable vascular disease and approximately 10,000 patients with recent acute ischemic events.
There were several key findings in this report: First, Lp-PLA2 mass and activity levels were found to be significantly associated with each other as well as with pro-atherogenic lipoprotein markers such as non-HDL-C and Apo-B. Second, Lp-PLA2 levels are significantly related to CVD risk in a continuous, log-linear association. Third, the CVD risk due to elevated Lp-PLA2 levels in this LSC analysis (10% per 1-SD increase in Lp-PLA2) is comparable to the elevated CVD risk associated with two other well established risk markers: non-HDL-C and blood pressure. Accordingly, Lp-PLA2 levels provide CVD risk assessment independent from, and on par with, other risk factors and could provide distinct insight into the relationship between inflammation, atherosclerosis and cardiovascular outcomes.
Novel biomarkers such as Lp-PLA2 have recently gained much attention in the literature, based upon their potential to be used as an adjunct to traditional risk factors to more precisely evaluate those at risk for future development of cardiovascular disease. The need to improve the accuracy of conventional risk prediction models is particularly important among patients with intermediate risk. This group is often comprised of persons with imprecisely identified cardiovascular risk for whom treatment decisions are often uncertain.
Improving the prediction of cardiovascular disease risk: value of Lp-PLA2 as an adjunct to traditional risk factors
A recently published paper examining area under the curve (AUC) of receiver operating characteristic curves (ROC) reported that novel markers such as hs-CRP, MPO and Lp-PLA2 provide little or no additional value to traditional risk factors in improving the prediction of future cardiovascular disease risk [31]. This statistical approach, commonly referred to as the c-statistic, is not the optimal test in assessing a biomarker’s ability to predict an individual’s future CVD risk. For example, studies have shown that even well accepted traditional risk factors such as smoking, dyslipidemia, and hypertension have only marginal impact on the c statistic [28]. As pointed out by Cook [32], a better technique to determine the value of novel biomarkers in a clinical setting is to determine whether a biomarker or series of biomarkers added to traditional risk factors more accurately stratify individuals into higher or lower risk categories, where therapeutic treatment strategies are dictated based upon determined risk level. Nambi and co-workers examined the ability to reclassify individuals into low-, moderate-, or high-risk categories compared to traditional risk factors, based upon Lp-PLA2 and hs-CRP levels [33]. Low risk was defined as less than a 2% risk of suffering an ischemic stroke in 5 years, moderate risk 2–5%, and high risk greater than 5%. Traditional risk factors used to initially classify risk level included age, gender, smoking status, systolic blood pressure, use of hypertensive medication, total cholesterol, HDL-cholesterol, and diabetes status. Initial classification demonstrated that 86% of the participants examined were low risk, 11% were intermediate and 3% were classified as high risk. Most revealing, the addition of hs-CRP and Lp-PLA2 reclassified approximately 39% of the intermediate-risk category (28% reclassified to a lower risk and 11% reclassified to a higher risk).
Clinical use of Lp-PLA2 measurements: expert recommendations
Consensus expert panel recommendations advocate for the measurement of Lp-PLA2 as an adjunct to traditional risk factor assessment to improve the prediction of cardiovascular risk [34]. It is recommended that Lp-PLA2 testing be performed in patients at moderate risk, those defined as apparently healthy with two or more traditional risk factors or a 10-year Framingham risk score of 10–20%, as well as high risk, those with established CHD or CHD risk equivalents, who will benefit from more aggressive lifestyle changes and lipid-modifying therapies (Fig. 3). The LpPLA2 cut point, or clinical decision threshold, for risk reassignment is >200 ng/mL. The consensus panel agreed to this threshold based upon review of Lp-PLA2 studies [28, 35, 36] which demonstrated a sufficient increase in the risk of cardiovascular events above this level to warrant more aggressive patient management. Patients at moderate risk with Lp-PLA2 levels >200 ng/mL are reclassified as having high CHD risk status, while those persons with known CHD or CHD risk equivalent and elevated levels of Lp-PLA2 are reclassified as very high risk (Fig. 4).
Fig. 4.
