

Abstract
Background
Fetal microchimerism (F-MC), the persistence of fetal cells in the mother, is frequently encountered following pregnancy. The high prevalence of F-MC in autoimmune disease prompts consideration of the role for immune tolerance and regulation. This study examines the association between F-MC and multiple sclerosis (MS), an autoimmune disorder, of undetermined etiology.
Results
21 out of 51 MS-positive subjects (41%) were classified as positive for F-MC; 4 of 22 (18%) of MS-negative sibling controls, were also positive for MC (p = 0.066). Unanticipated F-MC in controls lead to re-evaluation using 30 female singleton cord blood units (CBUs) as a biological control. Four CBUs were low-level positive.
Study Design and Methods:
Seventy-three female subjects were assigned to three groups according to disease status and pregnancy history: (1) MS positive (+) women with a history of one male pregnancy before symptom onset (n = 27); (2) MS negative (−) female siblings of MS+ women with a history of one male pregnancy (n = 22); and (3) MS+ women that reported never having been pregnant (n = 24). Ten micrograms of genomic DNA obtained from peripheral blood leukocytes of each subject were analyzed for F-MC using allele-specific real-time PCR targeting the SR-Y sequence on the Y-chromosome. MC classification was dichotomous (positive vs. negative) based on PCR results.
Conclusion
The association between F-MC and MS warrants further study to define this relationship. F-MC in women self-reporting as nulligravid, supports previous findings that a significant proportion of pregnancies go undetected. This lead to re-validation of a Y-chromosome based assay for F-MC detection.
Key words: multiple sclerosis, microchimerism, fetal cells, autoimmune disease, twinning, pregnancy
Introduction
Multiple Sclerosis (MS) is a progressive degenerative neurologic disease, characterized by chronic, inflammatory demyelination and axonal damage.1 Despite 150 years of investigation, the precise etiology of MS still remains largely unknown.2 Although widely regarded as an autoimmune disease, there still remains a divergence of opinion that challenges the role of central dysregulaton in disease pathogenesis.3 A diverse array of associations has been described; this may allude to a phenotypically similar group of diseases rather than a single, distinct entity. Immune dysregulaton appears to be central to MS as evidenced by antibodies and autoreactive T cells against components of the myelin sheath; there is also shared pathology with experimental autoimmune encephalitis and a downstream clinical response to immunomodulatory therapy.4,5 Host genetics have a strong contribution with defined linkage to the major histocompatibility complex (MHC).1 Finally, the contribution of an environmental trigger has yet to be definitively excluded as a means of either initiating or maintaining disease progression.
Within this complex interplay the phenomenon of leukocyte chimerism has further been hypothesized to harbor a role, both broadly in autoimmune disease, as well as specifically in MS.6 Chimerism refers to the enduring co-existence of genetically disparate populations of cells within a single host. This phenomenon is well demonstrated in a number of clinical settings, notably pregnancy,7 twinning, transplantation6 and blood transfusion.8,9 Chimeric populations of allogeneic or non-self cells elude the host immune system and persist at low levels. Given that chimeric populations typically account for less than 5% of host cellular burden the term microchimerism (MC) is often used to describe this phenomenon. The clinical significance of MC is only beginning to be appreciated and extends to both adverse effect e.g., Graft versus host disease (GVHD), Autoimmune disease7,10,11 as well as the potential for therapeutic benefit e.g., Graft versus malignancy effect, adoptive immunotherapy, tissue regeneration and repair.12
MC has gained increasing attention through its plausible link to autoimmune disease. MC is well described in pregnancy through bidirectional trafficking of cells between mother and fetus, a proportion of which may persist following separation at delivery. An MC-linked autoimmune hypothesis builds on the notion that sustained occult populations of fetal cells, expressing paternal-derived “foreign” antigens, could be stimulating an alloimmune response, masquerading as an “autoimmune” disease. This hypothesis followed the observation of shared histopathological overlap between scleroderma, an autoimmune disease of, again, undetermined cause, with GVHD, a predictable adverse effect of chimerism in the setting of transplantation. This questions the existing paradigm of central immune dysregulation underlying autoimmune disease. The aim to understand whether MC indeed has a role in this complex process, forged the basis for our study to evaluate whether there is an association between MS and the presence of MC.
