

Abstract
Mitochondria play essential roles in cellular metabolism, redox homeostasis, and regulation of cell death. Emerging evidences suggest that cancer cells exhibit various degrees of mitochondrial dysfunctions and metabolic alterations, which may serve as a basis to develop therapeutic strategies to preferentially kill the malignant cells. Mitochondria as a therapeutic target for cancer treatment is gaining much attention in the recent years, and agents that impact mitochondria with anticancer activity have been identified and tested in vitro and in vivo using various experimental systems. Anticancer agents that directly target mitochondria or indirectly affect mitochondrial functions are collectively classified as mitocans. This review article focuses on several natural compounds that preferentially kill cancer cells with mitochondrial dysfunction, and discusses the possible underlying mechanisms and their therapeutic implications in cancer treatment. Mitocans that have been comprehensively reviewed recently are not included in this article. Important issues such as therapeutic selectivity and the relevant biochemical basis are discussed in the context of future perspectives.
Keywords: Mitochondrial dysfunction, Energy metabolism, Reactive oxygen species, Apoptosis, 2-Phenethyl isothiocyanate, Honokiol
1. Introduction
Mitochondria are double membrane-enclosed organelles that play essential roles in the complex network of cellular metabolism, redox signaling, calcium homeostasis, and regulation of cell proliferation and cell death. These cellular organelles are the only ones that have their own genetic material (mtDNA) and replication system independent of the nuclear DNA replication system. Under physiological conditions, mitochondria keep dynamic processes of fusion and fission upon exo-stimulus. The replication (reproduction) of these organelles is not synchronous with the cell cycle events, and thus mitochondrial biogenesis may occur without cell division. Due to their multiple roles in production of ATP, generation of reactive oxygen species (ROS), and regulation of apoptosis, mitochondria are involved in many pathological processes including neurodegenerative diseases, cancer, ischemia/reperfusion injury, aging, diabetes, obesity, and other disease conditions. The complex regulatory mechanisms involved in mitochondrial energetics, epigenetics, and genetics have been comprehensively reviewed recently (Wallace and Fan, 2010).
Due to their ability to effectively generate ATP to meet the energy requirements for cellular metabolism and proliferation, mitochondria are considered as the cellular power plant. Generation of ATP in the mitochondria is accomplished by a series of biochemical processes known as oxidative phosphorylaion. The mitochondrial electron transport complexes (I–V) are the key molecular machineries that execute the biochemical processes of mitochondria respiration and ATP synthesis. The mitochondrial respiratory chain accepts electrons from NADH and succinate via complex I and complex II, respectively, and delivers them to the terminal electron acceptor oxygen, forming water. This process generates a proton gradient cross the mitochondrial inner-membrane, and the energy potential stored in this transmembrane electrochemical gradient can be used for the ATP synthesis at complex V (ATP synthase). Under physiological condition, mitochondria supply the majority of the ATP needed to maintain normal cellular functions. Associated with the electron transport process during oxidative phosphorylation, some of the electrons may leak or “escape” from complex I or III, and be captured by molecular oxygen to form superoxide radical, which can then be converted to other forms of reactive oxygen species such as hydrogen peroxide or react with nitric oxide to form other reactive nitrogen species (RNS) such as peroxyl nitride. It is estimated that about 1–2% O2 consumed by cells may be converted to superoxide radicals. ROS/RNS play important roles in regulation of physiological functions. At moderate levels, ROS/RNS may affect many cell biological processes by transcriptional regulation through certain redox-sensitive transcription factors including NF-κB, P53, Nrf-2, and HIF, or by directly modifying protein/enzyme molecules such as TRX, AKt, PTEN, Bcl-2 involved in cell proliferation, transformation or survival (Trachootham et al., 2008a). At highly elevated levels, ROS/RNS can cause severe oxidative damage to biolipid membranes, DNA, and protein, leading to cell injury and cell death (Trachootham et al., 2009).
In addition to their essential role in cellular energy metabolism, mitochondria also function as a hub of the cell death signaling pathways including apoptosis, necrosis and autophagy. Cell death is the evolutionary conserved and genetically regulated process to maintain the normal homeostasis in mammal cells. In the intrinsic apoptotic pathway, the death signals directly or indirectly act on mitochondria through the Bcl-2 family or permeability pore to release the apoptosis factors such as cytochrome c, AIF, and Smac/DIABLO leading to activation of the caspase cascades, nuclear DNA cleavage, and cell death. In the extrinsic apoptotic pathway, the death signal/ligands bind to the death receptors, which trigger the activation of caspase-8, followed by activation of caspase-3 and other downstream proteases. The activated proteases can then cause destruction of cellular proteins leading to cell death. This cell death process can be further amplified by caspase-mediated cleavage of Bid, which is then translocated to mitochondria to induce the release of apoptosis factors such as cytochrome c, AIF, and Smac/DIABLO from mitochondria into cytosol. Severe oxidative stress may cause acute damage to the mitochondrial membranes and other biomembranes, leading to acute cell death. During autophagy, inclusion of mitochondria within the lysosomal structure is a characteristic change. Considering the important roles of mitochondria in various cell death pathways, it is not surprising that compounds that target mitochondria may significantly affect cell viability by inducing apoptosis, necrosis, or autophagy.
2. Mitochondrial dysfunction in cancer cells
Mitochondrial dysfunction has been observed in cancer cells. Several factors, including mitochondrial DNA mutations, oncogenic stress, loss of p53 tumor suppressor, and aberrant expression of metabolic enzymes, are thought to contribute to mitochondrial abnormalities. Since mitochondria play major roles in energy metabolism, ROS generation and redox signaling, and regulation of cell death, dysfunction of these cellular organelles would cause alterations in these important cellular processes, which may provide a biochemical basis for preferentially impacting cancer cells. The following sections describe important alterations as a consequence of mitochondrial dysfunction in cancer cells and their relevance to cancer therapeutics.
2.1. Mitochondrial dysfunction and alterations in energy metabolism
A prominent alteration in energy metabolism in cancer cells is the increase in aerobic glycolysis, a phenomenon known as the Warburg effect, first observed by Otto Warburg in the 1920s and subsequently demonstrated in various types of cancer cells including leukemia cells in the presence of abundant oxygen (Warburg, 1956). Although the exact mechanisms responsible for the elevated glycolysis in cancer cells still remain elusive, recent genomic and metabolic studies have provided substantial new supporting evidence (Flier et al., 1987; Shim et al., 1997; Altenberg and Greulich, 2004). Warburg attributed such metabolic alterations to “respiration injury,” and considered this as the origin of cancer (Warburg, 1956). The glycolytic pathway produces only two ATP molecules per glucose, while 36 ATP can be generated per glucose in the mitochondria through oxidative phosphorylation. Thus, a small loss in mitochondrial respiration due to mitochondrial dysfunction would require a substantial increase in glycolysis to compensate the energy production. Importantly, active glycolytic activity may also provide essential metabolic intermediates as a source of essential biosynthesis for cancer cell growth and proliferation. This might in part explain why most cancer cells exhibit highly active glucose uptake and utilization. Indeed, the development of positron emission tomography (PET), a highly sensitive and non-invasive imaging technology for cancer diagnosis and monitoring of therapeutic responses, is largely based on high glucose uptake by cancer cells in patients (Gambhir, 2002).
The reasons why cancer cells are addicted to aerobic glycolysis and the underlying molecular mechanisms still remain controversial. Mitochondrial dysfunction caused by mitochondrial DNA mutations, oncogenic signals, and ROS stress may be an important event that forces cancer cells to be more rely on the glycolytic pathway for energy production and for generation of metabolic intermediates for biogenesis. Glycolysis may be still highly active even in cancer cells with competent mitochondria function and activate oxidative phosphorylation, and thus blocking a single energy metabolic pathway might not be effect in killing cancer cells (Moreno-Sanchez et al., 2010). Although glycolysis has a lower yield of ATP production, the higher rate of glycolytic activity in cancer cells may gain certain selective advantage over normal cells, which are more vulnerable to the lack of nutrition and oxygen (Pfeiffer et al., 2001). In addition, metabolic intermediates from glycolysis provide cells with an important source of precursors for ribose, glycerol, citrate, NADPH, and amino acids which are needed for biosynthesis of nucleotides and lipids (DeBerardinis et al., 2008). Therefore, the persistent increase in aerobic glycolysis in cancer cells may confer cell survival and proliferation advantage. In addition, the acidification of microenvironment due to lactate production during glycolysis may facilitate tumor invasion and metastasis (Gatenby and Gillies, 2004; Weinberg and Chandel, 2009).
It has been found that certain mitochondrial proteins encoded by nuclear genes can be tumor suppressors and their abnormalities are involved in tumor development (Gottlieb and Tomlinson, 2005; Weinberg and Chandel, 2009). Succinate dehydrogenase (SDH) and fumarate hyratase (FH) are involved in the tricarboxylic acid (TCA) cycle that metabolically connects glycolysis in the cytosol to oxidative phosphorylation in the mitochondria (Gottlieb and Tomlinson, 2005). Mutations in those genes are linked to phaeochromocytoma, paraganglioma or leiomyoma. Interestingly, tumors such as paraganglioma that contain this type of mutations are relatively benign.
Recent studies suggest that upregulation of glucose transporters and hexokinases may be involved in promoting the Warburg effect. Elevated expression of glucose transporters (GLUTs) especially GLUT1, which has been correlated with tumor invasiveness and metastasis, is induced by oncogenic transformation caused by c-Myc (Osthus et al., 2000), ras or scr (Flier et al., 1987). C-Myc also activates lactate dehydrogenase A (LDH-A) overexpression, which seems required for c-Myc-mediated transformation (Shim et al., 1997). Another key regulator that controls glycolysis and mediates cellular adaptation to hypoxia is the transcription factor HIF-1α (hypoxia inducible factor 1 alpha). Activation of HIF-1α seems able to induce the “Warburg effect” and promote aggressiveness of the malignant cells (Robey et al., 2005). HIF-1α deregulation or sustained stability under normoxia condition has been related to many cancer types (Semenza, 2007). The HIF-1α level is high in RCC4 renal carcinoma cell line due to a mutation in von Hippel-Lindau (VHL) ubiqutin ligase. By reintroducing VHL into RCC4 cells, HIF-1α is destabilized, which leads to a substantial decrease in aerobic glucose consumption (Robey et al., 2005). In addition to its role in the aerobic glycolysis, HIF-1α can also be stabilized by the accumulation of lactate and pyruvate, and in turn promote the expression of several genes including GLUT3, vascular endothelial growth factor (VEGF), and erythropoietin (Lu et al., 2002). Therefore, HIF-1α may contribute to a vicious cycle of up-regulated glycolysis leading to the accumulation of lactate and pyruvate, which further stimulate HIF-1α and promote further increases in glycolytic enzymes expression (Ristow and Schulz, 2009).
The tumor suppressor p53 has been shown to be an important molecule that affects glucose metabolism, and loss of p53 function in cancer cells may contribute to the glycolytic phenotype. Wild-type p53 represses GLUT1 and GLUT4 gene transcription, while mutations within the DNA binding domain of p53 impair the repressive effect on GLUT transcription, leading to increased glucose metabolism (Schwartzenberg-Bar-Yoseph et al., 2004). In hepatoma cells, over-expression of mutant p53 protein binds to the p53 response elements on the type II hexokinase promoter region and activates its expression (Mathupala et al., 1997). P53 also indirectly regulates glycolysis through the IKK-NF-κB pathway (Kawauchi et al., 2008). Recently, TP53-induced glycolysis and apoptosis regulator (TIGAR) was identified as an important molecule that attenuates glycolysis (Bensaad et al., 2006). When p53 is mutated, glycolysis is increased due to a lack of TIGAR. Importantly, the wild-type p53 function seems required to maintain the normal mitochondrial respiratory function via its transcriptional regulation of SCO2, which is important for the proper assembly of the respiratory chain complex IV (Matoba et al., 2006). A loss of p53 function leads to decrease of mitochondrial respiration and increased glycolysis. In addition, a role of Akt in promoting glycolysis has been observed. Activation of Akt seems sufficient to stimulate a switch to aerobic glycolysis without affecting the rate of oxidative phosphorylation in transformed cells (Elstrom et al., 2004). Glioblastoma cells with high Akt activity displayed an increased rate of glycolysis, while those cells lacking Akt show low glycolytic activity. Consistently, Akt-expressing cells are more susceptible to glucose withdrawal than the control cells.
The Warburg effect has been used as a biochemical basis for developing therapeutic strategy to preferentially target cancer cells. It has been shown that in glioblastoma cells which mainly rely on glycolysis for ATP generation, glucose withdrawal induced extensive apoptosis, whereas normal astrocytes seemed able to shift to other sources for energy metabolism (Jelluma et al., 2006). Several agents inhibiting glycolytic enzymes have been in various stages of preclinical and clinical studies (Pelicano et al., 2006; Pathania et al., 2009; Wang et al., 2010). These compounds inhibit different steps in the glycolysis pathway and appear able to preferentially kill cancer cells with high glycolysis. For example, a glucose analog 2-deoxy-d-glucose (2DG) and a hexokinase inhibitor 3-bromopyruvate (3BrPA) can cause ATP depletion and death in cancer cells, which is even effective in multi drug resistant cells (Ko et al., 2001; Xu et al., 2005). As mentioned above, HIF-1α promotes glycolysis and cancer cell survival, and has been regarded as a target for cancer therapy. Agents have been designed to target the HIF-1α regulatory mechanisms, its interactions with DNA, or its activity (Pathania et al., 2009). Likewise, attenuation of LDH-A expression has been shown to compromise glycolysis, increase mitochondrial respiration, and diminish tumorigenicity (Fantin et al., 2006). Taken together, the metabolism difference between normal cells and cancer cells associated with mitochondrial respiration may provide a biological basis for developing novel and effective therapeutic approaches to cancer treatment.