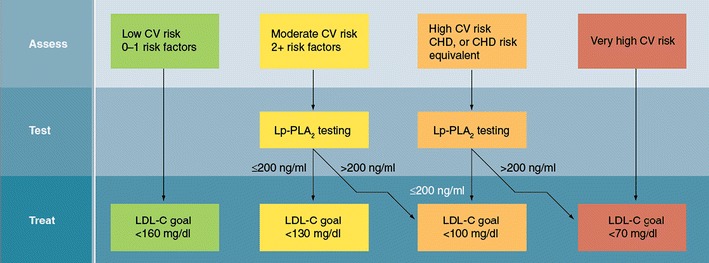
Algorithm for Lp-PLA2 screening and utility for refining cardiovascular risk estimation. It is not recommended that Lp-PLA2 be measured in patients at low risk for cardiovascular disease (one or fewer risk factors). Patients with two or more risk factors (10 year Framingham risk score of 10–20%) are optimal candidates for Lp-PLA2 screening. If the serum level of this enzyme is <200 ng/mL, then their level of risk requires no further adjustment. However, if the serum level of Lp-PLA2 is >200 ng/mL, then the patient is reassigned to high risk status and the LDL-C and non-HDL-C targets are adjusted to <100 mg/dL and <130 mg/dL, respectively. Among patients who are high risk (established CHD, diabetes mellitus, abdominal aortic aneurysm, peripheral vascular disease, symptomatic carotid artery disease, or a 10 year Framingham risk score >20%), consideration can be given to further refining risk estimation with an Lp-PLA2 measurement. If the patient’s serum Lp-PLA2 measurement >200 ng/ml, then the patient can be reclassified as very high risk, and the LDL-C and non-HDL-C targets should be <70 mg/dL and <100 mg/dL, respectively. Reproduced with permission from [34]
In clinical “real world” scenarios and general practice, it has been found that until such time that a much larger database is evaluated, it is perhaps more appropriate to also add “borderline” Lp-PLA2 levels to the continuum of clinical decision-making. Many clinical labs utilize 200–235 ng/mL as the borderline range, based on the consensus panel’s 200 ng/mL and the 235 ng/mL median from reference interval studies of normal healthy individuals. Values obtained in this range would then be weighed with other patient data, such as clinical history, physical findings and cardiovascular test results to more appropriately classify a patient’s cardiovascular risk and help decide on appropriate intensity of therapy.
Lp-PLA2 levels and lipid lowering therapies
A clinician can use Lp-PLA2 data to take action and effect change in a patient’s risk factors and vascular inflammatory status. Once an elevated Lp-PLA2 result is identified, the modifiable risk factors that contribute to vascular inflammation should and can be significantly improved. These include physical inactivity, excess body fat, smoking, lipids and high blood pressure, and they can be modified through lifestyle, diet and exercise changes, as well as through the use of prescription and other medications to reduce future CVD risk [37, 38]. Tracking the changes in these modifiable risk factors provides a good indication of future CVD risk. While Lp-PLA2 values do not change drastically over short time periods, they are beneficially affected by treatment regimens over several weeks to months. While more research is required to definitively demonstrate Lp-PLA2 as a therapeutic target, changes in Lp-PLA2 as surrogate marker of plaque stability and vascular inflammation provides potentially one of the best indications of efficacy of treatment and reduction in CVD risk. In most major statin trials, even a very aggressive reduction of LDL levels left a substantial residual risk of CV events [39]. Assessment of post-treatment Lp-PLA2 levels may help identify those patients with the greatest remaining residual risk. Studies are underway to determine how to best use serial measurements in the course of patient assessment and treatment.
In published reference interval studies, Lp-PLA2 levels range from 120 to 342 ng/mL for women and 131 to 376 ng/mL for men, in the central 90th percentile [40]. While more research is required to demonstrate Lp-PLA2 as a therapeutic target, traditional lipid lowering medications such as statins, fibrates, niacin, and omega-3 fish oil have been shown to significantly reduce plasma concentrations of Lp-PLA2. For example, statins and fibrates reduce Lp-PLA2 by as much as 30%. Among persons already treated with a statin, omega-3 fish oil therapy and extended release niacin reduce Lp-PLA2 by 13% and 20%, respectively [41–44]. While the mechanism by which these compounds reduce Lp-PLA2 levels remains somewhat speculative, it is thought that the drug induced reductions in plasma concentrations of apoB containing lipoproteins as well as a transfer of the Lp-PLA2 enzyme from apoB containing lipoproteins to HDL contribute to the overall reductions that are routinely observed [43].
While lipid modifying medications can have a profound effect on circulating lipoprotein levels, and it is known that Lp-PLA2 is predominantly associated with LDL, the therapeutic reduction in LDL does not fully explain the reduction in Lp-PLA2. Data from the Pravastatin Inflammation CRP Evaluation (PRINCE) trial demonstrated that Lp-PLA2 reduction by statin therapy is only associated with LDL lowering by statin therapy by about 6% (R 2 = 0.06) [45]. It should be reinforced that while Lp-PLA2 is not currently FDA-approved as a direct target of therapy, changes in Lp-PLA2 potentially provides one of the best indications of efficacy of treatment in improving plaque stability and reduction of vascular inflammation.
Darapladib, an orally available, small molecule drug that specifically inhibits Lp-PLA2 in a dose-dependent fashion, is currently in advanced stages of clinical development. Preclinically, it has been shown to reduce atheroma lysophosphatidylcholine content and expression of multiple genes associated with macrophage and T-lymphocyte functioning, with considerable decrease in plaque and necrotic core area [46]. The goal for this drug is to become an anti-atherosclerotic therapy complementary to current lipid-modifying therapies (e.g. statins), that addresses residual cardiovascular risk beyond traditional targets and therapies [47]. While enzyme activity is rapidly and directly inhibited by darapladib, it is still unclear what effects this may have on enzyme mass. Longer term follow up of patients on inhibitor therapy will be required to ascertain its quantitative effect on the Lp-PLA2 protein.
