

Abstract
Cardiovascular complications such as hypertension are a continuous concern in patients with autosomal dominant polycystic kidney disease (ADPKD). The PKD2 encoding for polycystin-2 is mutated in ≈15% of ADPKD patients. Here, we show that polycystin-2 is localized to the cilia of mouse and human vascular endothelial cells. We demonstrate that the normal expression level and localization of polycystin-2 to cilia is required for the endothelial cilia to sense fluid shear stress through a complex biochemical cascade, involving calcium, calmodulin, Akt/PKB, and protein kinase C. In response to fluid shear stress, mouse endothelial cells with knockdown or knockout of Pkd2 lose the ability to generate nitric oxide (NO). Consistent with mouse data, endothelial cells generated from ADPKD patients do not show polycystin-2 in the cilia and are unable to sense fluid flow. In the isolated artery, we further show that ciliary polycystin-2 responds specifically to shear stress and not to mechanical stretch, a pressurized biomechanical force that involves purinergic receptor activation. We propose a new role for polycystin-2 in transmitting extracellular shear stress to intracellular NO biosynthesis. Thus, aberrant expression or localization of polycystin-2 to cilia could promote high blood pressure because of inability to synthesize NO in response to an increase in shear stress (blood flow).
Keywords: biophysical force, endothelia, mechanotransduction, primary cilium, shear stress
Autosomal dominant polycystic disease (ADPKD) (Online Mendelian Inheritance in Man 173900) is characterized by bilateral cyst formation in the kidneys. The only effective treatments presently available for ADPKD patients are renal dialysis and transplantation. Although the cystic kidney phenotype is the hallmark of ADPKD, a wide range of cardiovascular complications also affect a large number of ADPKD patients. In particular, hypertension occurs frequently and is an early manifestation of ADPKD.1–4 Aside from hypertension, other vascular complications may include left ventricular hypertrophy, cerebral aneurysm, thoracic/abdominal aortic aneurysm, and prolapse of the mitral valve.1,2 Although the etiology of these cardiovascular abnormalities is currently unclear, ADPKD patients are generally placed on antihypertensive therapy.
ADPKD is a genetic disease caused by the mutation of either PKD1 or PKD2, which encode for polycystin-1 or polycystin-2, respectively. Whereas polycystin-1 is an 11-transmembrane protein with a long extracellular domain, polycystin-2 is a cation channel with 6-transmembrane domains and belongs to a superfamily of transient receptor potential (TRP) ion channels. To advance the understanding of the cellular and molecular mechanisms of ADPKD, several Pkd2 mouse models have been generated. They, too, present degrees of cardiovascular abnormalities.5 In particular, expression of polycystin-2 has been detected in vascular systems of mice6,7 and humans.7,8 In addition to focal hemorrhage, the Pkd2 mouse exhibits progressive total body edema, a feature of cardiac failure.5
Polycystin-2 is a calcium channel whose activity and/or localization is modulated by polycystin-1.9 Through antibody mediated inhibition of polycystin-2, we have previously shown that polycystin-2 may function as a mechanosensitive calcium channel in renal epithelial cells.10 Polycystin-2 functioning as a fluid flow sensor has been further suggested in mouse nodal cells.11 In the present study, we expand this observation and investigate the roles of polycystin-2 in sensing mechanical fluid shear stress in Pkd2 knockdown and knockout mouse aortic and human endothelial cells. Mutations in PKD2 have been suggested to contribute to vascular hypertension,3,4 probably because of failure to convert an increase in mechanical blood flow into cellular nitric oxide (NO) biosynthesis to control the vascular tone, ie, blood pressure. To examine polycystin-2 roles in hypertension, we also measured intracellular NO in endothelial cells generated from ADPKD patients. We show that endothelial cells depend on polycystin-2 and a cascade of intracellular signaling molecules to synthesize NO in response to fluid shear stress.
Materials and Methods
Signed and informed consent to collect disposed ADPKD human kidneys was obtained from the patients, and kidney collection protocols were approved by the Department for Human Research Protections of the Biomedical Institutional Review Board of The University of Toledo. The use of animal tissues was approved by The University of Toledo animal care and use committee.