2.2. Mitochondrial dysfunction and redox alterations in cancer
Compelling evidence suggests that cancer cells tend to have elevated levels of ROS, compared to the normal cells of the same tissue origins (Szatrowski and Nathan, 1991). Although the exact causes of the ROS stress in cancer are still not entirely clear, mitochondrial dysfunction has been proposed as one of the possible reasons (Brandon et al., 2006; Pani et al., 2009). Several lines of evidences support this hypothesis. For example, deficiency in functional p53 resulting in instability of mitochondrial genome was associated with the increased cellular ROS stress (Achanta et al., 2005). Mitochondrial DNA (mtDNA) mutations have also been shown to be correlated with increased ROS levels in several types of cancer cells both in solid tumors and leukemia (Carew et al., 2003; Indo et al., 2007; Ishikawa et al., 2008). Interestingly, in ρ0 cells, whose mitochondrial DNA is depleted and mitochondrial respiration is defective, ROS alterations seem to follow a more complex pattern (Pelicano et al., 2003). The association between mitochondrial DNA defect and redox alteration is likely due to alterations in the mitochondrial DNA-encoded protein components of the electron transport chain complexes (Brandon et al., 2006). Mutation of mitochondrial DNA could result in malformation of electron transport chain and thus compromise the electron transporting process, leading to an increased leakage of electrons and superoxide formation (Adam-Vizi and Chinopoulos, 2006; Brandon et al., 2006). On the other hand, a total depletion of mtDNA would lead to a lack of mitochondrial respiration, and thus a decrease in superoxide generation in the mitochondria. In additional, increased mitochondrial transmembrane potential in cancer cells is another potential mechanism that can enhances ROS production from the electron transport chain.
It has been recognized that the increased ROS stress in cancer cells is correlated with the aggressiveness of tumors and poor prognosis (Patel et al., 2007; Kumar et al., 2008). It seems that oxidative stress may play a significant role in the acquisition of the hallmarks of cancer (Hanahan and Weinberg, 2000), immortalization and transformation (Behrend et al., 2003), cancer cell proliferation (Achanta et al., 2005), mitogenic signaling (Irani et al., 1997), cell survival and disruption of cell death signaling (Pervaiz and Clement, 2004; Clerkin et al., 2008; Trachootham et al., 2008a), epithelial–mesenchymal transition and metastasis (Radisky et al., 2005; Wu, 2006; Nishikawa, 2008), angiogenesis (Komatsu et al., 2008; Ushio-Fukai and Nakamura, 2008), and chemoresistance (Pervaiz and Clement, 2004; Achanta et al., 2005; Sullivan and Graham, 2008). Nevertheless, ROS is a double-edged sword and may impose different effects on the cells, depending on the degree and duration of the ROS elevation. When the increase of ROS reaches a certain threshold level that exceeds the cellular antioxidant capacity, ROS may exert a cytotoxic effect leading to oxidative damage and the death of tumor cells and thus suppress cancer progression (Fruehauf and Meyskens, 2007). To maintain cell viability and proliferation under intrinsic ROS stress, cancer cells must acquire adaptive mechanisms to adequately counteract the potential toxic effects of the elevated ROS and to promote cell survival pathways (Irmak et al., 2003). These adaptive mechanisms may include stimulating the redox-sensitive transcriptional factors and upregulating the expression of antioxidant molecules (Sullivan and Graham, 2008). This redox adaptation which confers cell survival advantage may play important roles not only in cancer development but also in drug resistance (Pervaiz and Clement, 2004; Tiligada, 2006; Sullivan and Graham, 2008).
Redox alterations associated with mitochondrial dysfunction in cancer cells may be exploited for therapeutic purpose. As cancer cells have elevated ROS generation and are under increased intrinsic oxidative stress, it is conceivable that these malignant cells would be more dependent on the cellular antioxidant systems and the redox-sensitive pro-survival signal to maintain viability, and therefore more vulnerable to further oxidative insults induced by ROS-generating agents or by compounds that abrogate the key antioxidant systems in the cells. As such, manipulating ROS levels by redox modulation seems to be a feasible way to selectively kill cancer cells with less toxicity to normal cells (Schumacker, 2006). Indeed, several ROS-modulating agents seem to exhibit promising therapeutic activity in preclinical and clinical studies (Trachootham et al., 2009). For example, Arsenic trioxide, which is capable of impairing the respiratory chain function and promote superoxide generation, has been shown to effectively kill leukemia cells (Pelicano et al., 2003). However, some cancer cells, especially those in advanced disease stages, have become highly adapted to intrinsic oxidative stress with up-regulated antioxidant capacity. This redox adaptation not only enables the cancer cells to survive under increased ROS stress, but also provides a mechanism of resistance to many anticancer agents due to the increased ability to tolerate exogenous stress, an upregulation of survival molecules, and high capacity for drug inactivation. For example, resistance to arsenic trioxide was found to be associated with an upregulation of hemoxygenase I, SOD1 and glutathione (Hour et al., 2004; Zhou et al., 2005a). To achieve both therapeutic selectivity and overcome drug resistance associated with redox adaptation, it is important to design new strategies that exploit the redox difference between normal and cancer cells and also disable the redox adaptation mechanism in cancer cells (Trachootham et al., 2009). One such approach is to target the key redox regulatory mechanisms that control both the level of ROS and the function of redox-sensitive survival proteins (Trachootham et al., 2009). The thiol-based antioxidants, including glutathione, thioredoxin, and peroxiredoxin, can be considered as potential targets for such a redox intervention. The natural compound PEITC is one of such agents that kill cancer cells by disabling the glutathione antioxidant system, which will be described in detail under Section 3.
2.3. Mitochondrial dysfunction and resistance to apoptosis
Proper balance between cell proliferation and cell death is essential to maintain tissue homeostasis, and the failure to eliminate cells by apoptosis may play an important role in carcinogenesis. Abnormal decrease in apoptosis has been considered as a mechanism responsible for the accumulation of cancer cells, especially in certain malignancies such as chronic lymphocytic leukemia (Reed, 1998). Mitochondria play a pivotal role in regulating apoptosis. Among the important molecules that affect the intrinsic apoptotic pathway through mitochondria, the Bcl-2 family proteins play a major role in cell survival and drug sensitivity. Dysregulation of Bcl-2 family is often observed in various types of human cancer, including renal, ovarian, stomach, and brain tumors and leukemia (Sharief et al., 2003; Heiser et al., 2004; Takeuchi et al., 2005). Over expression of the anti-apoptotic Bcl-2 family members seems to correlate with poor prognosis and therapeutic responses. Interestingly, certain oncogenes and tumor suppressor genes implicated in the regulation of apoptosis are known to affect the expression of Bcl-2 (Hemann et al., 2005; Herbst et al., 2005; Mestre-Escorihuela et al., 2007). For instance, c-Myc may affect apoptosis by altering the ratio of anti-apoptotic factors (Bcl-2, Bcl-xl) and pro-apoptotic factors (Bax, Bak). The tumor suppressor p53 induces apoptosis in part through regulating Bax or the BH3-only protein Bim, NOXA and PUMA. In the extrinsic apoptotic pathway, the CD95/TRAIL death receptors trigger the cell death process through activation of caspase-8 upon ligand binding, leading to activation of the downstream caspases and cleavage of the pro-apoptotic molecule Bid to the active form tBid, which then amplifies the cell death signal by initiating the mitochondrial apoptotic pathway (Stegehuis et al., 2010).
The detail molecular mechanisms that govern the mitochondrial apoptotic events still remain to be elucidated. The opening of the mitochondrial permeability transition pore (PTP) is considered as a major even that can lead to mitochondrial depolarization and the release of apoptotic factors. PTP is thought to be a multi-protein complex, although its exact composition is still a matter of debate. It has been proposed that PTP contains certain core molecules including voltage-dependent anion channel (VDAC, outer-membrane), adenine nucleotide translocase (ANT, inner-membrane), and cyclophilin D (CypD, inner-membrane) with other regulatory proteins such as peripheral benzodiazepine receptor (PBR), creatine kinase (CK), and hexokinase (HK) (Halestrap, 2009), and may play important roles in the regulation of mitochondrial homeostasis and apoptosis. Under physiological conditions, PTP allows the proper passage of small molecules in and out of the mitochondria. Under apoptotic stimuli, the opening of PTP may lead to the loss of cytochrome c, malfunction of the mitochondrial respiratory chain, depletion of ATP, and possibly the inflow of p53 (Berridge et al., 2009). As such, the opening of PTP seems to integrate multiple death signals. The outflow of apoptotic factors such as cytochrome c, AIF, endoG, and Smac can trigger the downstream cell death processes. Abnormalities in PTP have been implicated in pathological conditions such as cancer, neurodegeneration, ischemia/reperfusion and aging (Corsi et al., 2008; Cavalieri et al., 2009). Over expression of PBR has been observed in a number of neoplastic tissues such as breast cancer, hepatoma, ovarian and colon carcinomas (Corsi et al., 2008). HKII and CK are upregulated in various tumors (Meffert et al., 2005; Palmieri et al., 2009). VDAC, ANT and CypD are also reported to increase in tumor tissues and seem to suppress apoptosis (Kim et al., 2006b; Pedersen, 2008; Chen et al., 2009a).
Many Bcl-2 family proteins, including pro-apoptotic and anti-apoptotic members have transmembrane domains, which insert into the outer-membrane of the mitochondria. The pro-apoptotic factors Bax and Bak induce the mitochondria outer-membrane permeabilization by forming oligomers, leading to the release of cytochrome c into the cytosol to initiate the activation of caspases cascades. The anti-apoptotic Bcl-2 members seem to protect mitochondrial membrane integrity by blocking the action of Bax and Bak. It seems that most of the Bcl-2 family members can affect the mitochondria membrane integrity either by inducing pore formation or by sequestering the pore-forming molecules (Knudson and Brown, 2008). Depending on the strength and duration of the apoptosis signal, mitochondria may either exhibit transient/reversible alterations in transmembrane potential, or reach a “point of no return” with a massive opening of the permeability transition pore, collapse of membrane integrity, and release of apoptotic factors (Kroemer et al., 2007; Jourdain and Martinou, 2009). The important role of PTP in apoptosis suggests that it may be a potential therapeutic target. In fact, many anticancer agents may affect PTP directly or indirectly.
3. Natural compounds that preferentially kill cancer cells with mitochondrial dysfunction
Several natural compounds have been shown to be able to selectively kill cancer cells with mitochondrial dysfunction. Some of these compounds directly target mitochondria and affect the mitochondrial metabolism or apoptotic process, while other compounds do not exert their impact directly on mitochondria but preferentially interfere with the metabolic alterations as the consequence of mitochondrial dysfunction in cancer cells. Table 1 shows examples of such compounds, their modes of action, and their current status in drug development.
Table 1.
Natural compounds and analogues that preferentially impact cancer cells with mitochondrial dysfunction.
| Compound | Source | Mode of action | Cancer type | Drug status |
|---|---|---|---|---|
| β-phenylethyl isothiocyanate (PEITC) |
Cruciferous vegetables |
Depleting GSH; inhibiting GPX; modifying Prx3 and tubulin. |
Leukemia, breast, prostate, lung, ovarian, bladder, hepatic cancer |
Phase II cancer prevention trial |
| Honokiol |
Magnolia
grandifloris |
Targeting CypD and inducing mitochondrial apoptosis. |
Esophagus, ovarian, breast, lung cancer, myeloma, leukemia |
Preclinical |
| α-Tocopheryl succinate (α-TOS) |
Synthetic vitamin E analog |
Targeting ubiquinone- binding site of complex II |
Melanoma, prostate, colorectal, breast cancer, mesothelioma |
Preclinical |
| Epigallocatechin- 3-gallate (EGCG) |
Green tea | Accumulation in mitochondria, inducting apoptosis |
Lymphoma, myeloma, melanoma, pancreatic, colon, breast cancer |
Phase II clinical trial |
| Curcumin | Turmeric (Curcuma longa) |
Inducing apoptotic via multiple mechanisms |
Lymphoma, Gastric, skin, cervical, pancreatic non- small cell lung cancer |
Phase II clinical trial |
| Pancratistatin (PST) |
Spider lily Pancratium littorale |
Inducing ROS stress, loss of mitochondrial potential, apoptosis |
Lymphoma, breast, colon cancer |
Preclinical |
| OSW-1 |
Ornithogalum
saudersiae |
Damaging mitochondrial membranes and cristae; Ca2+- dependent apoptosis. |
Leukemia, malignant brain tumor, pancreatic, ovarian cancer |
Preclinical |
| Resveratrol | Red grapes, blueberries, mulberries, etc. |
Competing with coenzyme Q, inhibiting complex III activity. |
Colon rectal, breast, liver, lung, gastric cancer, myeloma, follicular lymphoma |
Phase II clinical trial |
| Vitamin K3 | Synthetic vitamin K precursor |
Inhibiting mitochondrial pol γ; causing ROS stress |
Leukemia and various solid tumors |
Phase II clinical trial |
3.1. β-Phenylethyl isothiocyanate (PEITC)
As described above, mitochondrial respiratory chain is a major site of ROS generation, and mitochondrial dysfunction may lead to a significant elevation of ROS production due to increased leak of electrons from the respiratory chain. The intrinsic increase of ROS generation in cancer cells with mitochondrial dysfunction may render them more vulnerable to further oxidative insult, compared to the normal cells with lower ROS output. Thus, the redox difference between normal and cancer cells may serve as a biochemical basis for selective killing of cancer cells by exogenous ROS-generating agents (Huang et al., 2000; Achanta et al., 2005; Schumacker, 2006). However, cancer cells under intrinsic stress may activate the stress adaptive mechanisms by upregulation of the cellular antioxidant capacity, which counteract the harmful effect of ROS (Kim et al., 2006a; Ogasawara and Zhang, 2008). As such, these cancer cells may become resistant to exogenous agents such as H2O2 and arsenic trioxide which enhance intracellular ROS (Lenehan et al., 1995; Zhou et al., 2005a). One way to overcome such drug resistance is to use compounds capable of disabling the up-regulated antioxidant system (Trachootham et al., 2009). β-Phenylethyl isothiocyanate (PEITC) is one such compound that shows promising anticancer activity through a ROS-modulating mechanism.