One of the first published clinical studies performed in humans with darapladib evaluated subjects with stable CHD or a CHD risk equivalent who were on aggressive lipid-lowering therapy with atorvastatin 20 or 80 mg per day and were randomized to placebo or darapladib 40, 80, or 160 mg/d [48]. Darapladib treatment resulted in a dose-dependent decrease in Lp-PLA2 activity by up to 66% in the 160 mg group as compared with placebo. Furthermore, treatment with darapladib (160 mg) resulted in additional lowering of other inflammatory biomarkers: C-reactive protein (CRP) levels were lowered by 20%, despite already modest baseline CRP levels, and darapladib also reduced interleukin-6 (IL-6) levels. However, levels of myeloperoxidase (MPO) and matrix metalloproteinase-9 (MMP-9) were not affected by darapladib at the doses tested. In addition, darapladib was shown to produce substantial additional reductions in Lp-PLA2 activity even when added to intensive atorvastatin therapy [49]. Since the drug was well tolerated and the initial study did not reveal any adverse clinical events or unexpected laboratory values, the inhibitor was moved into further clinical development.
The Integrated Biomarker and Imaging Study-2 (IBIS-2) randomized 330 patients with angiographically demonstrated coronary disease to 160 mg/d of darapladib versus placebo for 12 months and then reassessed coronary atheroma volume and plaque characteristics by intravascular ultrasound with virtual histology [50]. In the IBIS-2 study, a 59% reduction in Lp-PLA2 activity was shown, but without change in hs-CRP. Importantly, the necrotic core volume increased significantly (p = 0.009) in the placebo group, whereas this increase was halted in the darapladib group (p = 0.71 from baseline). This progression versus stabilization of necrotic core volume resulted in a significant treatment difference of −5.2 mm3 (p = 0.01). However, these compositional changes within the plaque occurred without a significant treatment difference in total atheroma volume or degree of calcification (P = 0.95). Based on these results, it was decided to move forward with a large outcomes trial: the Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy (STABILITY) trial.
The STABILITY trial is well underway to enrolling 15,500 patients with stable coronary disease, already taking statins, and randomizing them to 160 mg/d of darapladib orally versus placebo for 3 years [51, 52]. The primary end point is a composite of major adverse cardiovascular events, including cardiovascular death, nonfatal myocardial infarction, and nonfatal stroke. Another pivotal clinical trial with darapladib, the Stabilization of Plaques Using Darapladib-Thrombolysis in Myocardial Infarction 52 (SOLID-TIMI 52) trial, has just begun recruitment and will enroll 11,500 acute coronary patients to evaluate similar dose and endpoints as STABILITY [53].
The Lp-PLA2 inhibitor trials underline the importance of going beyond traditional risk factor treatment in patients with CAD. In many previous outcomes studies, a large amount of residual risk was seen; this was true even in trials where these risk factors were treated intensively. While cardiovascular risk was often reduced by 25–35% in major statin trials (e.g. 4S, WOSCOPS, LIPID, PROSPER, ASCOT), 65–75% of events were not prevented. This is the residual risk not addressed by solely assessing traditional risk factors. Lp-PLA2 measurements constitute a valuable means by which to identify patients who warrant aggressive, comprehensive risk factor identification and management or further intensification of ongoing therapy.
Lp-PLA2 plays an important role in personalized clinical medicine today
Lp-PLA2 testing has demonstrated an important positive effect on management of patients with cardiovascular risk and associated therapeutic decisions. These therapeutic decisions may include lifestyle modifications and drug therapy, as well as the respective intensities of each therapy. These interventions by the healthcare practitioner can be personalized to the individual according to their Lp-PLA2 levels at presentation and followed over time with serial measurements.
The following case history is presented as illustrative of the role of Lp-PLA2 testing in preventive and personalized clinical medicine:
A new patient presented to her physician for a first visit on November 19 of last year. This nominally healthy 48 year old woman presented with the chief complaint of desiring her blood pressure checked because she had noted that since the previous summer it had been high on numerous self-checks (i.e. 140/90, 147/90, 155/101, 156/99, 146/96 148/98, 152/103 mmHg). She had been checking her blood pressure twice per day since then, finding it ranged from 126–155/87–104 mmHg (systolic/diastolic), despite her reporting that it had always been “good” in the past. She is a regular and moderate exerciser with a BMI under 25, nonsmoker, non-diabetic, has an excellent and healthy diet, has one cup of coffee per day, does not drink alcohol, has no immune disease, no sleep apnea, and takes no medications. She works as an office manager, is married with two children ages 18 and 22. She sees her gynecologist annually for recommended screening and exams. Her family history is significant for CAD, with her father having had two MI’s in his late fifties and then triple coronary artery bypass surgery in his sixties, although it should be noted this does not meet NCEP criteria for the patient. Her father and sister have been diagnosed with hypertension, her maternal grandmother had a stroke, and the patient’s sister, who was overweight and smoked, had a stroke the prior year at age 50. The patient’s nephew has diabetes mellitus. Review of systems revealed an otherwise healthy woman, but her heart “feels fluttery” at times. Physical exam was normal, revealing height of 61 in., weight 112 lb, BMI 21.2, blood pressure (BP) 142/82 mmHg, respiratory rate (RR) 16 per minute, heart rate (HR) 64 beats/minute. Her electrocardiogram was normal, and her chest x-ray from the prior year was normal.