An expanded Materials and Methods section is available in the online data supplement at http://circres.ahajournals.org.
Results
Polycystin-2 Is Localized to Endothelial Cilia and Functions in Mechanosensing
We previously showed that cilia are microsensory compartments that house sensory molecules and that polycystin-1 is among the sensory molecules.12 Because polycystin-1 and -2 interact with each other,9,13 we examined whether polycystin-2 could also localize to endothelial cilia like polycystin-1. We show here, for the first time, that polycystin-2 is localized to the cilia of the vascular endothelia in the femoral artery from an adult mouse (Figure 1a). Polycystin-2 is colocalized with acetylated α-tubulin, a well-recognized marker for cilia. Using high-resolution differential interference contrast imaging, we also show the presence of cilia in the arterial lumen. To study the mechanosensory function of polycystin-2, we first identified its presence in previously characterized endothelial cells from mouse embryonic aorta.12 Polycystin-2 is clearly present in the cilia of cultured endothelial cells (Figure 1b). The cells were further validated to retain endothelial marker, CD144.
Figure 1.

Polycystin-2 localization in vivo and in vitro. Localization of polycystin-2 (PC2) was examined with immunofluorescence staining. a, High-resolution differential interference contrast (DIC) image shows a section of femoral artery with a thickness of 10 μm. The inset in the top right corner shows the full section of the artery. The red box magnifies an endothelial cell, which shows localization of PC2 to endothelial cilia. Acetylated α-tubulin (α-tub) was used as a ciliary marker. b, Cultured endothelial cells also show the presence of polycystin-2 in cilia, and VE-cadherin (CD144) was used as an endothelial marker. Nuclear marker (DAPI) is shown in the merged images. Scale bar=25 μm.
Because endothelial cell lines with Pkd2 mutation do not exist, we used a small interfering (si)RNA approach to inhibit the expression level of polycystin-2. We designed several siRNA probes to target a series of Pkd2 mRNA sites (supplemental Table I). The efficiency of transfection was verified by examining the transcript and expression levels of polycystin-2 (Figure 2a and 2b). We noted that the efficiency of siRNA approach on Pkd2 depends largely on the siRNA probes; siRNA1 and siRNA4 appear to be more effective than siRNA2 and siRNA3. To assay mechanosensory cilia function, we perfused the cells with an optimal shear stress of 7 dyn/cm2. This magnitude of shear stress provides the greatest increase in cytosolic calcium and NO production in endothelial cells, as determined previously.12 When changes in cytosolic calcium in response to fluid flow were examined, cells transfected with siRNA1 and siRNA4 were less responsive toward fluid shear (Figure 2c). To confirm our calcium readout, we also monitored changes in cytosolic NO as an indication of NO biosynthesis (Figure 2d). Although variations in cytosolic calcium and NO were observed within a cell population, we consistently observed that in 3 independent experiments, cells transfected with siRNA1 or siRNA4 were less responsive to fluid shear (Figure 2e). Their calcium and NO responses were significantly repressed compared to corresponding calcium and NO in control groups.
Figure 2.

Effects of polycystin-2 expression in mouse endothelial cells. The expression level of polycystin-2 corresponds to the cytosolic calcium increase and NO biosynthesis in response to fluid shear stress. Mouse endothelial cells were transfected with Lipofectin only (control, C), scramble siRNA (S), siRNA1, siRNA2, siRNA3, or siRNA4 of polycystin-2. a, The transfected cells were collected to examine the transcript levels of polycystin-2 (PC2) and polycystin-1 (PC1), and α-tubulin (tub) was used as a control. b, The transfected cells were analyzed for polycystin-2 expression, and the membrane was reblotted for α-tubulin as a loading control. c, Transfected cells were challenged with fluid shear stress of 7 dyn/cm2 (indicated by arrows), and 5 individual responses were randomly selected and analyzed for changes in cytosolic calcium levels. d, Biosynthesis of NO in response to shear stress was plotted in 5 individual cells. e, Peaks of cytosolic calcium and NO in response to shear stress were averaged. Asterisks indicate statistically significant against control at P<0.05 (N=3).