Results
The results of SRY MC assays are summarized in Tables 2–4. Tables 2 and 3 present the initial results of two contingency table analyses for the prevalence of MC in the three groups. Table 4 presents the summary findings following introduction of the CBU controls and presents the different rates of positivity between the groups. Among the 27 women in group 1 (MS with a history of male pregnancy), 10 (37.0%) had evidence of MC. Among the 24 women in group 3 (MS without known male pregnancy), 11 (45.8%) had male MC. Overall, of 51 women with MS, 21 (41.2%) had MC. Among 22 MS-negative siblings, 4 (18.2%) had male MC. All positive MC assays were near the lower limit of detection consistent with the presence of a single copy or small number of copies of target sequence. There was no occurrence of a positive amplification in any notemplate control.
Table 1.
Clinical data
| Description | Number of subjects | Mean age of onset (Range) | Mean age of entry into study (Range) | Mean number of live births | ||
| Male | Female | |||||
| Group 1 | MS Pos Women with one Male Pregnancy | 27 | 39.46 (23–39) | 52.96 (39–74) | 1.52 | 0.85 |
| Group 2 | MS Neg Women with one Male Pregnancy | 22 | N/A | 51.24 (37–68) | 1.45 | 0.91 |
| Group 3 | MS Pos Women who Have Never Been Pregnant | 24 | 28.58 (13–48) | 37.79 (16–61) | N/A | N/A |
Table 4.
Summary comparison of average rates of positivity for F-MC between MS patients, siblings and CBU controls
| Group | N | Average rate* (95% CI) | Relative rate** (95% CI) | p-value** |
| CBU | 30 | 0.009 (0.003, 0.02) | - | |
| MS-prior birth of a male child | 27 | 0.04 (0.03, 0.06) | 5.56 (1.40, 21.97) | 0.015 |
| MS-reported never pregnant | 24 | 0.04 (0.03, 0.06) | 5.87 (1.49, 23.16) | 0.012 |
| SIB | 22 | 0.02 (0.01, 0.03) | 2.27 (0.45, 11.46) | 0.32 |
Average positivity rates for each group (defined for an individual as the proportion of positive results observed in 22 amplification cycles), with exact 95% Poisson confidence intervals.
Relative positivity rates for each group, using the CBU group as the reference. (Estimates from Poisson regression).
p-values for comparison of estimated positivity rates in each group to rate in CBU group.
Table 2.
Comparison of microchimerism in MS-positive subjects with male pregnancy versus MS-negative siblings with male pregnancy*
| MS pos | MS neg | Totals | |
| N (%) | N (%) | ||
| Microchimerism neg | 17 (0.49) | 18 (0.51) | 35 |
| Microchimerism pos | 10 (0.71) | 4 (0.29) | 14 |
| Totals | 27 | 22 | 49 |
p = 0.207 by Fisher's exact test (2-tailed).
Table 3.
Comparison of microchimerism prevalence in all MS-positive versus MS-negative subjects*
| MS pos | MS neg | Totals | |
| N (%) | N (%) | ||
| Microchimerism neg | 30 (0.63) | 18 (0.38) | 48 |
| Microchimerism pos | 21 (0.84) | 4 (0.16) | 25 |
| Totals | 51 | 22 | 73 |
p = 0.066 by Fisher's exact test (2-tailed).
Cord blood results.
Each of the 30 samples underwent 22 amplification cycles using the same protocol employed for the initial study subjects. Summarizing each individual's results as the number of positive results observed over all cycles provides an alternate outcome to the binary indicator of positivity summarized above. A Poisson regression model fitted to these results for all individuals, taking the indicator of membership in the four groups as the predictor variable provided estimates of the group-specific positivity rates as well as estimated relative rates comparing groups, taking the CBU group as the reference. Results (Table 4) indicate significantly higher rates in both groups of women with MS. Additional comparisons of the two MS groups with the MS- siblings revealed no significant differences.