PEITC is a natural product found in commonly consumed cruciferous vegetables such as watercress, cabbage, and broccoli in the form of glucosinotes as its precursors. The endogenous mirosinase enzyme in the vegetables cleaves glucosinolates, yielding multiple metabolites including PEITC. This compound has been considered as a chemopreventive agent in part due to its ability to stimulate or activate phase II detoxification enzymes (Hecht, 1999; Zhang et al., 2005). In the recent years, studies from different groups suggest that this compound has potent anticancer activity with low toxicity to normal cells, and that the mode of action seems to involve a ROS-mediated mechanism (Xu and Thornalley, 2001; Hu et al., 2003; Rose et al., 2003; Zhang et al., 2003, 2008a; Wu et al., 2005; Satyan et al., 2006; Trachootham et al., 2006, 2008b, 2009). Furthermore, PEITC effectively kills cancer cells resistant to standard chemotherapeutic agents such as fludarabine, cisplatin and gleevec. The potent anticancer activity of PEITC was demonstrated in vitro, in tumor xenograft models, and in primary cancer cells from leukemia patients (Trachootham et al., 2006, 2008b; Zhang et al., 2008a; Matoba et al., 2006).
The biochemical basis for the anticancer activity and selectivity of PEITC may be attributed to its ability to disable the glutathione (GSH) antioxidant system, and to the dependency of cancer cells on GSH to maintain redox balance. Since GSH system is a key redox regulator of both the cellular ROS levels and the function of redox-sensitive proteins (Estrela et al., 2006), this system plays a major role in neutralizing the toxic effect of ROS and in redox adaptation. The increased GSH levels promote cancer cell survival and resistance to certain anticancer agents due to the increased ability to scavenge ROS and to stabilize survival molecules through thiol modifications (Trachootham et al., 2009). Importantly, the increase in basal ROS generation renders cancer cells highly dependent on GSH antioxidant system and thus vulnerable to agents that abrogate this antioxidant system. PEITC effectively disables the GSH antioxidant system by depletion of cellular GSH and inhibition of the redox-modulating enzyme glutathione peroxidase (Xu and Thornalley, 2001; Trachootham et al., 2009). Inhibition of the GSH antioxidant system by PEITC in cancer cells may cause a severe accumulation of ROS due to the high basal ROS output in these cells, especially in those cells with mitochondrial dysfunction, and trigger massive cell death either by c-jun kinase-induced apoptosis and/or by direct damage to cellular components, such as membranes, proteins and DNA (Xu and Thornalley, 2001; Hu et al., 2003; Trachootham et al., 2006). As GSH also has a crucial role in regulating the function of proteins through thiol modifications (Hurd et al., 2005; Trachootham et al., 2008a), the abrogation of the GSH system can also inactivate the pro-survival signals, such as nuclear factor κB (NF-κB) and MCL-1, and thus further facilitate cell death (Trachootham et al., 2006, 2008b). Inhibition of ROS-scavenging systems and inactivation of pro-survival pathways can together effectively kill cancer cells. In contrast, normal cells which have lower levels of basal ROS output and are less dependent on the GSH system, may tolerate suppression of the GSH system by PEITC at the concentrations that effectively kill cancer cells (Trachooootham et al., 2009).
Abrogation of glutathione system seems to be a key mechanism of action of PEITC. It is important to understand how PEITC disrupts the glutathione system and why it requires a relatively lower concentration and shorter time to exert its selective cytotoxic effect against cancer cells compared to the common glutathione-depleting agent buthionine sulfoximine (BSO) (Trachootham et al., 2009). A previous study showed that PEITC can conjugate with glutathione and cause its export from the cells, thus leading to a depletion of intracellular glutathione pool (Xu and Thornalley, 2001). A study by Zhang et al. suggested that the conjugation between GSH and PEITC as an electrophilic–nucleophilic interaction owing to the fact that the central carbon in the isothiocyanate group (N=C=S) of PEITC is highly electrophilic (Zhang and Talalay, 1994). Further studies showed that the phenyl ring and the short alkyl chain of PEITC may also be important for its cytotoxic effect since the other derivatives lacking the ring or the alkyl chain exhibited less anticancer activity (Zhang et al., 2005).
The chemical properties of PEITC and its mechanisms of action may explain why this compound seems to have more potent and rapid action against cancer cells compared to BSO. BSO inhibits the glutathione synthesis enzyme (Meister, 1991), while PEITC directly removes the existing GSH pool by direct conjugation and export (Xu and Thornalley, 2001). Since GSH could be replenished by either the de novo synthesis, the recycling of the oxidized glutathione (GSSG) through reduction by glutathione reductase, or by deglutathionylation of thiol proteins (Estrela et al., 2006), cells whose glutathione synthesis enzyme is inhibited by BSO can still utilize the recycling mechanism to generate GSH. In contrast, cells treated with PEITC will have a net loss of glutathione due to the export of GSH-PEITC conjugate. Thus, PEITC can cause a rapid depletion of GSH through the export mechanism while glutathione depletion by BSO is a slow process due to the presence of the GSSG–GSH recycling system. In addition, the ability of PEITC to inhibit glutathione peroxidase (GPX) enzyme activity further makes this compound a potent agent that can effectively disable the glutathione antioxidant system (Fig. 1). As such, when cells are exposed to PEITC, they may not have sufficient time to activate the adaptive response pathways to counteract ROS stress, while the slow action of BSO might allow time for the cancer cells upregulate various antioxidant mechanisms to survive.
Fig. 1.
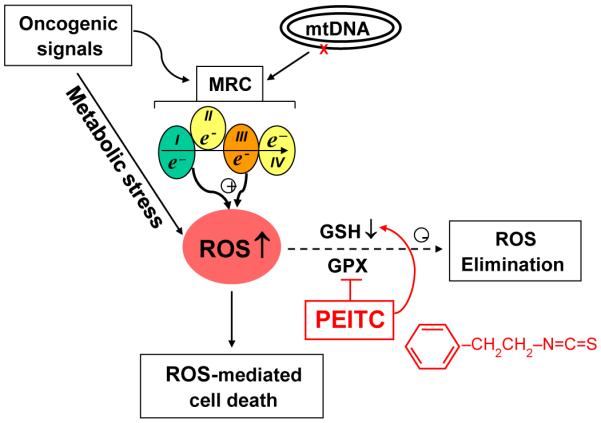
Schematic illustration of the possible mechanisms of action of PEITC in preferential killing of cancer cells with mitochondrial dysfunction. Multiple factors including mitochondrial mutations, oncogenic signals, and metabolic stress may cause dysfunction of the mitochondrial respiratory chain (MRC), leading to increased leakage of electrons, which promote formation of reactive oxygen species (ROS). Such increased oxidative stress renders the cancer cells more dependent on the cellular antioxidant systems such as glutathione (GSH) and glutathione peroxidase (GPX) to keep redox balance. PEITC (chemical structure shown in red) potently disables the glutathione antioxidant system by causing depletion of GSH and inhibition of GPX activity (see text for detail), and thus induces severe ROS accumulation in the cancer cells due to their high ROS output, leading to oxidative damage and cell death. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The keap-1/Nrf-2 system is a major pathway that mediates cellular defensive responses to oxidative stress by promoting the expression of numerous detoxifying and antioxidant (Itoh et al., 2004). Nrf2 is sequestered by keap-1, but such sequestration can be abrogated by electrophiles leading to release of Nrf2 as an active transcriptional factor to enhance the expression of antioxidant molecules (Itoh et al., 2004). Because PEITC is an electrophile, it may stimulate the cellular stress response through the keap-1/Nrf-2 system at low concentrations tolerable by the cells. This may explain why PEITC can enhance detoxifying capacity as a chmoprevention agent (Xu et al., 2006). However, when cancer cells are exposed to a sufficient concentration of PEITC, the rapid depletion of GSH and severe ROS accumulation induced by this compound will cause cancer cell death. Induction of cancer cell death is thought to contribute to the cancer preventive effect of PEITC (Cheung et al., 2010).
Recent studies suggest that PEITC may also directly target certain thiol proteins such as peroxiredoxin 3 (Prx3) and tubulin. Analysis of proteins from cells treated with PEITC showed that this compound may cause irreversible modifications of Prx3 and tubulin, and that such protein modification may be important for the cytotoxic action of PEITC (Mi et al., 2007; Brown et al., 2008). Due to its lipophilicity, PEITC can readily penetrate cell membrane and be accumulated intracellularly to a high concentration. HPLC analysis of cell extracts from cancer cells incubated with 10 μM PEITC showed that this compound could be accumulated inside the cells up to 250–500 fold greater than that in the culture medium, yielding the intracellular concentration of PEITC in the range of mM (Xu and Thornalley, 2001; Trachootham et al., 2009). This may explain why the use of PEITC at the μM concentration in the culture medium can effectively deplete cellular GSH, which is in the mM range. It is worth noting that that in human the plasma concentration of PEITC could reach the micromolar range 2–8 h after oral ingestion (Liebes et al., 2001; Ji and Morris, 2003). PEITC is metabolized in liver and excreted in urine within 24 h, and there was no accumulative effect after multiple doses of this compound. These pharmacokinetic data suggest that effective plasma concentrations of PEITC can be achieved in human, and that multiple doses at proper intervals may be required to maintain the therapeutic concentrations in vivo.
3.2. Honokiol
Honokiol is a natural product that exhibits anticancer effects in experimental models with various types of cancer cells, including esophageal, ovarian, breast, and lung cancer, as well as myeloma and leukemia. It is speculated that this compound causes cancer cell death in part through targeting mitochondria (Munroe et al., 2007; Chen et al., 2009a; Fried and Arbiser, 2009). Honokiol is thought to be a key active component of the Chinese herb medicine known as houpo, which is from the bark of Magnolia officinalis and has been used clinically in traditional oriental medicine for treatment of various indications. Although honokiol was originally considered as an antioxidant, its antioxidant role still remains controversial. For instance, this compound has been reported to act as scavengers of hydroxyl radicals and lipid peroxides, and seems to have suppressive effect on NADPH oxidase (Sheu et al., 2008). Paradoxically, other studies suggested that honokiol could induce an increase in ROS levels in various cell lines (Li et al., 2007; Chen et al., 2009a). Since honokiol is a polyphenolic compound, it may function as an antioxidant or as a pro-oxidant depending on the redox environment. The anticancer activity of honokiol as a single agent or in combination with other chemotherapy agents has been consistently demonstrated in experimental cancer models (Liu et al., 2008) (for review, see Fried and Arbiser, 2009). Several mechanisms of action have been suggested, including induction of apoptosis by causing mitochondrial dysfunction and endoplasmic reticulum stress (Chen et al., 2009b), arrest of cell cycle by interfering Rb function and inhibition of the E2F1 transcriptional activity (Hahm and Singh, 2007), down-regulation of Ras and Akt/mTOR pathways (Garcia et al., 2008; Crane et al., 2009), and inhibition of angiogenesis and tumor invasion through modulating NF-κB pathway (Tse et al., 2005; Deng et al., 2008). Among these possible mechanisms, the impact on mitochondria and induction of the intrinsic apoptotic pathway through mitochondria are important for the cytotoxic action of honokiol.
It has been reported that honokiol may overcome drug resistance in human multiple myeloma by caspase-dependent and -independent apoptosis (Ishitsuka et al., 2005). Although caspase 3, 7, 8, 9 activation is triggered by honokiol, the pan-caspase inhibitor z-VAD-fmk could not block the cell death process. In different cell lines, however, low expression of caspase-3 conferred resistance to honokiol. In the same study, the authors observed that honokiol treatment triggered the cleavage of the anti-apoptotic protein Mcl-1 and upregulation of the pro-apoptotic protein Bad, leading to the release of AIF from mitochondria to initiate apoptosis. In another study using lung cancer cells, honokiol was shown to upregulate Bad and downregulate Bcl-xl, leading to cytochrome c release into cytosol and activation of the caspase cascade. Similar effect of honokiol was also observed in breast and ovarian cancers (Yang et al., 2002). Thus, it seems that effect of honokiol on mitochondria is mediated, at least in part, through the Bcl-2 family proteins.
Recently, it was reported that the mitochondrial permeability transition pore (PTP) might be an important target of HNK in various cancer cells. In particular, cyclophilin D (CypD), a component of the VDAC/ANT/CypD complex involved in PTP, seems to play a pivotal role in mediating honokiol-induced apoptosis (Chen et al., 2009a). Upon incubation of human esophageal carcinoma cells with honokiol, the mitochondrial membrane potential dropped dramatically, followed by apoptotic cell death. Interestingly, the cytotoxic effect of honokiol could not be blocked by z-VAD-fmk, but could be partially prevented by pre-treatment with cyclosporine A (CsA), an inhibitor of CypD that can attenuate the opening of PTP. Consistently, siRNA knockdown of CypD also reduced the sensitivity to honokiol (Chen et al., 2009a,b). Associated with the opening of PTP induced by honokiol, cellular ROS and cytosolic cytochorome c increase was observed. However, the ROS scavengers N-acetylcysteine and catalase could not block cytochrome c release and apoptosis, although cellular ROS level was reduced. These observations suggest that the ROS increase in honokiol-treated cells was a downstream event following PTP opening (Li et al., 2007; Chen et al., 2009a). Interestingly, the expression of CypD seems elevated in certain cancer cells such as esophageal carcinoma. This may in part contribute to their higher sensitivity to honokiol, which seems to have low toxicity to non-malignant esophageal epithelial cells (Chen et al., 2009a,b).