Ciliary Polycystin-2 Is Functionally Relevant in Human Endothelial Cells
To examine the clinical relevance of polycystin-2, we isolated endothelial cells from interlobar arteries of nine ADPKD kidneys. Interestingly, we observed either a normal or null response within a diseased kidney. For example, within 5 successful endothelial isolations from a kidney of patient 5, endothelial cells from segment 7 consistently showed neither calcium nor NO responses (Figure 3a). On the other hand, cells from other segments responded to fluid shear stress by showing cytosolic calcium increases and NO biosynthesis. Surprisingly, the ciliary expression of polycystin-2 correlates with our fluid shear assays (Figure 3b). Similar findings from patient 6 are also presented (Figure I in the online data supplement). Although ADPKD kidneys that we obtained more likely had PKD1 mutations than PKD2 mutations, ≈85% compared to 15% of the ADPKD cases, respectively, we could confidently suggest that the failure of 5–7 endothelial cells (from patient 5, segment 7) to respond to fluid shear stress is attributable, in part, to an absence of ciliary polycystin-2. In addition, we could show that 5–2 and 5–7 cells possessed endothelial markers CD144 and endothelial NO synthase (eNOS) (Figure 3c). Although we were not able to further analyze these cells because of the short passages of primary cultures, our Western blot analysis from pooled endothelial cells of patients 7, 8, and 9 confirms our cell isolation technique (supplemental Figure I).
Figure 3.

Effects of fluid shear stress in vascular endothelial cells of an ADPKD patient. Vascular endothelial cells were isolated from several interlobar arteries of an ADPKD kidney. a, Endothelial cells (5-2 and 5-7) from segmental arteries 2 and 7 of patient 5 were cultured and challenged with fluid shear stress, and their cytosolic calcium and NO changes were recorded. Arrows indicate the start of fluid flow. b, The primary culture was then subjected to immunolocalization studies for polycystin-2 (PC2). Acetylated α-tubulin (α-tub) was used as a ciliary marker, and nuclear marker (DAPI) is shown in the merged images. Arrows indicate the absence of polycystin-2. c, Immunofluorescence studies indicated the presence of endothelial markers CD144 and eNOS in both 5-2 and 5-7 cells. N=3 with passages 2, 3, and 4. Scale bar=5 μm.
To delineate the roles of polycystin-2 in human cells independently from polycystin-1 function, we used the siRNA approach on cultured human umbilical vein endothelial cells. Several siRNA probes were made against a series of PKD2 mRNA sites (supplemental Table I). The efficiency of transfection was verified by quantifying the transcript and expression levels of polycystin-2 (Figure 4a and 4b). Similar to results from the mouse Pkd2, we noted that the efficiency of siRNA approach on human PKD2 depends largely on the siRNA probes; siRNA2 and siRNA3 appear to be more effective than siRNA1 and siRNA4. To assay mechanosensory cilia function, we perfused the cells and measured changes in cytosolic calcium and NO in response to fluid flow. Cells transfected with siRNA2 and siRNA3 did not respond to fluid shear (Figure 4c). To complement our calcium readout, we also monitored changes in cytosolic NO (Figure 4d). Although variations in cytosolic calcium and NO were observed in individual cells, especially with siRNA4, we consistently observed that in 3 independent experiments, cells transfected with siRNA2 or siRNA3 did not respond or showed less response to fluid shear (Figure 4d). Their calcium and NO responses were statistically repressed, compared to corresponding calcium and NO in control groups.
Figure 4.

Effects of polycystin-2 expression in human endothelial cells. The expression level of polycystin-2 corresponds with the cytosolic calcium increase and NO biosynthesis in response to fluid shear stress. Human umbilical vein endothelial cells were transfected with Lipofectin only (control, C), scramble siRNA (S), siRNA1, siRNA2, siRNA3, or siRNA4 of polycystin-2. a, The transfected cells were collected to examine the transcript levels of polycystin-2 (PC2) and polycystin-1 (PC1), and α-tubulin (tub) was used as a control. b, The transfected cells were analyzed for polycystin-2 expression, and the membrane was reblotted for α-tubulin as a loading control. c, Transfected cells were challenged with fluid shear stress of 7 dyn/cm2 (indicated by arrows), and 5 individual responses were randomly selected and analyzed for changes in cytosolic calcium levels. d, Biosynthesis of NO in response to shear stress was plotted in 5 individual cells. e, Peaks of cytosolic calcium and NO in response to shear stress were averaged. Asterisks indicate statistically significant against control at P<0.05 (N=3).