Discussion
The association between fetal derived MC and later development of MS, as suggested by our findings, is consistent with other studies examining this relationship.13
Our results are also similar to those reported for scleroderma in which the presence of rare allogeneic cells has been documented.14 However, the levels of MC observed in our study exceed that of others,13 particularly in subjects that were reportedly nulliparous.15,16 We can only speculate that alternative methodologies and reporting strategies in part account for this difference. More specifically, this may be attributable to the higher levels of DNA analyzed in our study. Given the limited sample size, we are, however, reluctant to overstate this finding.
The data from our study are based on 1,500,000 inputs for each sample in which approximately 1–12 male allogeneic microchimeric cells were detected in a positive subject. This is distinctly different from the comparatively robust levels of MC encountered in transfusion-associated MC in which the allogeneic, minor population may occupy as much as 1–4% of circulating host leukocyte burden. Sustained high-level MC following transfusion has been repeatedly described in subjects transfused following severe traumatic injury.17–19 The clinical significance of a quantitative difference between transfusion-associated MC and fetal associated MC is still not known.
Our study also draws attention to the deficiencies inherent to MC evaluation using Y-chromosome based platforms. This followed the unexpected finding in which a similar proportion of MS+ females displayed male MC independent of a known history of male pregnancy. Eleven of 24 (∼46%) of MS+ females without known pregnancy tested positive for MC. Although counterintuitive, similar unexpected findings have been reported in other studies,15,16 albeit at lower levels than that encountered in our study. This underscores that self-reported negative pregnancy history may not definitively exclude a history of pregnancy: up to 30% of pregnancies end in early fetus loss with up to 14% being occult and clinically unrecognized.20 It has further been suggested these early miscarriages may indeed convey greater risk to develop persistent fetal MC.21
Other sources of potential error were also addressed: spurious reporting and coding errors were primarily excluded upon audit of the results. PCR contamination, another necessary consideration, was deemed unlikely as both positive and negative controls were run in parallel with the assays. Furthermore, in the event that amplicon contamination occurs, it tends to be at a quantitatively higher level than that encountered in our study and is typically uniform across samples.22 One would not expect intermittent, exceedingly low-level contamination while still preserving the ability to discriminate between experimental groups such as MS and non-MS subjects as observed in the current study. Assay function and reliability is critical to MC analysis and our group has consequently studied different aspects of sample viability with concurrent PCR contamination in view of MC detection. The outlined results do not appear consistent with any known form of amplicon contamination.23 We have also conducted extensive technical validation of MC PCR assay performance, which includes both rigorous spiking studies, as well as direct sequencing to definitively verify the identity of reaction products.24,25
Although the unexpected findings were therefore postulated to be real, we employed a biological control in order to validate the results. Female neonatal cord bloods were used for this purpose, representing an ideal control having never been pregnant. Although still subject to trafficking of cells from the mother, maternal cells will elude capture by a Y-chromosome based assay. There does remain the rare possibility of an undetected or resorbed male twin contributing cells; this is, however, considered unlikely. There also remains the theoretical possibility of intergenerational chimerism whereby trafficking of cells from a prior pregnancy into the mother could lead to downstream exchange with the new fetus as reflected in the associated cord blood.26,27
The rationale for including both qualitative and quantitative results (see Table 4) is to present a balanced interpretation of the data. Simply reporting as positive vs. negative neglects a grey area where subjects test positive, but are near a threshold for positivity. Although categorized as being chimeric, these cases are more likely attributable to non-specific amplification and background noise. This was evident in the CBUs: despite selection of these samples as the closest approximation to a biological control, results demonstrate there were still qualitative positives. However, the quantitative data (number of positive wells) derived in this study demonstrated a more plausible negative interpretation, i.e., results approached an absolute negative, both in the proportion of samples affected when compared with the nulliparous MS-siblings, and also the observed rate of positivity (Table 4). The latter was not significantly different from zero, and also significantly lower than corresponding rates in the two microchimerism groups. Findings in the current study also emphasize the inherent limitations of using sex chromosome probes in evaluation of MC; this has lead to utilization of alternative platforms using HLA-based and Non HLA-Insertion-deletion (Indel) panels to impart greater precision for this purpose.