Chemically, honokiol is a biphenolic compound (C18H18O2) with the molecular weight of 266.33. This compound is found in the cones, bark, and leaves of Magnolia grandifloris, and exists naturally with its structural isomer magnolol. Honokiol can be purified from the natural sources by extraction and chromatographic methods such as HPLC, or chemically synthesized via the Muara–Suzuki coupling procedures (Fried and Arbiser, 2009). This compound has poor solubility in water, but is soluble in ethanol and seems to have good bioavailability with a sustained plasma drug concentration in mice and a two-compartment pharmacokinetics in rats (Chen et al., 2004; Tsai et al., 1994). Derivatives of honokiol have recently been synthesized as potential anticancer compounds (Amblard et al., 2007; Luo et al., 2008).
3.3. Vitamin E analogs
α-Tocopheryl succinate (α-TOS) is representative of the vitamin E analogs capable of targeting mitochondria and preferentially inhibiting cancer cells (Zhao et al., 2009). Recent studies showed that this compound induce cell death by targeting the ubiquinone-binding site of the mitochondrial respiratory complex II (Dong et al., 2008, 2009). In experimental systems using various cancer types, this compound exhibits promising anticancer activity both in vitro and in vivo. Several other vitamin E analogs and novel liposome-based formulations are currently under active development, aiming at improving therapeutic activity (Koudelka et al., 2009; Turanek et al., 2009; Zhao et al., 2009). The mechanisms of action of this class of compounds and their anticancer activity have been comprehensively reviewed recently (Neuzil et al., 2007a; Ralph and Neuzil, 2009; Zhao et al., 2009).
3.4. Epigallocatechin-3-gallate (EGCG)
Green tea has been recognized for a long time for its potential ability to reduce cancer risk. People in Asian countries who drank green tea daily showed lower cancer incidence, later onset age, and lower recurrence rates of various types of cancer (Imai et al., 1997; Nakachi et al., 1998; Jian et al., 2004). Epigallocatechin-3-gallate (EGCG) is an abundant polyphenol in green tea and considered to be a key component of green tea that contributes to its anticancer property. It has been shown that EGCG was able to rapidly cause apoptotic cell death in various malignant B-cell lines by triggering the mitochondria-related cell death events including loss of mitochondrial transmembrane potential (Δψm); release of various mitochondrial apoptogenic proteins (cytochrome c, Smac/DIABLO, and AIF), activation of caspases, and ROS generation (Nakazato et al., 2005). Apoptosis induced by EGCG could be reduced by antioxidant compounds, catalase and SOD2, suggesting that ROS may play a key role in mediating the cell death process. Similar results were found in pancreatic cancer, colon cancer and melanoma cell lines as well as breast cancer xenograft animal models (Chen et al., 2003; Baliga et al., 2005; Nihal et al., 2005; Qanungo et al., 2005). In addition, tumor cells lacking caspase-3 expression could escape apoptosis when treated by EGCG (Hsu et al., 2003). Interestingly, EGCG selectively killed melanoma cells without exerting toxic effect to normal melanocytes (Nihal et al., 2005). High concentration of EGCG treatment caused a severe ROS increase in oral malignant cells but led to a ROS decrease in normal epithelial cells (Yamamoto et al., 2003). Further analysis of cellular antioxidants in both cells showed that normal cells had a higher level of catalase than cancer cells, which might explain why EGCG caused an increase of ROS only in cancer cells (Yamamoto et al., 2003). When combined with chemotherapy drugs like arsenic acid and erlotinib, EGCG showed synergetic effect in killing cancer cells (Nakazato et al., 2005; Zhang et al., 2008b).
The precise mechanisms responsible for the preferential cytotoxic effect of EGCG against cancer cells remain to be investigated. Interestingly, a recent study showed that the vast majority of this compound (over 90%) is accumulated in the mitochondria after incubation of neurons with EGCG in culture, and that such accumulation seemed to confer a protective effect on normal neurons under oxidative stress conditions (Schroeder et al., 2009). In contrast, in vitro incubation of multiple brain tumor cells with EGCG resulted in significant growth inhibition (Pilorget et al., 2003). It is possible that the differences between normal and cancer cells in their mitochondrial function might be responsible for such different responses, and that mitochondrial dysfunction in cancer cells might render them vulnerable to EGCG. Currently, it is unclear if EGCG could directly bind to a specific target molecule in the mitochondria.
EGCG has entered clinical trials in cancer prevention and cancer treatment. A preliminary clinical study showed that three out of four patients with low grade B-cell malignancies had partial response after oral ingestion of EGCG (Shanafelt et al., 2006). Phase I clinical trial in chronic lymphocytic leukemia patients also yielded encouraging results (Shanafelt et al., 2009).
3.5. Curcumin
Curcumin (1,7-bis-(4-Hydroxy-3-methoxyphenyl)-1,6 hepta-diene-3,5-dione or diferuloyl methane) is a major constituent of turmeric powder from the plan Curcuma longa, has been widely used as a spice. Although turmeric has been used as a traditional herbal medicine in South Asia for thousands of years, it attracted much attention for its use in cancer treatment only in the recent years when its potential anticancer activity was reported in 1980s. The study by Kuttan et al. (1985) showed that curcumin inhibited cell growth in Chinese Hamster Ovary cells and was cytotoxic to lymphocytes and lymphoma cells. Soon afterward, they tested an ethanol extract of turmeric and an ointment of curcumin in patients with external cancerous lesions and obtained promising results (Kuttan et al., 1987). Further studies found that curcumin induced apoptosis in many cancer cell lines including gastric, cervical, and non-small cell lung cancers by upregulating pro-apoptotic molecule Bax, downregulating anti-apoptotic Bcl-2 and Bcl-XL, and causing the release of AIF and cytochrome c leading to activation of caspase-9 and caspase-3 (Koo et al., 2004; Sen et al., 2005; Madden et al., 2009). It has also been reported that curcumin exhibited synergistic effects with many cytotoxic drugs including fluorouracil, vinorelbine, and gemcitabine (Koo et al., 2004; Sen et al., 2005; Kunnumakkara et al., 2007). Due to its ability to inhibit cancer cell growth and suppress carcinogenesis, curcumin is also considered as a chempreventive agent (Amin et al., 2009). Among the mechanisms proposed, inhibition of the NF-κB survival pathway has been considered as a molecular basis for the drug action (Divya and Pillai, 2006; Singh and Khar, 2006). The effect of curcumin on cellular ROS and redox states is rather complex, since this compound has been shown to have antioxidant and pro-oxidant functions (Thayyullathil et al., 2008; Manikandan et al., 2009; Ortiz-Ortiz et al., 2009; Pinlaor et al., 2009). Interestingly, although curcumin could induced apoptosis in human skin cancer cells, its toxicity was significantly diminished in their respiration deficient (ρ0) clones, suggesting that mitochondrial respiration and redox status may be important in apoptosis signaling induced by curcumin (Subudhi et al., 2008). As such, mitochondrial dysfunction and increased ROS generation in cancer cells may render them more sensitive to curcumin.
3.6. Pancratistatin
Pancratistatin (PST) is a natural compound isolated from the spider lily Pancratium littorale (Pettit et al., 1984; Luduena et al., 1992). It seems to preferentially target mitochondria in cancer cells and induce apoptosis in multiple cancer cell lines while exhibiting little harmful effect to their non-cancerous counterparts (McLachlan et al., 2005; Pandey et al., 2005; Siedlakowski et al., 2008). A study using human breast cancer cells found that PST caused an increase in ROS levels and a decrease in cellular ATP and mitochondrial transmembrane potential (Siedlakowski et al., 2008). Another study revealed that caspase-3 activation and exposure of phophatidyl serine on the outer leaflet of the plasma membrane occurred 1 h after human lymphoma cells were treated with PST, and this was much earlier than ROS generation and DNA fragmentation (Kekre et al., 2005). PST exhibited synergistic effect with tamoxifen on breast cancer cells (Ray, 2008; Siedlakowski et al., 2008). Its derivative is also highly effective in a human colon cancer model (Shnyder et al., 2008).
3.7. OSW-1
OSW-1 (3β,16β,17α-trihydroxycholest-5-en-22-one16-O-{O-(2-O-(4-methoxybenzoyl)-β-d-xylopyranosyl)-(1 → 3)-2-O-acetyl-α-arabinopyranoside) is a steroidal glycoside found in the bulbs of Ornithogalum saudersiae and has been chemically synthesized (Deng et al., 1999). It has extremely potent cytotoxic activity against a broad spectrum of malignant cell lines in vitro at the concentrations in the sub-nanomolar range, and exhibits less toxicity to normal cells (Guo et al., 1999; Ma et al., 2000, 2001; Zhou et al., 2005b). A study using Chinese hamster ovary cells showed that OSW-1 induced apoptosis involving a caspase-8-dependent cleavage of Bcl-2 (Zhu et al., 2005). Further study revealed that OSW-1 caused significant damage to mitochondrial membranes and cristae, likely due to calcium overload, and this led to a loss of mitochondrial membrane integrity and activation of calcium-dependent apoptosis in human leukemia and pancreatic cancer cell lines (Zhou et al., 2005b). The same study also showed that OSW-1 effectively killed primary leukemia cells from patients with chronic lymphocytic leukemia that were refractory to fludarabine. Interestingly, clones of leukemia cells lacking mitochondrial DNA and defective in respiration (ρ0 cells) were resistant to OSW-1 (Zhou et al., 2005b), suggesting that mitochondria play an important role in mediating the cytotoxic action of this compound. Subsequent studies using OSW-1 and its analogues further confirm their potent anticancer activity (Kasai et al., 2007; Peng et al., 2007; Tang et al., 2007; Wojtkielewicz et al., 2007; Xue et al., 2008), although the molecular target of OSW-1 has not been identified.
3.8. Resveratrol
Resveratrol is a natural compound found in the skin of red grapes and blueberries. It has been shown to prevent skin cancer development in mice treated with a carcinogen (Jang et al., 1997) and induce apoptosis in various cancer cells (Filomeni et al., 2007; Juan et al., 2008). The mechanisms of action responsible for the cancer preventive effect and anticancer activity of resveratrol still remain elusive. An early study showed that this compound inhibited mitochondrial respiratory chain function and lowered ROS generation by competing with coenzyme Q thus decreasing complex III activity (Zini et al., 1999). A recent study found that resveratrol induce apoptosis in colon cancer cells seems to require nitric oxide production and caspase activation, and that colon cancer cell lacking p53 were not sensitive to this compound (Kim et al., 2009). In addition, resveratrol seems to induce apoptosis by interfering with the signaling pathways involving PI3K/AKT, JAK/STAT and MAPK (Filomeni et al., 2007; Juan et al., 2008; Madan et al., 2008; Roy et al., 2009).
3.9. Vitamin K3
Vitamin K3 (2-methyl-1,4-naphthoquinone; menadione) is a synthetic compound, which can function as a precursor of various types of vitamin K in the body. The anticancer activity of vitamin K3 has been shown in various cancer cells in vitro and in vivo (Prasad et al., 1981; Chlebowski et al., 1985; Gold, 1986; Ngo et al., 1991; Nutter et al., 1991). This compound is able to perturb calcium homeostasis and caused a dose-dependent decrease of intracellular glutathione (GSH), mainly through inducing its oxidation to glutathione disulfide GSSG (Di Monte et al., 1984). In cancer cells, vitamin K3 specifically inhibited DNA polymerase (pol)γ, caused impairment of mitochondrial DNA replication, and promoted ROS generation leading to apoptosis (Sasaki et al., 2008). A recent study also found that vitamin K3 could bind at the colchicines binding site of tubulin and inhibit microtubule polymerization (Acharya et al., 2009). This compound has been evaluated in clinical trials in patients with advanced malignancies. A phase I clinical trial showed that intravenous infusion of vitamin K3 starting at 40 mg/m2 every 3 weeks with escalation to 1360 mg/m2 produced no objective partial or complete responses (Lim et al., 2005). High dose vitamin K3 daily infusion has been evaluated in patients with advanced liver cancer, and produced objective response in 17.4% patients with improved survival in the responsive patients but did not affect the overall survival (Sarin et al., 2006).
4. Summary and future perspectives
Owing to their essential roles in energy metabolism and regulation of redox homeostasis and apoptosis, mitochondria are considered as an attractive target for cancer therapy (Ralph and Neuzil, 2009) Recent studies have identified agents that target mitochondria by directly interacting with the key molecules in the mitochondria or indirectly impacting the metabolic alterations as the consequence of mitochondrial dysfunction in cancer cells. Such anticancer agents that directly or indirectly impacting mitochondria are collectively classified as “mitocans” (Neuzil et al., 2006, 2007b; Ralph et al., 2010). Among the mitocans identified, several natural compounds, including vitamin E analogs, PEITC, and hohokiol, have been evaluated extensively in recent studies, and exhibit promising anticancer activity in vitro and in vivo. One common feature of these compounds is their preferential killing of cancer cells with low cytotoxicity to normal cells. These compounds are important in that they provide a new possibility to improve cancer therapeutic outcomes, and that the investigation of the underlying mechanisms may gain new insights into cancer biology in term of mitochondrial alterations and their interactions with therapeutic agents.