This patient’s Framingham Risk Score is very low, as are her traditional risk factors, and she was noted to already be maintaining a healthy lifestyle and diet. The physician’s plan included drawing blood for metabolic profile, TSH and comprehensive lipid panel (VAP, Atherotech), Lp-PLA2 (PLAC test), and AST/ALT, and follow up in a few weeks to go over the results. Additionally, the physician started the patient on the ACE inhibitor ramipril 5 mg daily for her hypertension.
On the patient’s second visit a month later to follow up on the labs and ramipril response, her blood pressure was found to have decreased, and her vital signs at that time were: BP 132/82, RR 16, HR 66. The VAP lipid panel showed: total cholesterol (TC) = 174, triglycerides (TG) = 78, direct LDL cholesterol = 97, non-HDL cholesterol = 112, HDL = 62, lipoprotein(a)-cholesterol = 7, IDL = 7, VLDL = 15, VLDL3 = 9, LDL density pattern A, LDL3–4 = 52, LDL1–2 = 32, apolipoprotein B (ApoB) = 77, apolipoprotein A1 (ApoA1) = 159, with ApoB/ApoA1 ratio = 0.48. These values from the VAP comprehensive lipid panel all were normal and favorable. Other labs: glucose = 97, hemoglobin A1c = 5.5%, insulin 9.5, TSH 1.5, ALT/AST 19/22, CK 70, BUN 10, and creatinine 0.9. Further advanced panel testing showed: hs-CRP = 0.9, homocysteine = 5.0, NT-proBNP = 36, vitamin D level = 34.8, ApoE3/E3 (most common), cystatin C = 0.66, and Lp-PLA2 = 269.8 (elevated).
The physician’s assessment and plan at this second visit were based on considering the patient’s positive family history of CVD (stroke in smoker, overweight sister, and father with MI, CABG) and notably her elevated Lp-PLA2 level. Her comprehensive lipid panel (VAP) was noted to be quite favorable, with lipoproteins and apoliporoteins all well into the desired range as recommended by clinical guidelines, and furthermore her blood glucose, thyroid function tests and hs-CRP were all normal. However, the one laboratory test result that was found to be abnormal was her elevated Lp-PLA2 (PLAC) concentration, with a value of 270 ng/mL. Lp-PLA2 values over 200 ng/mL and especially over 235 ng/mL are associated with enhanced cardiovascular risk, as discussed in the above sections. Although her Framingham Risk Score and Reynolds Risk Score, with CRP and family history additions, were still very low, it was called into question whether this patient was truly low risk, given the positive inflammatory plaque signal of significantly elevated Lp-PLA2. It was also noted that since her HDL (62 mg/dL) was favorable, the concept of “dysfunctional HDL” was considered, which is compatible with elevated Lp-PLA2 and vascular specific inflammation. As has been discussed above, at this point it would be reasonable to take more aggressive action to further assess this patient. The next step in the plan was to obtain a carotid ultrasound-carotid intima-media thickness assessment (CUS/CIMT).
The patient had a third visit about 6 weeks later to follow up on progress with antihypertensive medication and review the CUS/CIMT results. Her blood pressure had improved with ramipril 5 mg/day, with vital signs at this visit of: BP 112/72 mm/Hg, HR 64, RR 16. The CUS/CIMT was performed after her previous visit, resulting in the following data: average IMT = 0.585 mm, with intimal age suggesting 48 for population demographic (patient actual age is 48 years old); the left carotid bulb showed a 2.3 mm heterogeneous mixed plaque, which is of moderate size. The physician’s assessment and plan at this third visit noted that this was an abnormal CUS/CIMT with atherosclerosis, and the patient was started on a statin medication, with lab tests to be repeated in 3 months.
On the patient’s fourth visit approximately 3 months later in June of this year, her vitals were noted as BP 132/82, HR 80, RR 16, and her medications were noted as simvastatin 20 mg qhs and ramipril 5 mg/day. Her new lab results on statin therapy were: total cholesterol = 143, direct LDL-C = 59, non-HDL-C = 72, TG = 65, HDL-C = 71, VLDL = 8, LDL3–4 = 35, LDL1–2 = 10, pattern A, ApoB = 52, ApoA1 = 177, with ApoB/ApoA1 ratio = 0.29, and AST/ALT 27/23. Notably her Lp-PLA2 level decreased to 165.6 ng/mL from 269.8 ng/mL about 6 months earlier.