Results from our analyses raise the question of whether MS confers a higher risk of fetal loss. The literature asserts the contrary: MS confers neither increased risk of fetal loss nor other pregnancy related complication.28–30 Pregnancy is also associated with clinical improvement while disease relapse is frequently evident in the post-partum period. The age of MS onset was earlier in subjects that reported never having been pregnant. It is possible, however, that a diagnosis of MS may have influenced a decision to pursue pregnancy, at least in this small group of individuals.
In summary, these pilot results, while bound by certain limitations, do suggest that low-level MC is associated with MS. Prospective study of a larger subject population, using greater input of genomic DNA with serial blood samplings of subjects is needed. In addition, confirmation of MC by alternative assays is important. Of note the HLA-DR and InDel assays have already achieved remarkable results in the setting of transfusion associated MC.9,25 Through targeting selective, informative alleles expressed on a panel of somatic chromosomes, these assays both bypass the gender restriction of the Y-chromosome based assays as well as avoid the associated problems of a sex-chromosome based probe as illustrated by this study.9,25 Finally, it is important to note that blood may not be the ideal target tissue in which to evaluate MC; rather it is a tissue of convenience for both the present study as well as other studies seeking to gauge tissue MC. In view of these pilot findings, spinal fluid and affected neural tissue (brain) may provide a more representative sample for examination of MC. If MC is common in MS and involves target tissues specific to the disease, these findings could unravel new conceptual models for future investigation of MS.
Methods
Subjects.
The study was conducted on samples from 51 stringently ascertained MS-affected individuals and 22 unaffected family members, obtained through the UCSF Multiple Sclerosis DNA Bank (MSDB). All known ancestors were Non-Hispanic White, and of European descent. Diagnostic criteria and ascertainment protocols are summarized elsewhere in references 31 and 32. White blood cells were isolated by Ficoll gradient and high molecular weight DNA isolated using standard desalting procedures. The work was approved by the Committee of Human Research at The University of California San Francisco.
Subjects were selected according to disease status, reproductive history and availability of genomic DNA (see Table 1 for clinical data). Subjects were selected in three groups such that use of Y-chromosome analysis of MC would always be informative:
Group 1: MS+ women with a history of one male pregnancy before symptom onset (n = 27)
Group 2: MS− siblings of MS+ subjects with a history of male pregnancy (n = 22)
Group 3: MS+ women who reported they had never had a known pregnancy (n = 24).
Analysis of microchimerism.
Given that subjects were selected on the basis of having had a male pregnancy, the Y-chromosome marker was informative for allogeneic cells—microchimerism. An allele-specific quantitative PCR assay for a 73-bp region of the sex-determining region of the human Y chromosome sequence (SRY) was used as a marker for male MC in all subjects. The detailed methodology and technical validation of these MC assays has been described previously in reference 33 (see Fig. 1 for typical amplification curve). Briefly, 10 µg of genomic DNA representing approximately 1,500,000 cell equivalents, was analyzed; 600 µL of a 1:1 mixture of Solution A (0.1 M KCl, 0.01 M Tris Base, 0.0025 M MgCl2·6H20, pH 8.3) and Solution B (10 mM Tris, 2.5 mM MgCl2·6H20, 1% Tween-20, 1% NP40, pH 8.3) was added to the DNA preparation. Twenty-five µL of DNA was added to 50 µL of buffer consisting of 1 µM of the each primer SB (5′-GAG GCG CAA GAT GGC TCT AGA G-3′) and SC (5′-CCA CTG GTA TCC CAG CTG CTT GC-3′) (Integrated DNA Technologies, Coralville, IA), 6 mM Magnesium, 25× of SYBR Green (FMC BioProducts, Rockland, ME) and 1 mM of dNTPs (Roche). Real-time PCR was conducted using the GeneAmp 5700 machine (Applied Biosystems, Foster City, CA) and cycle conditions (10 min @ 95°C followed by 45 cycles of: 30 sec @ 95°C, 30 sec @ 68°C and 45 sec @ 72°C). All reagents were prepared and retained in a dedicated laboratory, separated from the sample preparation. A female technologist performed all procedures. In order to analyze the full 10 µg of genomic DNA without inhibition of amplification, we assayed multiple identical aliquots in parallel. An average of twenty-four reaction volumes, representing the total of 10 µg DNA, were carried out per subject. In two reaction tubes, we spiked 10 copies of Y-chromosome positive DNA as positive controls. Replicate no-template negative controls were included in each run. Results were evaluated for endpoint positivity. A count of one positive event (genomic equivalent) was attributed to each well exhibiting low-level positive amplification.