One important consideration in targeting mitochondria for cancer treatment is the issue of therapeutic selectivity. Since many of the mitochondrial functions are essential for the viability of the normal cells, a compound that directly and completely inhibits one of these essential functions may be detrimental to the normal cells and would likely to have a significant toxic side effect. Thus, a good mitocan should be a agent that is able to preferentially target the mitochondrial abnormalities in cancer cells, not a compound that has potent and general inhibitory effect on mitochondria without discrimination between normal and cancer cells. For instance, certain metabolic changes in cancer cells might render the mitochondrial complex II more vulnerable to inhibition by the vitamin E analog α-tocopheryl succinate (α-TOS), which exhibits promising anticancer activity with low toxicity to normal cells. Similarly, the increased ROS generation from the dysfunctional mitochondria in cancer cells may make them highly dependent on the GSH antioxidant system to maintain redox balance, and thus more vulnerable to PEITC that abrogates the glutathione system. Therefore, it is extremely important to investigate the differences between normal and cancer cells in their mitochondrial structure and functions, the downstream metabolic alterations, and the underlying mechanisms, and to exploit these differences for therapeutic purpose. Such mechanistic studies and the identification and testing of new agents capable of preferentially targeting the aberrant mitochondria in cancer cells and/or their metabolic alterations are important tasks in future research.
Acknowledgement
Preparation of this work was supported in part by grants CA085563, CA100428, and CA109041 from the National Institutes of Health.
References
- Achanta G, Sasaki R, Feng L, Carew JS, Lu W, Pelicano H, Keating MJ, Huang P. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA Pol gamma. The EMBO J. 2005;24(19):3482–3492. doi: 10.1038/sj.emboj.7600819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya BR, Choudhury D, Das A, Chakrabarti G. Vitamin K3 disrupts the microtubule networks by binding to tubulin: a novel mechanism of its antiproliferative activity. Biochemistry. 2009;48(29):6963–6974. doi: 10.1021/bi900152k. [DOI] [PubMed] [Google Scholar]
- Adam-Vizi V, Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006;27(12):639–645. doi: 10.1016/j.tips.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84(6):1014–1020. doi: 10.1016/j.ygeno.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Amblard F, Govindarajan B, Lefkove B, Rapp KL, Detorio M, Arbiser JL, Schinazi RF. Synthesis, cytotoxicity, and antiviral activities of new neolignans related to honokiol and magnolol. Bioorg. Med. Chem. Lett. 2007;17(16):4428–4431. doi: 10.1016/j.bmcl.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin AR, Kucuk O, Khuri FR, Shin DM. Perspectives for cancer prevention with natural compounds. J. Clin. Oncol. 2009;27(16):2712–2725. doi: 10.1200/JCO.2008.20.6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliga MS, Meleth S, Katiyar SK. Growth inhibitory and antimetastatic effect of green tea polyphenols on metastasis-specific mouse mammary carcinoma 4T1 cells in vitro and in vivo systems. Clin. Cancer Res. 2005;11(5):1918–1927. doi: 10.1158/1078-0432.CCR-04-1976. [DOI] [PubMed] [Google Scholar]
- Behrend L, Henderson G, Zwacka RM. Reactive oxygen species in oncogenic transformation. Biochem. Soc. Trans. 2003;31(Pt 6):1441–1444. doi: 10.1042/bst0311441. [DOI] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Berridge MV, Herst PM, Lawen A. Targeting mitochondrial permeability in cancer drug development. Mol. Nutr. Food Res. 2009;53(1):76–86. doi: 10.1002/mnfr.200700493. [DOI] [PubMed] [Google Scholar]
- Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25(34):4647–4662. doi: 10.1038/sj.onc.1209607. [DOI] [PubMed] [Google Scholar]
- Brown KK, Eriksson SE, Arner ES, Hampton MB. Mitochondrial peroxiredoxin 3 is rapidly oxidized in cells treated with isothiocyanates. Free Radic. Biol. Med. 2008;45(4):494–502. doi: 10.1016/j.freeradbiomed.2008.04.030. [DOI] [PubMed] [Google Scholar]
- Carew JS, Zhou Y, Albitar M, Carew JD, Keating MJ, Huang P. Mitochondrial DNA mutations in primary leukemia cells after chemotherapy: clinical significance and therapeutic implications. Leukemia. 2003;17(8):1437–1447. doi: 10.1038/sj.leu.2403043. [DOI] [PubMed] [Google Scholar]
- Cavalieri E, Bergamini C, Mariotto S, Leoni S, Perbellini L, Darra E, Suzuki H, Fato R, Lenaz G. Involvement of mitochondrial permeability transition pore opening in alpha-bisabolol induced apoptosis. FEBS J. 2009;276(15):3990–4000. doi: 10.1111/j.1742-4658.2009.07108.x. [DOI] [PubMed] [Google Scholar]
- Chen C, Shen G, Hebbar V, Hu R, Owuor ED, Kong AN. Epigallocatechin-3- gallate-induced stress signals in HT-29 human colon adenocarcinoma cells. Carcinogenesis. 2003;24(8):1369–1378. doi: 10.1093/carcin/bgg091. [DOI] [PubMed] [Google Scholar]
- Chen F, Wang T, Wu YF, Gu Y, Xu XL, Zheng S, Hu X. Honokiol: a potent chemotherapy candidate for human colorectal carcinoma. World J. Gastroenterol. 2004;10(23):3459–3463. doi: 10.3748/wjg.v10.i23.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Izzo J, Demizu Y, Wang F, Guha S, Wu X, Hung MC, Ajani JA, Huang P. Different redox states in malignant and nonmalignant esophageal epithelial cells and differential cytotoxic responses to bile acid and honokiol. Antioxid. Redox Signal. 2009a;11(5):1083–1095. doi: 10.1089/ars.2008.2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Wu CL, Liu JF, Fong YC, Hsu SF, Li TM, Su YC, Liu SH, Tang CH. Honokiol induces cell apoptosis in human chondrosarcoma cells through mitochondrial dysfunction and endoplasmic reticulum stress. Cancer Lett. 2009b doi: 10.1016/j.canlet.2009.08.032. [DOI] [PubMed] [Google Scholar]
- Cheung KL, Khor TO, Huang MT, Kong AN. Differential in vivo mechanism of chemoprevention of tumor formation in azoxymethane/dextran sodium sulfate mice by PEITC and DBM. Carcinogenesis. 2010;31(5):880–885. doi: 10.1093/carcin/bgp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlebowski RT, Dietrich M, Akman S, Block JB. Vitamin K3 inhibition of malignant murine cell growth and human tumor colony formation. Cancer Treat. Rep. 1985;69(5):527–532. [PubMed] [Google Scholar]
- Clerkin JS, Naughton R, Quiney C, Cotter TG. Mechanisms of ROS modulated cell survival during carcinogenesis. Cancer Lett. 2008;266(1):30–36. doi: 10.1016/j.canlet.2008.02.029. [DOI] [PubMed] [Google Scholar]
- Corsi L, Geminiani E, Baraldi M. Peripheral benzodiazepine receptor (PBR) new insight in cell proliferation and cell differentiation review. Curr. Clin. Pharmacol. 2008;3(1):38–45. doi: 10.2174/157488408783329878. [DOI] [PubMed] [Google Scholar]
- Crane C, Panner A, Pieper RO, Arbiser J, Parsa AT. Honokiol-mediated inhibition of PI3K/mTOR pathway: a potential strategy to overcome immunoresistance in glioma, breast, and prostate carcinoma without impacting T cell function. J. Immunother. 2009;32(6):585–592. doi: 10.1097/CJI.0b013e3181a8efe6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Deng S, Yu B, Lou Y, Hui Y. First Total Synthesis of an Exceptionally Potent Antitumor Saponin, OSW-1. J. Org. Chem. 1999;64(1):202–208. doi: 10.1021/jo981685c. [DOI] [PubMed] [Google Scholar]
- Deng J, Qian Y, Geng L, Chen J, Wang X, Xie H, Yan S, Jiang G, Zhou L, Zheng S. Involvement of p38 mitogen-activated protein kinase pathway in honokiol-induced apoptosis in a human hepatoma cell line (hepG2) Liver Int. 2008;28(10):1458–1464. doi: 10.1111/j.1478-3231.2008.01767.x. [DOI] [PubMed] [Google Scholar]
- Di Monte D, Ross D, Bellomo G, Eklow L, Orrenius S. Alterations in intracellular thiol homeostasis during the metabolism of menadione by isolated rat hepatocytes. Arch. Biochem. Biophys. 1984;235(2):334–342. doi: 10.1016/0003-9861(84)90206-6. [DOI] [PubMed] [Google Scholar]
- Divya CS, Pillai MR. Antitumor action of curcumin in human papillomavirus associated cells involves downregulation of viral oncogenes, prevention of NFkB and AP-1 translocation, and modulation of apoptosis. Mol. Carcinog. 2006;45(5):320–332. doi: 10.1002/mc.20170. [DOI] [PubMed] [Google Scholar]
- Dong LF, Low P, Dyason JC, Wang XF, Prochazka L, Witting PK, Freeman R, Swettenham E, Valis K, Liu J, Zobalova R, Turanek J, Spitz DR, Domann FE, Scheffler IE, Ralph SJ, Neuzil J. Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene. 2008;27(31):4324–4335. doi: 10.1038/onc.2008.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong LF, Freeman R, Liu J, Zobalova R, Marin-Hernandez A, Stantic M, Rohlena J, Valis K, Rodriguez-Enriquez S, Butcher B, Goodwin J, Brunk UT, Witting PK, Moreno-Sanchez R, Scheffler IE, Ralph SJ, Neuzil J. Suppression of tumor growth in vivo by the mitocan alpha-tocopheryl succinate requires respiratory complex II. Clin. Cancer Res. 2009;15(5):1593–1600. doi: 10.1158/1078-0432.CCR-08-2439. [DOI] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64(11):3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- Estrela JM, Ortega A, Obrador E. Glutathione in cancer biology and therapy. Crit. Rev. Clin. Lab. Sci. 2006;43(2):143–181. doi: 10.1080/10408360500523878. [DOI] [PubMed] [Google Scholar]
- Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9(6):425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- Filomeni G, Graziani I, Rotilio G, Ciriolo MR. trans-Resveratrol induces apoptosis in human breast cancer cells MCF-7 by the activation of MAP kinases pathways. Genes Nutr. 2007;2(3):295–305. doi: 10.1007/s12263-007-0059-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flier JS, Mueckler MM, Usher P, Lodish HF. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science. 1987;235(4795):1492–1495. doi: 10.1126/science.3103217. [DOI] [PubMed] [Google Scholar]
- Fried LE, Arbiser JL. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid. Redox Signal. 2009;11(5):1139–1148. doi: 10.1089/ars.2009.2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruehauf JP, Meyskens FL., Jr. Reactive oxygen species: a breath of life or death? Clin. Cancer Res. 2007;13(3):789–794. doi: 10.1158/1078-0432.CCR-06-2082. [DOI] [PubMed] [Google Scholar]
- Gambhir SS. Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer. 2002;2(9):683–693. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- Garcia A, Zheng Y, Zhao C, Toschi A, Fan J, Shraibman N, Brown HA, Bar-Sagi D, Foster DA, Arbiser JL. Honokiol suppresses survival signals mediated by Ras-dependent phospholipase D activity in human cancer cells. Clin. Cancer Res. 2008;14(13):4267–4274. doi: 10.1158/1078-0432.CCR-08-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer. 2004;4(11):891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- Gold J. In vivo synergy of vitamin K3 and methotrexate in tumor-bearing animals. Cancer Treat. Rep. 1986;70(12):1433–1435. [PubMed] [Google Scholar]
- Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat. Rev. Cancer. 2005;5(11):857–866. doi: 10.1038/nrc1737. [DOI] [PubMed] [Google Scholar]
- Guo C, LaCour TG, Fuchs PL. On the relationship of OSW-1 to the cephalostatins. Bioorg. Med. Chem. Lett. 1999;9(3):419–424. doi: 10.1016/s0960-894x(98)00743-4. [DOI] [PubMed] [Google Scholar]
- Hahm ER, Singh SV. Honokiol causes G0-G1 phase cell cycle arrest in human prostate cancer cells in association with suppression of retinoblastoma protein level/phosphorylation and inhibition of E2F1 transcriptional activity. Mol. Cancer Ther. 2007;6(10):2686–2695. doi: 10.1158/1535-7163.MCT-07-0217. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009;46(6):821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hecht SS. Chemoprevention of cancer by isothiocyanates, modifiers of carcinogen metabolism. J. Nutr. 1999;129(3):768S–774S. doi: 10.1093/jn/129.3.768S. [DOI] [PubMed] [Google Scholar]