Shear-Induced NO Biosynthesis Depends on Ciliary Polycystin-2 Calcium Channel
Together with mouse (Figure 2) and human (Figures 3 and 4) endothelial cells, we report here, for the first time, that endothelial cells ability to sense fluid shear stress depends on the expression level and/or ciliary localization of polycystin-2. In addition, a “2-hit” mechanism has been suggested in ADPKD.14,15 This mechanism explains that patients would inherit a germ line mutation from one of the parents, and a random second somatic mutation is required to facilitate the disease phenotypes. To examine this possibility in vascular hypertension in PKD and to further verify our data, especially those obtained from human ADPKD patients, we used a Pkd2 mouse model to compare fluid sensing ability of Pkd2+/− and Pkd2−/− primary endothelial cells. Unlike Pkd2+/− cells, Pkd2−/− endothelial cells did not respond to fluid shear stress (Figure 5a). The Pkd2+/−, but not Pkd2−/−, cells show cytosolic calcium increases in response to fluid shear. Extracellular and intracellular measurements of NO were in agreement with the results from the calcium readout. The subcellular ciliary localization of polycystin-2 was also examined (Figure 5b). As expected, we never observed ciliary polycystin-2 in Pkd2−/− cells. To examine differential expressions of polycystin-1 in Pkd2+/− and Pkd2−/− endothelial cells, we performed immunoprecipitation studies. When polycystin-1 was immunoprecipitated, no apparent difference was observed in polycystin-1 expressions between Pkd2+/− and Pkd2−/− endothelial cells (Figure 5c, i). Because we could reblot for polycystin-2 in Pkd2−/− cells, this study further indicates that polycystin-1 interacts with polycystin-2 in vascular endothelial cells. We next immunoprecipitated polycystin-2 and blotted for both polycystin-1 and -2 to demonstrate that polycystin-1 and -2 interaction could be confirmed reversibly (Figure 5c, ii). In all cases, polycystin-2 expression was not detected in Pkd2−/− endothelial cells. We further showed that Pkd2+/− and Pkd2−/− primary endothelial cells within passages 2, 3, and 4 consistently retain endothelial markers, such as eNOS, CD144, and Akt (Figure 5d).
Figure 5.
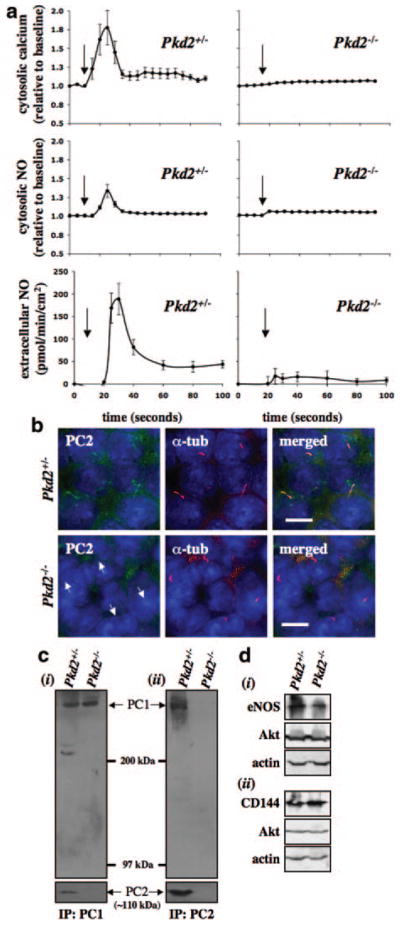
Polycystin-2 required in fluid-flow sensing. Vascular endothelial cells were isolated from Pkd2+/− and Pkd2−/− embryonic aortas. a, The endothelial cells were cultured and challenged with fluid shear stress, and their cytosolic calcium increase, intracellular NO production, and extracellular NO release were measured. Arrows indicate the start of fluid flow. b, The primary culture was then subjected to immunolocalization studies for polycystin-2 (PC2). Acetylated α-tubulin (α-tub) was used as a ciliary marker, and nuclear marker (DAPI) is shown in the merged images. Arrows indicate the absence of polycystin-2. c, Immunoprecipitation studies for polycystin-1 (i) and -2 (ii) confirmed interaction of polycystin-1 and -2 in vascular endothelial cells and absence of polycystin-2 in Pkd2−/− cells. d, The presence of endothelial markers eNOS (i), CD144 (ii), and Akt (i and ii) confirmed the cell type used in our study, and β-actin was used as a loading control. N=3 for calcium and NO measurements with passages 2, 3, and 4. Scale bar=5 μm.