Figure 1.
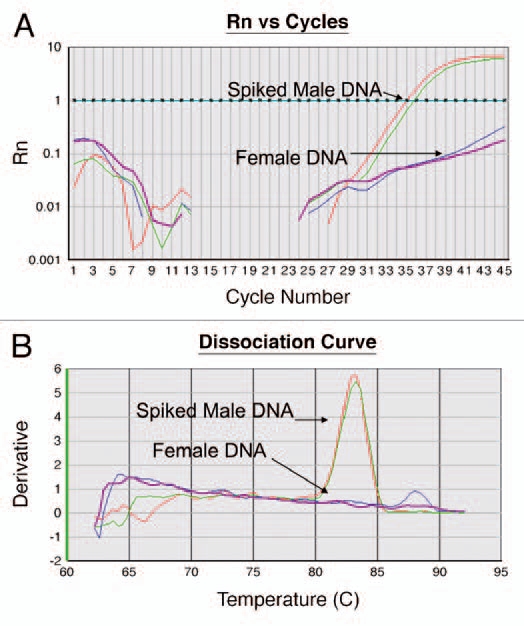
(A) Amplification Curve. The assay is performed in a 96 well plate where each well is monitored at every cycle for fluorescent intensity (Rn, y-axis). As PCR progresses and generation of new amplicons, Rn increases until reaching a plateau. At the end of each run, a user-defined threshold is set. This threshold is the level of fluorescence at which CTs or threshold cycle, is calculated. This threshold is set higher than the noise level in the baseline. During the reaction, the cycle number at which the fluorescent intensity crosses the threshold value is defined as CT. The CT represents the cycle at which a statistically significant increase in ΔRn is first detected. Therefore, samples with a low CT have an abundant target. In this figure, the threshold is set to 1 and the CT of the female DNA spiked with positive DNA is at an average of 35.5. The DNA from a female subject (negative) does not cross the threshold. (B) Dissociation Curve. At the end of the cycle, the generated amplicons were analyzed for specificity. The amplified products were melted by increasing the temperature to 95°C with melt temperature corresponding to the temperature at which half of the amplicons are denatured. The derivative (y-axis) is the slope of the curve generated by the melting curve. The peak of the dissociation curve is equal to the melting temperature. Temperature is labeled on x-axis (60°C to 95°C). In this figure the spiked positive DNA melts at 83°C. The DNA from a female subject (negative) predictably failed to generate any amplified product.
Data analysis.
Subjects were classified as positive or negative for MC on the basis of Y-chromosome PCR results. A subject was considered positive for MC if any positive reaction was present in any of the 24 parallel aliquots assayed. The proportion of subjects positive or negative for MC was compared in 2 × 2 contingency tables according to disease status and/or pregnancy history using 2-tailed Fisher's exact test. To investigate the possible influence of observed between-group variation in overall number of positive results, complementary analyses were based on regression models for individual specific “rates” of positivity for MC, defined for each patient as the number of positive reactions among aliquots assayed. Rates were compared between groups using Poisson regression models for count data, and inferences based on robust standard error estimates to account for possible overdispersion in observed counts. Results were summarized as relative rates for each of the MS groups and unaffected siblings, using cord blood unit (CBU) controls as the reference group. Analyses were performed using Stata (version 11.1).