- Heiser D, Labi V, Erlacher M, Villunger A. The Bcl-2 protein family and its role in the development of neoplastic disease. Exp. Gerontol. 2004;39(8):1125–1135. doi: 10.1016/j.exger.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, Cleveland JL, Tansey WP, Lowe SW. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436(7052):807–811. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst A, Hemann MT, Tworkowski KA, Salghetti SE, Lowe SW, Tansey WP. A conserved element in Myc that negatively regulates its proapoptotic activity. EMBO Rep. 2005;6(2):177–183. doi: 10.1038/sj.embor.7400333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hour TC, Huang CY, Lin CC, Chen J, Guan JY, Lee JM, Pu YS. Characterization of molecular events in a series of bladder urothelial carcinoma cell lines with progressive resistance to arsenic trioxide. Anticancer Drugs. 2004;15(8):779–785. doi: 10.1097/00001813-200409000-00007. [DOI] [PubMed] [Google Scholar]
- Hsu S, Lewis J, Singh B, Schoenlein P, Osaki T, Athar M, Porter AG, Schuster G. Green tea polyphenol targets the mitochondria in tumor cells inducing caspase 3-dependent apoptosis. Anticancer Res. 2003;23(2B):1533–1539. [PubMed] [Google Scholar]
- Hu R, Kim BR, Chen C, Hebbar V, Kong AN. The roles of JNK and apoptotic signaling pathways in PEITC-mediated responses in human HT-29 colon adeno-carcinoma cells. Carcinogenesis. 2003;24(8):1361–1367. doi: 10.1093/carcin/bgg092. [DOI] [PubMed] [Google Scholar]
- Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature. 2000;407(6802):390–395. doi: 10.1038/35030140. [DOI] [PubMed] [Google Scholar]
- Hurd TR, Costa NJ, Dahm CC, Beer SM, Brown SE, Filipovska A, Murphy MP. Glutathionylation of mitochondrial proteins. Antioxid. Redox Signal. 2005;7(7-8):999–1010. doi: 10.1089/ars.2005.7.999. [DOI] [PubMed] [Google Scholar]
- Imai K, Suga K, Nakachi K. Cancer-preventive effects of drinking green tea among a Japanese population. Prev. Med. 1997;26(6):769–775. doi: 10.1006/pmed.1997.0242. [DOI] [PubMed] [Google Scholar]
- Indo HP, Davidson M, Yen HC, Suenaga S, Tomita K, Nishii T, Higuchi M, Koga Y, Ozawa T, Majima HJ. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion. 2007;7(1-2):106–118. doi: 10.1016/j.mito.2006.11.026. [DOI] [PubMed] [Google Scholar]
- Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, Goldschmidt-Clermont PJ. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science (New York, NY) 1997;275(5306):1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- Irmak MB, Ince G, Ozturk M, Cetin-Atalay R. Acquired tolerance of hepatocellular carcinoma cells to selenium deficiency: a selective survival mechanism? Cancer Res. 2003;63(20):6707–6715. [PubMed] [Google Scholar]
- Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science (New York, NY) 2008;320(5876):661–664. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- Ishitsuka K, Hideshima T, Hamasaki M, Raje N, Kumar S, Hideshima H, Shiraishi N, Yasui H, Roccaro AM, Richardson P, Podar K, Le Gouill S, Chauhan D, Tamura K, Arbiser J, Anderson KC. Honokiol overcomes conventional drug resistance in human multiple myeloma by induction of caspase-dependent and - independent apoptosis. Blood. 2005;106(5):1794–1800. doi: 10.1182/blood-2005-01-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Tong KI, Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med. 2004;36(10):1208–1213. doi: 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275(5297):218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- Jelluma N, Yang X, Stokoe D, Evan GI, Dansen TB, Haas-Kogan DA. Glucose withdrawal induces oxidative stress followed by apoptosis in glioblastoma cells but not in normal human astrocytes. Mol. Cancer Res. 2006;4(5):319–330. doi: 10.1158/1541-7786.MCR-05-0061. [DOI] [PubMed] [Google Scholar]
- Ji Y, Morris ME. Determination of phenethyl isothiocyanate in human plasma and urine by ammonia derivatization and liquid chromatography-tandem mass spectrometry. Anal. Biochem. 2003;323(1):39–47. doi: 10.1016/j.ab.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Jian L, Xie LP, Lee AH, Binns CW. Protective effect of green tea against prostate cancer: a case-control study in southeast China. Int. J. Cancer. 2004;108(1):130–135. doi: 10.1002/ijc.11550. [DOI] [PubMed] [Google Scholar]
- Jourdain A, Martinou JC. Mitochondrial outer-membrane permeabilization and remodelling in apoptosis. Int. J. Biochem. Cell Biol. 2009;41(10):1884–1889. doi: 10.1016/j.biocel.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Juan ME, Wenzel U, Daniel H, Planas JM. Resveratrol induces apoptosis through ROS-dependent mitochondria pathway in HT-29 human colorectal carcinoma cells. J. Agric. Food Chem. 2008;56(12):4813–4818. doi: 10.1021/jf800175a. [DOI] [PubMed] [Google Scholar]
- Kasai HF, Tsubuki M, Matsuo S, Honda T. Analysis of antitumor active OSW-1 and its analogues by liquid chromatography coupled with electrospray and atmospheric pressure chemical ionization quadrupole mass spectrometry. Rapid Commun. Mass Spectrom. 2007;21(7):1100–1114. doi: 10.1002/rcm.2939. [DOI] [PubMed] [Google Scholar]
- Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat. Cell Biol. 2008;10(5):611–618. doi: 10.1038/ncb1724. [DOI] [PubMed] [Google Scholar]
- Kekre N, Griffin C, McNulty J, Pandey S. Pancratistatin causes early activation of caspase-3 and the flipping of phosphatidyl serine followed by rapid apoptosis specifically in human lymphoma cells. Cancer Chemother. Pharmacol. 2005;56(1):29–38. doi: 10.1007/s00280-004-0941-8. [DOI] [PubMed] [Google Scholar]
- Kim GJ, Chandrasekaran K, Morgan WF. Mitochondrial dysfunction, persistently elevated levels of reactive oxygen species and radiation-induced genomic instability: a review. Mutagenesis. 2006a;21(6):361–367. doi: 10.1093/mutage/gel048. [DOI] [PubMed] [Google Scholar]
- Kim R, Emi M, Tanabe K, Murakami S, Uchida Y, Arihiro K. Regulation and interplay of apoptotic and non-apoptotic cell death. J. Pathol. 2006b;208(3):319–326. doi: 10.1002/path.1885. [DOI] [PubMed] [Google Scholar]
- Kim MY, Trudel LJ, Wogan GN. Apoptosis induced by capsaicin and resveratrol in colon carcinoma cells requires nitric oxide production and caspase activation. Anticancer Res. 2009;29(10):3733–3740. [PubMed] [Google Scholar]
- Knudson CM, Brown NM. Mitochondria potential, bax “activation,” and programmed cell death. Methods Mol. Biol. 2008;414:95–108. doi: 10.1007/978-1-59745-339-4_9. [DOI] [PubMed] [Google Scholar]
- Ko YH, Pedersen PL, Geschwind JF. Glucose catabolism in the rabbit VX2 tumor model for liver cancer: characterization and targeting hexokinase. Cancer Lett. 2001;173(1):83–91. doi: 10.1016/s0304-3835(01)00667-x. [DOI] [PubMed] [Google Scholar]
- Komatsu D, Kato M, Nakayama J, Miyagawa S, Kamata T. NADPH oxidase 1 plays a critical mediating role in oncogenic Ras-induced vascular endothelial growth factor expression. Oncogene. 2008;27(34):4724–4732. doi: 10.1038/onc.2008.102. [DOI] [PubMed] [Google Scholar]
- Koo JY, Kim HJ, Jung KO, Park KY. Curcumin inhibits the growth of AGS human gastric carcinoma cells in vitro and shows synergism with 5-fluorouracil. J. Med. Food. 2004;7(2):117–121. doi: 10.1089/1096620041224229. [DOI] [PubMed] [Google Scholar]
- Koudelka S, Masek J, Neuzil J, Turanek J. Lyophilised liposome-based formulations of alpha-tocopheryl succinate: preparation and physico-chemical characterisation. J. Pharm. Sci. 2010;99(5):2434–2443. doi: 10.1002/jps.22002. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007;87(1):99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008;68(6):1777–1785. doi: 10.1158/0008-5472.CAN-07-5259. [DOI] [PubMed] [Google Scholar]
- Kunnumakkara AB, Guha S, Krishnan S, Diagaradjane P, Gelovani J, Aggarwal BB. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-kappaB-regulated gene products. Cancer Res. 2007;67(8):3853–3861. doi: 10.1158/0008-5472.CAN-06-4257. [DOI] [PubMed] [Google Scholar]
- Kuttan R, Bhanumathy P, Nirmala K, George MC. Potential anticancer activity of turmeric (Curcuma longa) Cancer Lett. 1985;29(2):197–202. doi: 10.1016/0304-3835(85)90159-4. [DOI] [PubMed] [Google Scholar]
- Kuttan R, Sudheeran PC, Josph CD. Turmeric and curcumin as topical agents in cancer therapy. Tumori. 1987;73(1):29–31. doi: 10.1177/030089168707300105. [DOI] [PubMed] [Google Scholar]
- Lenehan PF, Gutierrez PL, Wagner JL, Milak N, Fisher GR, Ross DD. Resistance to oxidants associated with elevated catalase activity in HL-60 leukemia cells that overexpress multidrug-resistance protein does not contribute to the resistance to daunorubicin manifested by these cells. Cancer Chemother. Pharmacol. 1995;35(5):377–386. doi: 10.1007/s002800050250. [DOI] [PubMed] [Google Scholar]
- Li L, Han W, Gu Y, Qiu S, Lu Q, Jin J, Luo J, Hu X. Honokiol induces a necrotic cell death through the mitochondrial permeability transition pore. Cancer Res. 2007;67(10):4894–4903. doi: 10.1158/0008-5472.CAN-06-3818. [DOI] [PubMed] [Google Scholar]
- Liebes L, Conaway CC, Hochster H, Mendoza S, Hecht SS, Crowell J, Chung FL. High-performance liquid chromatography-based determination of total isothiocyanate levels in human plasma: application to studies with 2-phenethyl isothiocyanate. Anal. Biochem. 2001;291(2):279–289. doi: 10.1006/abio.2001.5030. [DOI] [PubMed] [Google Scholar]
- Lim D, Morgan RJ, Jr., Akman S, Margolin K, Carr BI, Leong L, Odujinrin O, Doroshow JH. Phase I trial of menadiol diphosphate (vitamin K3) in advanced malignancy. Invest. New Drugs. 2005;23(3):235–239. doi: 10.1007/s10637-005-6731-2. [DOI] [PubMed] [Google Scholar]
- Liu H, Zang C, Emde A, Planas-Silva MD, Rosche M, Kuhnl A, Schulz CO, Elstner E, Possinger K, Eucker J. Anti-tumor effect of honokiol alone and in combination with other anti-cancer agents in breast cancer. Eur. J. Pharmacol. 2008;591(1–3):43–51. doi: 10.1016/j.ejphar.2008.06.026. [DOI] [PubMed] [Google Scholar]
- Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002;277(26):23111–23115. doi: 10.1074/jbc.M202487200. [DOI] [PubMed] [Google Scholar]
- Luduena RF, Roach MC, Prasad V, Pettit GR. Interaction of dolastatin 10 with bovine brain tubulin. Biochem. Pharmacol. 1992;43(3):539–543. doi: 10.1016/0006-2952(92)90576-5. [DOI] [PubMed] [Google Scholar]
- Luo H, Zhong Q, Chen LJ, Qi XR, Fu AF, Yang HS, Yang F, Lin HG, Wei YQ, Zhao X. Liposomal honokiol, a promising agent for treatment of cisplatin-resistant human ovarian cancer. J. Cancer Res. Clin. Oncol. 2008;134(9):937–945. doi: 10.1007/s00432-008-0375-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Yu B, Hui Y, Xiao D, Ding J. Synthesis of glycosides bearing the disaccharide of OSW-1 or its 1->4-linked analogue and their antitumor activities. Carbohydr. Res. 2000;329(3):495–505. doi: 10.1016/s0008-6215(00)00225-1. [DOI] [PubMed] [Google Scholar]
- Ma X, Yu B, Hui Y, Miao Z, Ding J. Synthesis of OSW-1 analogues and a dimer and their antitumor activities. Bioorg. Med. Chem. Lett. 2001;11(16):2153–2156. doi: 10.1016/s0960-894x(01)00389-4. [DOI] [PubMed] [Google Scholar]
- Madan E, Prasad S, Roy P, George J, Shukla Y. Regulation of apoptosis by resveratrol through JAK/STAT and mitochondria mediated pathway in human epidermoid carcinomaA431cells. Biochem. Biophys. Res. Commun. 2008;377(4):1232–1237. doi: 10.1016/j.bbrc.2008.10.158. [DOI] [PubMed] [Google Scholar]
- Madden K, Flowers L, Salani R, Horowitz I, Logan S, Kowalski K, Xie J, Mohammed SI. Proteomics-based approach to elucidate the mechanism of antitumor effect of curcumin in cervical cancer. Prostaglandins Leukot. Essent. Fatty Acids. 2009;80(1):9–18. doi: 10.1016/j.plefa.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Manikandan R, Thiagarajan R, Beulaja S, Sudhandiran G, Arumugam M. Curcumin prevents free radical-mediated cataractogenesis through modulations in lens calcium. Free Radic. Biol. Med. 2009 doi: 10.1016/j.freeradbiomed.2009.11.011. [DOI] [PubMed] [Google Scholar]
- Mathupala SP, Heese C, Pedersen PL. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J. Biol. Chem. 1997;272(36):22776–22780. doi: 10.1074/jbc.272.36.22776. [DOI] [PubMed] [Google Scholar]