This fascinating case history demonstrates how Lp-PLA2 testing can have great utility in the personalized approach to preventive cardiovascular medicine. It proved to be an abnormal indicator in an otherwise spotless record, suggesting further clinical workup was warranted, leading to more aggressive treatment and vigilance, culminating in an excellent response in an otherwise very healthy middle aged woman. While sometimes multiple atherosclerosis signals present themselves in concert to indicate progressing CVD, this particular case demonstrates how using Lp–PLA2 testing may identify the “hot signal” as manifest by the inflamed plaque in a key artery. This setting is initially a call for further assessment, then becoming actionable with drug and other therapy, and finally it can be monitored with follow up Lp-PLA2 testing.
Conclusions
Atherosclerosis is a chronic inflammatory disease. Inflammation promotes endothelial cell dysfunction, the influx of inflammatory white blood cells into the subendothelial space, and LDL oxidation. As atherosclerotic plaque becomes progressively more inflamed, it becomes unstable and prone to rupture. Plaque rupture is responsible for the acute manifestations of atherosclerosis, including myocardial infarction, unstable angina, and death. While a number of inflammatory markers may predict increased risk for cardiovascular events, LpPLA2 exhibits some key differences: Lp-PLA2 is a marker of vascular-specific inflammation, whereas others biomarkers such as hs-CRP indicate systemic inflammation. The causal role of CRP in the progression of atherosclerosis is still being debated. Serum levels of CRP can vary greatly in response to a variety of host characteristics, including adiposity/insulin resistance, infection, rheumatologic disorders, and other common conditions. Lp-PLA2 participates directly in atherogenesis by potentiating lipid modification and inflammation. Lp-PLA2 hydrolyzes phosphatidylcholine to form lysophosphatidylcholine and oxidized free fatty acids, both of which stimulate atherosclerosis. Within individuals, serum levels of this enzyme have low biovariability and reflect the presence of rupture prone atherosclerotic plaque in both men and women. Lp-PLA2 is a valuable discriminator of risk for cardiovascular disease and can be used to reclassify risk in patients at intermediate and high risk for cardiovascular events. Used together, elevations in the serum levels of both CRP and Lp-PLA2 aid in the refinement and reclassification of risk [54].
Efforts to identify patients at increased risk for, or with clinically silent but established, atherosclerotic disease will intensify, and serum biomarkers will continue to play a crucial role in the four major domains of screening, diagnosis, prognosis, and management. Biomarkers, such as Lp-PLA2 levels, may serve a broader role as a prognostic aid and therapeutic target in atherosclerotic disease management. The measured levels of the mass and activity of this enzyme appear to be directly linked to the pathogenesis and progression of atherosclerosis and, importantly, serum levels decline in response to therapeutic agents that have been shown to reduce CHD events, including statins, fibrates, nicotinic acid, and omega-3 fatty acids. Direct molecular inhibitors of Lp-PLA2 such as darapladib, if proven to reduce events, will solidify this marker along with LDL-C, as a key treatment target in the reduction of cardiovascular risk and the prevention of myocardial infarction, stroke, and cardiovascular death. At the present time clinicians tend to use one inflammatory marker over another when evaluating risk. It is possible that optimal risk prediction and assignment will involve a panel of biomarkers that encapsulate both systemic inflammation and evidence of inflamed, unstable atherosclerotic plaque prone to rupture. The growing body of evidence points to an important role and utility for Lp-PLA2 testing in preventive and personalized clinical medicine.