Cord blood units.
Following the unanticipated finding of significant prevalence of male MC among the group 3 subjects (MS+ women who reported they had never had a known pregnancy), 30 singleton pregnancy female CBUs were used as a biological control as well as to validate the test performance. WBC pellets were prepared from cord blood by lysing the RBCs with saponin lysis solution and digesting the cell pellets with proteinase K.34 The DNA was quantified by amplifying a region of HLA-DQα.34 Ten micrograms of DNA, equaling 1,500,000 genomic equivalents were used per cord blood unit. Results were analyzed using two-sample tests of proportion comparing the number of positive wells for each MS subject group to the CBU group. Calculations were performed using SAS software, Version 9.1.3.
Acknowledgements
This project was initiated through a Pilot Research award from the National Multiple Sclerosis Society and an NHLBI Special Center of Research (SCOR) grant in Transfusion Medicine #P50-HL-54476. Additional support has been received both from the UCSF NMSS DNA Bank through a grant of the National Multiple Sclerosis Society RG2899 as well as NIH/NIAID grant R01AI059829 that facilitated Dr. Barcellos' input. Finally, the study has been supported to completion through NIH Grant No. 5R01HL083388.
The authors are sincerely grateful to Dr. Michael Busch of Blood Systems Research Institute and Dr. Jorge Oksenberg of the University of California San Francisco for helpful discussion and a critical reading of this manuscript. We also thank UCSF CTSI for providing statistical support.
References
- 1.Steinman L, Martin R, Bernard C, Conlon P, Oksenberg JR. Multiple sclerosis: deeper understanding of its pathogenesis reveals new targets for therapy. Annu Rev Neurosci. 2002;25:491–505. doi: 10.1146/annurev.neuro.25.112701.142913. [DOI] [PubMed] [Google Scholar]
- 2.Murray TJ. The history of multiple sclerosis: the changing frame of the disease over the centuries. J Neurol Sci. 2009;277:3–8. doi: 10.1016/S0022-510X(09)70003-6. [DOI] [PubMed] [Google Scholar]
- 3.Chaudhuri A, Behan PO. Multiple sclerosis is not an autoimmune disease. Arch Neurol. 2004;61:1610–1612. doi: 10.1001/archneur.61.10.1610. [DOI] [PubMed] [Google Scholar]
- 4.Weiner HL. Multiple sclerosis is an inflammatory T-cell-mediated autoimmune disease. Arch Neurol. 2004;61:1613–1615. doi: 10.1001/archneur.61.10.1613. [DOI] [PubMed] [Google Scholar]
- 5.Racke MK, Lovett-Racke AE, Karandikar NJ. The mechanism of action of glatiramer acetate treatment in multiple sclerosis. Neurology. 2010;74:25–30. doi: 10.1212/WNL.0b013e3181c97e39. [DOI] [PubMed] [Google Scholar]
- 6.Willer CJ, Sadovnick AD, Ebers GC. Microchimerism in autoimmunity and transplantation: potential relevance to multiple sclerosis. J Neuroimmunol. 2002;126:126–133. doi: 10.1016/s0165-5728(02)00048-6. [DOI] [PubMed] [Google Scholar]
- 7.Bianchi DW. Fetal cells in the mother: from genetic diagnosis to diseases associated with fetal cell microchimerism. Eur J Obstet Gynecol Reprod Biol. 2000;92:103–108. doi: 10.1016/s0301-2115(00)00432-2. [DOI] [PubMed] [Google Scholar]
- 8.Utter GH, Owings JT, Lee TH, Paglieroni TG, Reed WF, Gosselin RC, et al. Blood transfusion is associated with donor leukocyte microchimerism in trauma patients. J Trauma. 2004;57:702–707. doi: 10.1097/01.ta.0000140666.15972.37. [DOI] [PubMed] [Google Scholar]