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312(5780):1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- McLachlan A, Kekre N, McNulty J, Pandey S. Pancratistatin: a natural anti-cancer compound that targets mitochondria specifically in cancer cells to induce apoptosis. Apoptosis. 2005;10(3):619–630. doi: 10.1007/s10495-005-1896-x. [DOI] [PubMed] [Google Scholar]
- Meffert G, Gellerich FN, Margreiter R, Wyss M. Elevated creatine kinase activity in primary hepatocellular carcinoma. BMC Gastroenterol. 2005;5:9. doi: 10.1186/1471-230X-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacol. Ther. 1991;51(2):155–194. doi: 10.1016/0163-7258(91)90076-x. [DOI] [PubMed] [Google Scholar]
- Mestre-Escorihuela C, Rubio-Moscardo F, Richter JA, Siebert R, Climent J, Fresquet V, Beltran E, Agirre X, Marugan I, Marin M, Rosenwald A, Sugimoto KJ, Wheat LM, Karran EL, Garcia JF, Sanchez L, Prosper F, Staudt LM, Pinkel D, Dyer MJ, Martinez-Climent JA. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood. 2007;109(1):271–280. doi: 10.1182/blood-2006-06-026500. [DOI] [PubMed] [Google Scholar]
- Mi L, Wang X, Govind S, Hood BL, Veenstra TD, Conrads TP, Saha DT, Goldman R, Chung FL. The role of protein binding in induction of apoptosis by phenethyl isothiocyanate and sulforaphane in human non-small lung cancer cells. Cancer Res. 2007;67(13):6409–6416. doi: 10.1158/0008-5472.CAN-07-0340. [DOI] [PubMed] [Google Scholar]
- Moreno-Sanchez R, Saavedra E, Rodriguez-Enriquez S, Gallardo-Perez JC, Quezada H, Westerhoff HV. Metabolic control analysis indicates a change of strategy in the treatment of cancer. Mitochondrion. 2010 doi: 10.1016/j.mito.2010.06.002. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Munroe ME, Arbiser JL, Bishop GA. Honokiol, a natural plant product, inhibits inflammatory signals and alleviates inflammatory arthritis. J. Immunol. 2007;179(2):753–763. doi: 10.4049/jimmunol.179.2.753. [DOI] [PubMed] [Google Scholar]
- Nakachi K, Suemasu K, Suga K, Takeo T, Imai K, Higashi Y. Influence of drinking green tea on breast cancer malignancy among Japanese patients. Jpn J. Cancer Res. 1998;89(3):254–261. doi: 10.1111/j.1349-7006.1998.tb00556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazato T, Ito K, Ikeda Y, Kizaki M. Green tea component, catechin, induces apoptosis of human malignant B cells via production of reactive oxygen species. Clin. Cancer Res. 2005;11(16):6040–6049. doi: 10.1158/1078-0432.CCR-04-2273. [DOI] [PubMed] [Google Scholar]
- Neuzil J, Wang XF, Dong LF, Low P, Ralph SJ. Molecular mechanism of ‘mitocan’-induced apoptosis in cancer cells epitomizes the multiple roles of reactive oxygen species and Bcl-2 family proteins. FEBS Lett. 2006;580(22):5125–5129. doi: 10.1016/j.febslet.2006.05.072. [DOI] [PubMed] [Google Scholar]
- Neuzil J, Dong LF, Ramanathapuram L, Hahn T, Chladova M, Wang XF, Zobalova R, Prochazka L, Gold M, Freeman R, Turanek J, Akporiaye ET, Dyason JC, Ralph SJ. Vitamin E analogues as a novel group of mitocans: anti-cancer agents that act by targeting mitochondria. Mol. Aspects Med. 2007a;28(5-6):607–645. doi: 10.1016/j.mam.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Neuzil J, Dyason JC, Freeman R, Dong LF, Prochazka L, Wang XF, Scheffler I, Ralph SJ. Mitocans as anti-cancer agents targeting mitochondria: lessons from studies with vitamin E analogues, inhibitors of complex II. J. Bioenerg. Biomembr. 2007b;39(1):65–72. doi: 10.1007/s10863-006-9060-z. [DOI] [PubMed] [Google Scholar]
- Ngo EO, Sun TP, Chang JY, Wang CC, Chi KH, Cheng AL, Nutter LM. Menadione-induced DNA damage in a human tumor cell line. Biochem. Pharmacol. 1991;42(10):1961–1968. doi: 10.1016/0006-2952(91)90596-w. [DOI] [PubMed] [Google Scholar]
- Nihal M, Ahmad N, Mukhtar H, Wood GS. Anti-proliferative and proapoptotic effects of (-)-epigallocatechin-3-gallate on human melanoma: possible implications for the chemoprevention of melanoma. Int. J. Cancer. 2005;114(4):513–521. doi: 10.1002/ijc.20785. [DOI] [PubMed] [Google Scholar]
- Nishikawa M. Reactive oxygen species in tumor metastasis. Cancer Lett. 2008;266(1):53–59. doi: 10.1016/j.canlet.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Nutter LM, Cheng AL, Hung HL, Hsieh RK, Ngo EO, Liu TW. Menadione: spectrum of anticancer activity and effects on nucleotide metabolism in human neoplastic cell lines. Biochem. Pharmacol. 1991;41(9):1283–1292. doi: 10.1016/0006-2952(91)90099-q. [DOI] [PubMed] [Google Scholar]
- Ogasawara MA, Zhang H. Redox Regulation and its Emerging Roles in Stem Cells and Stem-Like Cancer Cells. Antioxid. Redox Signal. 2008 doi: 10.1089/ars.2008.2308. [DOI] [PubMed] [Google Scholar]
- Ortiz-Ortiz MA, Moran JM, Bravosanpedro JM, Gonzalez-Polo RA, Niso-Santano M, Anantharam V, Kanthasamy AG, Soler G, Fuentes JM. Curcumin enhances paraquat-induced apoptosis of N27 mesencephalic cells via the generation of reactive oxygen species. Neurotoxicology. 2009;30(6):1008–1018. doi: 10.1016/j.neuro.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000;275(29):21797–21800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- Palmieri D, Fitzgerald D, Shreeve SM, Hua E, Bronder JL, Weil RJ, Davis S, Stark AM, Merino MJ, Kurek R, Mehdorn HM, Davis G, Steinberg SM, Meltzer PS, Aldape K, Steeg PS. Analyses of resected human brain metastases of breast cancer reveal the association between up-regulation of hexokinase 2 and poor prognosis. Mol. Cancer Res. 2009;7(9):1438–1445. doi: 10.1158/1541-7786.MCR-09-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S, Kekre N, Naderi J, McNulty J. Induction of apoptotic cell death specifically in rat and human cancer cells by pancratistatin. Artif. Cells Blood Substit. Immobil. Biotechnol. 2005;33(3):279–295. doi: 10.1081/bio-200066621. [DOI] [PubMed] [Google Scholar]
- Pani G, Koch OR, Galeotti T. The p53-p66shc-Manganese Superoxide Dismutase (MnSOD) network: a mitochondrial intrigue to generate reactive oxygen species. Int. J. Biochem. Cell Biol. 2009;41(5):1002–1005. doi: 10.1016/j.biocel.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Patel BP, Rawal UM, Dave TK, Rawal RM, Shukla SN, Shah PM, Patel PS. Lipid peroxidation, total antioxidant status, and total thiol levels predict overall survival in patients with oral squamous cell carcinoma. Integr. Cancer Ther. 2007;6(4):365–372. doi: 10.1177/1534735407309760. [DOI] [PubMed] [Google Scholar]
- Pathania D, Millard M, Neamati N. Opportunities in discovery and delivery of anticancer drugs targeting mitochondria and cancer cell metabolism. Adv. Drug Deliv. Rev. 2009;61(14):1250–1275. doi: 10.1016/j.addr.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Pedersen PL. Voltage dependent anion channels (VDACs): a brief introduction with a focus on the outer mitochondrial compartment’s roles together with hexokinase-2 in the “Warburg effect” in cancer. J. Bioenerg. Biomembr. 2008;40(3):123–126. doi: 10.1007/s10863-008-9165-7. [DOI] [PubMed] [Google Scholar]
- Pelicano H, Feng L, Zhou Y, Carew JS, Hileman EO, Plunkett W, Keating MJ, Huang P. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J. Biol. Chem. 2003;278(39):37832–37839. doi: 10.1074/jbc.M301546200. [DOI] [PubMed] [Google Scholar]
- Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25(34):4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- Peng W, Tang P, Hu X, Liu JO, Yu B. Synthesis of the A, B-ring-truncated OSW saponin analogs and their antitumor activities. Bioorg. Med. Chem. Lett. 2007;17(20):5506–5509. doi: 10.1016/j.bmcl.2007.08.060. [DOI] [PubMed] [Google Scholar]
- Pervaiz S, Clement MV. Tumor intracellular redox status and drug resistance- serendipity or a causal relationship? Curr. Pharm. Des. 2004;10(16):1969–1977. doi: 10.2174/1381612043384411. [DOI] [PubMed] [Google Scholar]
- Pettit GR, Gaddamidi V, Cragg GM. Antineoplastic agents, 105. Zephyranthes grandiflora. J. Nat. Prod. 1984;47(6):1018–1020. doi: 10.1021/np50036a020. [DOI] [PubMed] [Google Scholar]
- Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292(5516):504–507. doi: 10.1126/science.1058079. [DOI] [PubMed] [Google Scholar]
- Pilorget A, Berthet V, Luis J, Moghrabi A, Annabi B, Beliveau R. Medulloblastoma cell invasion is inhibited by green tea (-)epigallocatechin-3-gallate. J. Cell. Biochem. 2003;90(4):745–755. doi: 10.1002/jcb.10667. [DOI] [PubMed] [Google Scholar]
- Pinlaor S, Yongvanit P, Prakobwong S, Kaewsamut B, Khoontawad J, Pinlaor P, Hiraku Y. Curcumin reduces oxidative and nitrative DNA damage through balancing of oxidant-antioxidant status in hamsters infected with Opisthorchis viverrini. Mol. Nutr. Food Res. 2009;53(10):1316–1328. doi: 10.1002/mnfr.200800567. [DOI] [PubMed] [Google Scholar]
- Prasad KN, Edwards-Prasad J, Sakamoto A. Vitamin K3 (menadione) inhibits the growth of mammalian tumor cells in culture. Life Sci. 1981;29(13):1387–1392. doi: 10.1016/0024-3205(81)90683-4. [DOI] [PubMed] [Google Scholar]
- Qanungo S, Das M, Haldar S, Basu A. Epigallocatechin-3-gallate induces mitochondrial membrane depolarization and caspase-dependent apoptosis in pancreatic cancer cells. Carcinogenesis. 2005;26(5):958–967. doi: 10.1093/carcin/bgi040. [DOI] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z, Bissell MJ. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436(7047):123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph SJ, Neuzil J. Mitochondria as targets for cancer therapy. Mol. Nutr. Food Res. 2009;53(1):9–28. doi: 10.1002/mnfr.200800044. [DOI] [PubMed] [Google Scholar]
- Ralph SJ, Rodriguez-Enriquez S, Neuzil J, Moreno-Sanchez R. Bioenergetic pathways in tumor mitochondria as targets for cancer therapy and the importance of the ROS-induced apoptotic trigger. Mol. Aspects Med. 2009 doi: 10.1016/j.mam.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Ray S. Synergy of Pancratistatin and Tamoxifen in inducing apoptosis in breast cancer cells. Cancer Biol. Ther. 2008;7(3):385–386. doi: 10.4161/cbt.7.3.5797. [DOI] [PubMed] [Google Scholar]
- Reed JC. Molecular biology of chronic lymphocytic leukemia. Semin. Oncol. 1998;25(1):11–18. [PubMed] [Google Scholar]
- Robey IF, Lien AD, Welsh SJ, Baggett BK, Gillies RJ. Hypoxia-inducible factor-1alpha and the glycolytic phenotype in tumors. Neoplasia. 2005;7(4):324–330. doi: 10.1593/neo.04430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose P, Whiteman M, Huang SH, Halliwell B, Ong CN. beta-Phenylethyl isothiocyanate-mediated apoptosis in hepatoma HepG2 cells. Cell. Mol. Life Sci. 2003;60(7):1489–1503. doi: 10.1007/s00018-003-3150-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy P, Kalra N, Prasad S, George J, Shukla Y. Chemopreventive potential of resveratrol in mouse skin tumors through regulation of mitochondrial and PI3K/AKT signaling pathways. Pharm. Res. 2009;26(1):211–217. doi: 10.1007/s11095-008-9723-z. [DOI] [PubMed] [Google Scholar]
- Sarin SK, Kumar M, Garg S, Hissar S, Pandey C, Sharma BC. High dose vitamin K3 infusion in advanced hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2006;21(9):1478–1482. doi: 10.1111/j.1440-1746.2006.04383.x. [DOI] [PubMed] [Google Scholar]
- Sasaki R, Suzuki Y, Yonezawa Y, Ota Y, Okamoto Y, Demizu Y, Huang P, Yoshida H, Sugimura K, Mizushina Y. DNA polymerase gamma inhibition by vitamin K3 induces mitochondria-mediated cytotoxicity in human cancer cells. Cancer Sci. 2008;99(5):1040–1048. doi: 10.1111/j.1349-7006.2008.00771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyan KS, Swamy N, Dizon DS, Singh R, Granai CO, Brard L. Phenethyl isothiocyanate (PEITC) inhibits growth of ovarian cancer cells by inducing apoptosis: role of caspase and MAPK activation. Gynecol. Oncol. 2006;103(1):261–270. doi: 10.1016/j.ygyno.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Schroeder EK, Kelsey NA, Doyle J, Breed E, Bouchard RJ, Loucks FA, Harbison RA, Linseman DA. Green tea epigallocatechin 3-gallate accumulates in mitochon- dria and displays a selective antiapoptotic effect against inducers of mitochondrial oxidative stress in neurons. Antioxid. Redox Signal. 2009;11(3):469–480. doi: 10.1089/ars.2008.2215. [DOI] [PubMed] [Google Scholar]
- Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10(3):175–176. doi: 10.1016/j.ccr.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64(7):2627–2633. doi: 10.1158/0008-5472.can-03-0846. [DOI] [PubMed] [Google Scholar]
- Semenza GL. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J. Bioenerg. Biomembr. 2007;39(3):231–234. doi: 10.1007/s10863-007-9081-2. [DOI] [PubMed] [Google Scholar]
- Sen S, Sharma H, Singh N. Curcumin enhances Vinorelbine mediated apoptosis in NSCLC cells by the mitochondrial pathway. Biochem. Biophys. Res. Commun. 2005;331(4):1245–1252. doi: 10.1016/j.bbrc.2005.04.044. [DOI] [PubMed] [Google Scholar]
- Shanafelt TD, Lee YK, Call TG, Nowakowski GS, Dingli D, Zent CS, Kay NE. Clinical effects of oral green tea extracts in four patients with low grade B-cell malignancies. Leuk. Res. 2006;30(6):707–712. doi: 10.1016/j.leukres.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Shanafelt TD, Call TG, Zent CS, LaPlant B, Bowen DA, Roos M, Secreto CR, Ghosh AK, Kabat BF, Lee MJ, Yang CS, Jelinek DF, Erlichman C, Kay NE. Phase I trial of daily oral Polyphenon E in patients with asymptomatic Rai stage 0 to II chronic lymphocytic leukemia. J. Clin. Oncol. 2009;27(23):3808–3814. doi: 10.1200/JCO.2008.21.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharief MK, Matthews H, Noori MA. Expression ratios of the Bcl-2 family proteins and disease activity in multiple sclerosis. J. Neuroimmunol. 2003;134(1–2):158–165. doi: 10.1016/s0165-5728(02)00400-9. [DOI] [PubMed] [Google Scholar]
- Sheu ML, Chiang CK, Tsai KS, Ho FM, Weng TI, Wu HY, Liu SH. Inhibition of NADPH oxidase-related oxidative stress-triggered signaling by honokiol suppresses high glucose-induced human endothelial cell apoptosis. Free Radic. Biol. Med. 2008;44(12):2043–2050. doi: 10.1016/j.freeradbiomed.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. U. S. A. 1997;94(13):6658–6663. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shnyder SD, Cooper PA, Millington NJ, Gill JH, Bibby MC. Sodium pancratistatin 3, 4-o-cyclic phosphate, a water-soluble synthetic derivative of pancratistatin, is highly effective in a human colon tumor model. J. Nat. Prod. 2008;71(3):321–324. doi: 10.1021/np070477p. [DOI] [PubMed] [Google Scholar]
- Siedlakowski P, McLachlan-Burgess A, Griffin C, Tirumalai SS, McNulty J, Pandey S. Synergy of Pancratistatin and Tamoxifen on breast cancer cells in inducing apoptosis by targeting mitochondria. Cancer Biol. Ther. 2008;7(3):376–384. doi: 10.4161/cbt.7.3.5364. [DOI] [PubMed] [Google Scholar]
- Singh S, Khar A. Biological effects of curcumin and its role in cancer chemoprevention and therapy. Anticancer Agents Med. Chem. 2006;6(3):259–270. doi: 10.2174/187152006776930918. [DOI] [PubMed] [Google Scholar]
- Stegehuis JH, de Wilt LH, de Vries EG, Groen HJ, de Jong S, Kruyt FA. TRAIL receptor targeting therapies for non-small cell lung cancer: Current status and perspectives. Drug Resist. Updat. 2009 doi: 10.1016/j.drup.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Subudhi U, Das K, Paital B, Bhanja S, Chainy GB. Alleviation of enhanced oxidative stress and oxygen consumption of L-thyroxine induced hyperthyroid rat liver mitochondria by vitamin E and curcumin. Chem. Biol. Interact. 2008;173(2):105–114. doi: 10.1016/j.cbi.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Sullivan R, Graham CH. Chemosensitization of cancer by nitric oxide. Curr. Pharm. Des. 2008;14(11):1113–1123. doi: 10.2174/138161208784246225. [DOI] [PubMed] [Google Scholar]
- Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51(3):794–798. [PubMed] [Google Scholar]
- Takeuchi O, Fisher J, Suh H, Harada H, Malynn BA, Korsmeyer SJ. Essential role of BAX, BAK in B cell homeostasis and prevention of autoimmune disease. Proc. Natl. Acad. Sci. U. S. A. 2005;102(32):11272–11277. doi: 10.1073/pnas.0504783102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang P, Mamdani F, Hu X, Liu JO, Yu B. Synthesis of OSW saponin analogs with modified sugar residues and their antiproliferative activities. Bioorg. Med. Chem. Lett. 2007;17(4):1003–1007. doi: 10.1016/j.bmcl.2006.11.032. [DOI] [PubMed] [Google Scholar]
- Thayyullathil F, Chathoth S, Hago A, Patel M, Galadari S. Rapid reactive oxygen species (ROS) generation induced by curcumin leads to caspase-dependent and -independent apoptosis in L929 cells. Free Radic. Biol. Med. 2008;45(10):1403–1412. doi: 10.1016/j.freeradbiomed.2008.08.014. [DOI] [PubMed] [Google Scholar]
- Tiligada E. Chemotherapy: induction of stress responses. Endocr.-Relat. Cancer. 2006;13(Suppl 1):S115–S124. doi: 10.1677/erc.1.01272. [DOI] [PubMed] [Google Scholar]
- Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, Huang P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta- phenylethyl isothiocyanate. Cancer Cell. 2006;10(3):241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008a;10(8):1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachootham D, Zhang H, Zhang W, Feng L, Du M, Zhou Y, Chen Z, Pelicano H, Plunkett W, Wierda WG, Keating MJ, Huang P. Effective Elimination of Fludarabine-Resistant CLL Cells by PEITC through a Redox-Mediated Mechanism. Blood. 2008b;112(5):1912–1922. doi: 10.1182/blood-2008-04-149815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discov. 2009;8(7):579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- Tsai TH, Chou CJ, Cheng FC, Chen CF. Pharmacokinetics of honokiol after intravenous administration in rats assessed using high-performance liquid chromatography. J. Chromatogr. B Biomed. Appl. 1994;655(1):41–45. doi: 10.1016/0378-4347(94)00031-x. [DOI] [PubMed] [Google Scholar]
- Tse AK, Wan CK, Shen XL, Yang M, Fong WF. Honokiol inhibits TNF-alpha-stimulated NF-kappaB activation and NF-kappaB-regulated gene expression through suppression of IKK activation. Biochem. Pharmacol. 2005;70(10):1443–1457. doi: 10.1016/j.bcp.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Turanek J, Wang XF, Knotigova P, Koudelka S, Dong LF, Vrublova E, Mahdavian E, Prochazka L, Sangsura S, Vacek A, Salvatore BA, Neuzil J. Liposomal formulation of alpha-tocopheryl maleamide: in vitro and in vivo toxicological profile and anticancer effect against spontaneous breast carcinomas in mice. Toxicol. Appl. Pharmacol. 2009;237(3):249–257. doi: 10.1016/j.taap.2009.01.027. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M, Nakamura Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008;266(1):37–52. doi: 10.1016/j.canlet.2008.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Fan W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion. 2010;10(1):12–31. doi: 10.1016/j.mito.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Ogasawara MA, Huang P. Small mitochondria-targeting molecules as anti-cancer agents. Mol. Aspects Med. 2009 doi: 10.1016/j.mam.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Weinberg F, Chandel NS. Mitochondrial metabolism and cancer. Ann. NY Acad. Sci. 2009;1177:66–73. doi: 10.1111/j.1749-6632.2009.05039.x. [DOI] [PubMed] [Google Scholar]
- Wojtkielewicz A, Dlugosz M, Maj J, Morzycki JW, Nowakowski M, Renkiewicz J, Strnad M, Swaczynova J, Wilczewska AZ, Wojcik J. New analogues of the potent cytotoxic saponin OSW-1. J. Med. Chem. 2007;50(15):3667–3673. doi: 10.1021/jm0613572. [DOI] [PubMed] [Google Scholar]
- Wu WS. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006;25(4):695–705. doi: 10.1007/s10555-006-9037-8. [DOI] [PubMed] [Google Scholar]
- Wu SJ, Ng LT, Lin CC. Effects of antioxidants and caspase-3 inhibitor on the phenylethyl isothiocyanate-induced apoptotic signaling pathways in human PLC/PRF/5 cells. Eur. J. Pharmacol. 2005;518(2–3):96–106. doi: 10.1016/j.ejphar.2005.06.021. [DOI] [PubMed] [Google Scholar]
- Xu K, Thornalley PJ. Involvement of glutathione metabolism in the cytotoxicity of the phenethyl isothiocyanate and its cysteine conjugate to human leukaemia cells in vitro. Biochem. Pharmacol. 2001;61(2):165–177. doi: 10.1016/s0006-2952(00)00526-8. [DOI] [PubMed] [Google Scholar]
- Xu RH, Pelicano H, Zhang H, Giles FJ, Keating MJ, Huang P. Synergistic effect of targeting mTOR by rapamycin and depleting ATP by inhibition of glycolysis in lymphoma and leukemia cells. Leukemia. 2005;19(12):2153–2158. doi: 10.1038/sj.leu.2403968. [DOI] [PubMed] [Google Scholar]
- Xu C, Yuan X, Pan Z, Shen G, Kim JH, Yu S, Khor TO, Li W, Ma J, Kong AN. Mechanism of action of isothiocyanates: the induction of ARE-regulated genes is associated with activation of ERK and JNK and the phosphorylation and nuclear translocation of Nrf2. Mol. Cancer Ther. 2006;5(8):1918–1926. doi: 10.1158/1535-7163.MCT-05-0497. [DOI] [PubMed] [Google Scholar]
- Xue J, Liu P, Pan Y, Guo Z. A total synthesis of OSW-1. J. Org. Chem. 2008;73(1):157–161. doi: 10.1021/jo7018812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Hsu S, Lewis J, Wataha J, Dickinson D, Singh B, Bollag WB, Lockwood P, Ueta E, Osaki T, Schuster G. Green tea polyphenol causes differential oxidative environments in tumor versus normal epithelial cells. J. Pharmacol. Exp. Ther. 2003;307(1):230–236. doi: 10.1124/jpet.103.054676. [DOI] [PubMed] [Google Scholar]
- Yang SE, Hsieh MT, Tsai TH, Hsu SL. Down-modulation of Bcl-XL, release of cytochrome c and sequential activation of caspases during honokiol-induced apoptosis in human squamous lung cancer CH27 cells. Biochem. Pharmacol. 2002;63(9):1641–1651. doi: 10.1016/s0006-2952(02)00894-8. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Talalay P. Anticarcinogenic activities of organic isothiocyanates: chemistry and mechanisms. Cancer Res. 1994;54(7 Suppl):1976s–1981s. [PubMed] [Google Scholar]
- Zhang Y, Tang L, Gonzalez V. Selected isothiocyanates rapidly induce growth inhibition of cancer cells. Mol. Cancer Ther. 2003;2(10):1045–1052. [PubMed] [Google Scholar]
- Zhang Y, Li J, Tang L. Cancer-preventive isothiocyanates: dichotomous modulators of oxidative stress. Free Radic. Biol. Med. 2005;38(1):70–77. doi: 10.1016/j.freeradbiomed.2004.09.033. [DOI] [PubMed] [Google Scholar]
- Zhang H, Trachootham D, Lu W, Carew J, Giles FJ, Keating MJ, Arlinghaus RB, Huang P. Effective killing of Gleevec-resistant CML cells with T315I mutation by a natural compound PEITC through redox-mediated mechanism. Leukemia. 2008a;22(6):1191–1199. doi: 10.1038/leu.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang H, Tighiouart M, Lee JE, Shin HJ, Khuri FR, Yang CS, Chen ZG, Shin DM. Synergistic inhibition of head and neck tumor growth by green tea (-)-epigallocatechin-3-gallate and EGFR tyrosine kinase inhibitor. Int. J. Cancer. 2008b;123(5):1005–1014. doi: 10.1002/ijc.23585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Neuzil J, Wu K. Vitamin E analogues as mitochondria-targeting compounds: from the bench to the bedside? Mol. Nutr. Food Res. 2009;53(1):129–139. doi: 10.1002/mnfr.200800045. [DOI] [PubMed] [Google Scholar]
- Zhou P, Kalakonda N, Comenzo RL. Changes in gene expression profiles of multiple myeloma cells induced by arsenic trioxide (ATO): possible mechanisms to explain ATO resistance in vivo. Br. J. Haematol. 2005a;128(5):636–644. doi: 10.1111/j.1365-2141.2005.05369.x. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Garcia-Prieto C, Carney DA, Xu RH, Pelicano H, Kang Y, Yu W, Lou C, Kondo S, Liu J, Harris DM, Estrov Z, Keating MJ, Jin Z, Huang P. OSW-1: a natural compound with potent anticancer activity and a novel mechanism of action. J. Natl Cancer Inst. 2005b;97(23):1781–1785. doi: 10.1093/jnci/dji404. [DOI] [PubMed] [Google Scholar]
- Zhu J, Xiong L, Yu B, Wu J. Apoptosis induced by a new member of saponin family is mediated through caspase-8-dependent cleavage of Bcl-2. Mol. Pharmacol. 2005;68(6):1831–1838. doi: 10.1124/mol.105.015826. [DOI] [PubMed] [Google Scholar]
- Zini R, Morin C, Bertelli A, Bertelli AA, Tillement JP. Effects of resveratrol on the rat brain respiratory chain. Drugs Exp. Clin. Res. 1999;25(2-3):87–97. [PubMed] [Google Scholar]