Acknowledgments
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Contributor Information
Kenneth J. Colley, Phone: +1-650-2466400, Email: kcolley@diadexus.com
Robert L. Wolfert, Phone: +1-650-2466400, Email: bwolfert@diadexus.com
Michael E. Cobble, Phone: +1-801-5721616, Email: mcobble@canyonsmedical.com
References
- 1.Toth PP. Making a case for quantitative assessment of cardiovascular risk. J Clin Lipidol. 2007;1:234–241. doi: 10.1016/j.jacl.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Ridker PM. Clinical application of C-reactive protein for cardiovascular disease detection and prevention. Circulation. 2003;107:363–369. doi: 10.1161/01.CIR.0000053730.47739.3C. [DOI] [PubMed] [Google Scholar]
- 3.Blake GJ, Dada N, Fox JC, Manson JE, Ridker PM. A prospective evaluation of lipoproteinassociated phospholipase A(2) levels and the risk of future cardiovascular events in women. J Am Coll Cardiol. 2001;38:1302–1306. doi: 10.1016/S0735-1097(01)01554-6. [DOI] [PubMed] [Google Scholar]
- 4.Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2005;25:1102–1111. doi: 10.1161/01.ATV.0000163262.83456.6d. [DOI] [PubMed] [Google Scholar]
- 5.Libby P. What have we learned about the biology of atherosclerosis? The role of inflammation. Am J Cardiol. 2001;88:3J–6J. doi: 10.1016/s0002-9149(01)01879-3. [DOI] [PubMed] [Google Scholar]
- 6.Libby P. Act local, act global: inflammation and the multiplicity of “vulnerable” coronary plaques. J Am Coll Cardiol. 2005;45:1600–1602. doi: 10.1016/j.jacc.2005.02.058. [DOI] [PubMed] [Google Scholar]
- 7.Davies MJ, Gordon JL, Gearing AJ, et al. The expression of the adhesion molecules ICAM-1, VCAM-1, PECAM, and E-selectin in human atherosclerosis. J Pathol. 1993;171:223–229. doi: 10.1002/path.1711710311. [DOI] [PubMed] [Google Scholar]
- 8.Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–2264. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- 9.Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–1844. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 10.Ridker P, Bassuk S, Toth PP. C-reactive protein and risk of cardiovascular disease: evidence and clinical application. Curr Atheroscler Rep. 2003;5:341–349. doi: 10.1007/s11883-003-0004-3. [DOI] [PubMed] [Google Scholar]
- 11.Brennan ML, Hazen SL. Emerging role of myeloperoxidase and oxidant stress markers in cardiovascular risk assessment. Curr Opin Lipidol. 2003;14:353–359. doi: 10.1097/00041433-200308000-00003. [DOI] [PubMed] [Google Scholar]
- 12.Ballantyne CM, Hoogeveen RC, Bang H, et al. Lipoprotein-associated phospholipase A2, highsensitivity C-reactive protein, and risk for incident coronary heart disease in middle-aged men and women in the Atherosclerosis Risk in Communities (ARIC) study. Circulation. 2004;109:837–842. doi: 10.1161/01.CIR.0000116763.91992.F1. [DOI] [PubMed] [Google Scholar]
- 13.Ballantyne CM, Hoogeveen RC, Bang H, et al. Lipoprotein-associated phospholipase A2, highsensitivity C-reactive protein, and risk for incident ischemic stroke in middle-aged men and women in the Atherosclerosis Risk in Communities (ARIC) study. Arch Intern Med. 2005;165:2479–2484. doi: 10.1001/archinte.165.21.2479. [DOI] [PubMed] [Google Scholar]
- 14.Biomarkers Definitions Working Group Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- 15.Garza CA, Montori VM, McConnell JP, Somers VK, Kullo IJ, Lopez-Jimenez F. Association between lipoprotein-associated phospholipase A2 and cardiovascular disease: a systematic review. Mayo Clin Proc. 2007;82:159–165. doi: 10.4065/82.2.159. [DOI] [PubMed] [Google Scholar]
- 16.Corson MA, Jones PH, Davidson MH. Review of the evidence for the clinical utility of lipoproteinassociated phospholipase A2 as a cardiovascular risk marker. Am J Cardiol. 2008;101:41F–50F. doi: 10.1016/j.amjcard.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 17.Ali M, Madjid M. Lipoprotein-associated phospholipase A2: a cardiovascular risk predictor and a potential therapeutic target. Future Cardiol. 2009;5:159–173. doi: 10.2217/14796678.5.2.159. [DOI] [PubMed] [Google Scholar]
- 18.Kolodgie FD, Burke AP, Skorija KS, et al. Lipoproteinassociated phospholipase A2 protein expression in the natural progression of human coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26:2523–2529. doi: 10.1161/01.ATV.0000244681.72738.bc. [DOI] [PubMed] [Google Scholar]
- 19.Lavi S, McConnell JP, Rihal CS, et al. Local production of lipoprotein-associated phospholipase A2 and lysophosphatidylcholine in the coronary circulation: association with early coronary atherosclerosis and endothelial dysfunction in humans. Circulation. 2007;115:2715–2721. doi: 10.1161/CIRCULATIONAHA.106.671420. [DOI] [PubMed] [Google Scholar]