- 9.Utter GH, Reed WF, Lee TH, Busch MP. Transfusion-associated microchimerism. Vox Sang. 2007;93:188–195. doi: 10.1111/j.1423-0410.2007.00954.x. [DOI] [PubMed] [Google Scholar]
- 10.Nelson JL. Microchimerism in human health and disease. Autoimmunity. 2003;36:5–9. doi: 10.1080/0891693031000067304. [DOI] [PubMed] [Google Scholar]
- 11.Bianchi DW. Fetomaternal cell trafficking: a new cause of disease? Am J Med Genet. 2000;91:22–28. doi: 10.1002/(sici)1096-8628(20000306)91:1<22::aid-ajmg4>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 12.Cirello V, Recalcati MP, Muzza M, Rossi S, Perrino M, Vicentini L, et al. Fetal cell microchimerism in papillary thyroid cancer: a possible role in tumor damage and tissue repair. Cancer Res. 2008;68:8482–8488. doi: 10.1158/0008-5472.CAN-08-0672. [DOI] [PubMed] [Google Scholar]
- 13.Willer CJ, Herrera BM, Morrison KM, Sadovnick AD, Ebers GC. Association between microchimerism and multiple sclerosis in Canadian twins. J Neuroimmunol. 2006;179:145–151. doi: 10.1016/j.jneuroim.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 14.Nelson JL, Furst DE, Maloney S, Gooley T, Evans PC, Smith A, et al. Microchimerism and HLA-compatible relationships of pregnancy in scleroderma. Lancet. 1998;351:559–562. doi: 10.1016/S0140-6736(97)08357-8. [DOI] [PubMed] [Google Scholar]
- 15.Yan Z, Lambert NC, Guthrie KA, Porter AJ, Loubiere LS, Madeleine MM, et al. Male microchimerism in women without sons: quantitative assessment and correlation with pregnancy history. Am J Med. 2005;118:899–906. doi: 10.1016/j.amjmed.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 16.Lambert NC, Pang JM, Yan Z, Erickson TD, Stevens AM, Furst DE, Nelson JL. Male microchimerism in women with systemic sclerosis and healthy women who have never given birth to a son. Ann Rheum Dis. 2005;64:845–848. doi: 10.1136/ard.2004.029314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reed W, Lee TH, Norris PJ, Utter GH, Busch MP. Transfusion-associated microchimerism: a new complication of blood transfusions in severely injured patients. Semin Hematol. 2007;44:24–31. doi: 10.1053/j.seminhematol.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 18.Lee TH, Paglieroni T, Utter GH, Chafets D, Gosselin RC, Reed W, et al. High-level long-term white blood cell microchimerism after transfusion of leukoreduced blood components to patients resuscitated after severe traumatic injury. Transfusion. 2005;45:1280–1290. doi: 10.1111/j.1537-2995.2005.00201.x. [DOI] [PubMed] [Google Scholar]
- 19.Lee TH, Paglieroni T, Ohto H, Holland PV, Busch MP. Survival of donor leukocyte subpopulations in immunocompetent transfusion recipients: frequent long-term microchimerism in severe trauma patients. Blood. 1999;93:3127–3139. [PubMed] [Google Scholar]
- 20.Wang X, Chen C, Wang L, Chen D, Guang W, French J. Conception, early pregnancy loss and time to clinical pregnancy: a population-based prospective study. Fertil Steril. 2003;79:577–584. doi: 10.1016/s0015-0282(02)04694-0. [DOI] [PubMed] [Google Scholar]
- 21.Khosrotehrani K, Johnson KL, Lau J, Dupuy A, Cha DH, Bianchi DW. The influence of fetal loss on the presence of fetal cell microchimerism: a systematic review. Arthritis Rheum. 2003;48:3237–3241. doi: 10.1002/art.11324. [DOI] [PubMed] [Google Scholar]