- 20.Mannheim D, Herrmann J, Versari D, et al. Enhanced expression of Lp-PLA2 and lysophosphatidylcholine in symptomatic carotid atherosclerotic plaque. Stroke. 2008;39:1445–1455. doi: 10.1161/STROKEAHA.107.503193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vickers KC, Maguire CT, Wolfert R, et al. Relationship of lipoprotein-associated phospholipase A 2 and oxidized low density lipoprotein in carotid atherosclerosis. J Lipid Res. 2009;50:1735–1743. doi: 10.1194/jlr.M800342-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macphee CH, Moores KE, Boyd HF, et al. Lipoprotein-associated phospholipase A2, platelet-activating factor acetylhydrolase, generates two bioactive products during the oxidation of low-density lipoprotein: use of a novel inhibitor. Biochem J. 1999;338:479–487. doi: 10.1042/0264-6021:3380479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Safaya R, Chai H, Kougias P, et al. Effect of lysophosphatidylcholine on vasomotor functions of porcine coronary arteries. J Surg Res. 2005;126:182–188. doi: 10.1016/j.jss.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 24.Zalewski A, Macphee C. Role of lipoprotein-associated phospholipase A2 in atherosclerosis: biology, epidemiology, and possible therapeutic target. Arterioscler Thromb Vasc Biol. 2005;25:923–931. doi: 10.1161/01.ATV.0000160551.21962.a7. [DOI] [PubMed] [Google Scholar]
- 25.Tuczu EM, Kapadia SR, Tutar E, et al. High prevalence of coronary atherosclerosis in asymptomatic teenagers and young adults: evidence from intravascular ultrasound. Circulation. 2001;103:2705–2710. doi: 10.1161/01.cir.103.22.2705. [DOI] [PubMed] [Google Scholar]
- 26.Wassertheil-Smoller S, Kooperberg C, McGinn AP, et al. Lipoprotein-associated phospholipase A2, hormone use, and the risk of ischemic stroke in postmenopausal women. Hypertension. 2008;51:1115–1122. doi: 10.1161/HYPERTENSIONAHA.107.103721. [DOI] [PubMed] [Google Scholar]
- 27.Sabatine MS, Morrow DA, O’Donoghue M, PEACE Investigators et al. Prognostic utility of lipoprotein-associated phospholipase A2 for cardiovascular outcomes in patients with stable coronary artery disease. Arterioscler Thromb Vasc Biol. 2007;27:2463–2469. doi: 10.1161/ATVBAHA.107.151670. [DOI] [PubMed] [Google Scholar]
- 28.Elkind MS, Tai W, Coates K, Paik MC, Sacco RL. High-sensitivity C-reactive protein, lipoprotein-associated phospholipase A2, and outcome after ischemic stroke. Arch Intern Med. 2006;166:2073–2080. doi: 10.1001/archinte.166.19.2073. [DOI] [PubMed] [Google Scholar]
- 29.Brilakis ES, McConnell JP, Lennon RJ, Elesber AA, Meyer JG, Berger PB. Association of lipoprotein-associated phospholipase A2 levels with coronary artery disease risk factors, angiographic coronary artery disease, and major adverse events at follow-up. Eur Heart J. 2005;26:137–144. doi: 10.1093/eurheartj/ehi010. [DOI] [PubMed] [Google Scholar]
- 30.Lp-PLA2 Studies Collaboration Lipoprotein-associated phospholipase A2 and risk of coronary disease, stroke, and mortality: collaborative analysis of 32 prospective studies. Lancet. 2010;375:1536–1544. doi: 10.1016/S0140-6736(10)60319-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melander O, Newton-Cheh C, Almgren P, et al. Novel and conventional biomarkers for prediction of incident cardiovascular events in the community. JAMA. 2009;302:49–57. doi: 10.1001/jama.2009.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cook N. Use and misuse of the receiver operating characteristic curve in risk prediction. Circulation. 2007;115:928–935. doi: 10.1161/CIRCULATIONAHA.106.672402. [DOI] [PubMed] [Google Scholar]
- 33.Nambi V, Hoogeveen RC, Chambliss L, et al. Lipoprotein associated phospholipase A2 and high-sensitivity C-reactive protein improve the stratification of ischemic stroke risk in the atherosclerosis risk in communities (ARIC) study. Stroke. 2009;40:376–381. doi: 10.1161/STROKEAHA.107.513259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davidson MH, Alberts MJ, Anderson JL, et al. Consensus panel recommendation for incorporating Lp-PLA2 testing into cardiovascular disease risk assessment guidelines. Am J Cardiol. 2008;101:51F–57F. doi: 10.1016/j.amjcard.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 35.Winkler K, Hoffmann MM, Winkelmann BR, et al. Lipoprotein-associated phospholipase A2 predicts 5-year cardiac mortality independently of established risk factors and adds prognostic information in patients with low and medium high-sensitivity C-reactive protein (the Ludwigshafen risk and cardiovascular health study) Clin Chem. 2007;53:1440–1447. doi: 10.1373/clinchem.2007.086298. [DOI] [PubMed] [Google Scholar]
- 36.Gerber Y, McConnell JP, Jaffe AS, Weston SA, Killian JM, Roger VL. Lipoprotein-associated phospholipase A2 and prognosis after myocardial infarction in the community. Arterioscler Thromb Vasc Biol. 2006;26:2517–2522. doi: 10.1161/01.ATV.0000240406.89440.0c. [DOI] [PubMed] [Google Scholar]
- 37.Hatoum IJ, Nelson JJ, Cook NR, Hu FB, Rimm EB. Dietary, lifestyle, and clinical predictors of lipoprotein-associated phospholipase A2 activity in individuals without coronary artery disease. Am J Clin Nutr. 2010;91:786–793. doi: 10.3945/ajcn.2009.28870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reddy KJ, Singh M, Batsell RR, et al. Effects of lifestyle counseling and combination lipid-modifying therapy on lipoprotein-associated phospholipase A2 mass concentration. J Clin Lipidol. 2009;3:275–280. doi: 10.1016/j.jacl.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 39.Cannon CJ, Braunwald E, McCabe CH, for the Pravastatin or Atorvastatin Evaluation and Infection Therapy—Thrombolysis in Myocardial Infarction 22 Investigators Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–1504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- 40.Lanman RB, Wolfert RL, Fleming JK, et al. Lipoprotein-associated phospholipase A2: review and recommendation of a clinical cut point for adults. Prev Cardiol. 2006;9:138–143. doi: 10.1111/j.1520-037X.2006.05547.x. [DOI] [PubMed] [Google Scholar]
- 41.Rosenson RS. Fenofibrate reduces lipoprotein associated phospholipase A2 mass and oxidative lipids in hypertriglyceridemic subjects with the metabolic syndrome. Am Heart J. 2008;155:499.e9–499.e16. doi: 10.1016/j.ahj.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 42.Kuvin JT, Dave DM, Sliney KA, et al. Effects of extended-release niacin on lipoprotein particle size, distribution, and inflammatory markers in patients with coronary artery disease. Am J Cardiol. 2006;98:743–745. doi: 10.1016/j.amjcard.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 43.Saougos VG, Tambaki AP, Kalogirou M, et al. Differential effect of hypolipidemic drugs on lipoprotein-associated phospholipase A2. Arterioscler Thromb Vasc Biol. 2007;27:2236–2243. doi: 10.1161/ATVBAHA.107.147280. [DOI] [PubMed] [Google Scholar]
- 44.Shalwitz RA, Maki KC, Doyle RT, Ballantyne CM. Lipoprotein subfraction responses differentially predict changes in lipoprotein-associated phospholipase A2 during prescription omega-3 therapy. Arterioscler Thromb Vasc Biol. 2007;abstract P328.
- 45.Albert MA, Glynn RJ, Wolfert RL, Ridker PM. The effect of statin therapy on lipoprotein associated phospholipase A2 levels. Atherosclerosis. 2005;182:193–198. doi: 10.1016/j.atherosclerosis.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 46.Wilensky RL, Shi Y, Mohler ER, et al. Inhibition of lipoprotein-associated phospholipase A2 reduces complex coronary atherosclerotic plaque development. Nat Med. 2008;14:1059–1066. doi: 10.1038/nm.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCullough PA. Darapladib and atherosclerotic plaque: should lipoprotein-associated phospholipase A2 be a therapeutic target? Curr Atheroscler Rep. 2009;11:334–337. doi: 10.1007/s11883-009-0050-6. [DOI] [PubMed] [Google Scholar]
- 48.Shi Y, Zalewski A, Macphee C, Dawson M. Selective inhibition of lipoprotein-associated phospholipase A2 attenuates markers of plaque vulnerability in humans. Circulation. 2007;116(suppl):II_108. [Google Scholar]
- 49.Mohler ER, Ballantyne CM, Davidson MH, Darapladib Investigators et al. The effect of darapladib on plasma lipoprotein-associated phospholipase A2 activity and cardiovascular biomarkers in patients with stable coronary heart disease or coronary heart disease risk equivalent: the results of a multicenter, randomized, double-blind, placebo-controlled study. J Am Coll Cardiol. 2008;51:1632–1641. doi: 10.1016/j.jacc.2007.11.079. [DOI] [PubMed] [Google Scholar]
- 50.Serruys PW, García-García HM, Buszman P, Integrated Biomarker and Imaging Study-2 Investigators et al. Effects of the direct lipoprotein-associated phospholipase A2 inhibitor darapladib on human coronary atherosclerotic plaque. Circulation. 2008;118:1172–1182. doi: 10.1161/CIRCULATIONAHA.108.771899. [DOI] [PubMed] [Google Scholar]
- 51.Riley RF, Corson MA. Darapladib, a reversible lipoprotein-associated phospholipase A2 inhibitor, for the oral treatment of atherosclerosis and coronary artery disease. IDrugs. 2009;12:648–655. [PubMed] [Google Scholar]
- 52.www.clinicaltrials.gov, NCT00799903.
- 53.www.clinicaltrials.gov, NCT01000727.
- 54.Koenig W, Khuseyinova N, Löwel H, Trischler G, Meisinger C. Lipoprotein-associated phospholipase A2 adds to risk prediction of incident coronary events by C-reactive protein in apparently healthy middle-aged men from the general population: results from the 14-year follow-up of a large cohort from southern Germany. Circulation. 2004;110:1903–1908. doi: 10.1161/01.CIR.0000143377.53389.C8. [DOI] [PubMed] [Google Scholar]