- 22.Kwok S, Higuchi R. Avoiding false positives with PCR. Nature. 1989;339:237–238. doi: 10.1038/339237a0. [DOI] [PubMed] [Google Scholar]
- 23.Reed W, Lee TH, Vichinsky EP, Lubin BH, Busch MP. Sample suitability for the detection of minor white cell populations (microchimerism) by polymerase chain reaction. Transfusion. 1998;38:1041–1045. doi: 10.1046/j.1537-2995.1998.38111299056314.x. [DOI] [PubMed] [Google Scholar]
- 24.Montalvo L, Walker P, Wen L, Lim W, Reed W, Busch MP, Lee TH. Clinical investigation of posttransfusion Kidd blood group typing using a rapid normalized quantitative polymerase chain reaction. Transfusion. 2004;44:694–702. doi: 10.1111/j.1537-2995.2004.03303.x. [DOI] [PubMed] [Google Scholar]
- 25.Lee TH, Chafets D, Reed W, Wen L, Yang Y, Chen J, et al. Enhanced ascertainment of microchimerism with real-time quantitative PCR amplification of insertion/deletion polymorphisms. Transfusion. 2006;46:1870–1878. doi: 10.1111/j.1537-2995.2006.00992.x. [DOI] [PubMed] [Google Scholar]
- 26.Nelson JL. Naturally acquired microchimerism: for better or for worse. Arthritis Rheum. 2009;60:5–7. doi: 10.1002/art.24217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guettier C, Sebagh M, Buard J, Feneux D, Ortin-Serrano M, Gigou M, et al. Male cell microchimerism in normal and diseased female livers from fetal life to adulthood. Hepatology. 2005;42:35–43. doi: 10.1002/hep.20761. [DOI] [PubMed] [Google Scholar]
- 28.Ferrero S, Pretta S, Ragni N. Multiple sclerosis: management issues during pregnancy. Eur J Obstet Gynecol Reprod Biol. 2004;115:3–9. doi: 10.1016/j.ejogrb.2003.10.020. [DOI] [PubMed] [Google Scholar]
- 29.Sandberg-Wollheim M, Frank D, Goodwin TM, Giesser B, Lopez-Bresnahan M, Stam-Moraga M, et al. Pregnancy outcomes during treatment with interferon beta-1a in patients with multiple sclerosis. Neurology. 2005;65:802–806. doi: 10.1212/01.wnl.0000168905.97207.d0. [DOI] [PubMed] [Google Scholar]
- 30.Weber-Schoendorfer C, Schaefer C. Multiple sclerosis, immunomodulators and pregnancy outcome: a prospective observational study. Mult Scler. 2009;15:1037–1042. doi: 10.1177/1352458509106543. [DOI] [PubMed] [Google Scholar]
- 31.Oksenberg JR, Barcellos LF, Cree BA, Baranzini SE, Bugawan TL, Khan O, et al. Mapping multiple sclerosis susceptibility to the HLA-DR locus in African Americans. Am J Hum Genet. 2004;74:160–167. doi: 10.1086/380997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barcellos LF, Oksenberg JR, Begovich AB, Martin ER, Schmidt S, Vittinghoff E, et al. HLA-DR2 dose effect on susceptibility to multiple sclerosis and influence on disease course. Am J Hum Genet. 2003;72:710–716. doi: 10.1086/367781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ariga H, Ohto H, Busch MP, Imamura S, Watson R, Reed W, Lee TH. Kinetics of fetal cellular and cell-free DNA in the maternal circulation during and after pregnancy: implications for noninvasive prenatal diagnosis. Transfusion. 2001;41:1524–1530. doi: 10.1046/j.1537-2995.2001.41121524.x. [DOI] [PubMed] [Google Scholar]
- 34.Lee TH, Wen L, Chrebtow V, Higuchi R, Watson RM, Sninsky JJ, Busch MP. Quantitation of residual WBCs in filtered blood components by high-throughput, real-time kinetic PCR. Transfusion. 2002;42:87–93. doi: 10.1046/j.1537-2995.2002.00009.x. [DOI] [PubMed] [Google Scholar]