Polycystin-2–Dependent NO Production Involves a Cascade of Signaling Molecules
In this study, we propose that ciliary polycystin-2 is a shear-sensitive calcium channel that is required to activate a biochemical cascade for NO production. To confirm that our biophysical calcium and biochemical NO readouts are biologically and technically relevant, we applied various inhibitors to block the molecular functions that are supposedly involved in shear-induced NO production.16 Removing extra-cellular calcium with EGTA abolished both calcium and NO readouts in wild-type endothelial cells, indicating that extra-cellular calcium influx is a prerequisite for both cytosolic increase in calcium and NO production (Figure 6). We also confirmed that eNOS inhibitor NG-nitro-L-arginine methyl ester (L-NAME) could block shear-induced NO biosynthesis but not cytosolic calcium increase. To explore calcium-dependent mechanisms of NO production, we used calphostin C and W7 to inhibit calmodulin and calcium-dependent protein kinase (PK)C and calmodulin, respectively. When these calcium-binding proteins were inhibited, calcium, but not NO, readout showed an increase, indicating that PKC and calmodulin act downstream of the calcium pathway and that inhibition of either molecule will block NO synthesis.
Figure 6.

Molecular cascade involved in shear stress–induced calcium and NO signaling. The left and right graphs represent the response to fluid shear stress in mouse endothelial cells for cytosolic calcium ([Ca2+]cyt) and NO ([NO]cyt), respectively. Effects of various inhibitors on response to fluid shear stress, as indicated by arrows, are presented in the line graphs. The extra-cellular calcium chelator EGTA abolished both calcium and NO increases, indicating extracellular calcium influx is required in shear-induced NO biosynthesis. NO synthase inhibitor L-NAME indicated that shear-induced NO production involves a rapid activation of NO synthase. Whereas PKC (calphostin C), calmodulin (W7), and Akt inhibitors played a role in mechanical fluid sensing, PI3K inhibitor (LY-294,002) did not affect shear-induced NO production. N≥4.
To explore the possibility of Akt or PKB contribution to shear stress–induced NO production,16 we treated wild-type cells with Akt inhibitor II. Inhibition of Akt/PKB resulted in blockage of NO readout but did not alter calcium signaling. In addition to calmodulin, phosphoinositide 3-kinase (PI3K) is also a major regulator for the Akt/PKB pathway.16 To further examine the roles of PI3K in Akt/PKB function, we treated the cells with either LY-294,002 or wortmannin (not shown). Interestingly, neither of these inhibitors significantly inhibited calcium signaling or NO production in response to fluid shear stress. Together, our data suggest that calcium is an important messenger for relaying extracellular fluid flow stimuli to intracellular NO production through ciliary polycystin-2 calcium channel.
Ciliary Polycystin-2 Is a Shear Stress–Specific Molecule
To investigate mechanosensory polycystin-2 function in more detail, we perfused isolated artery that had been transfected with either scrambled or Pkd2 siRNA. Artery with scrambled siRNA was either used as a control or further treated with apyrase. In a freely placed artery, a flow rate of 164 μL/sec resulted in cytosolic calcium increases (Figure 7a). In a control artery, a continuous fluid flow resulted in sustained increase in cytosolic calcium (Figure 7a and 7c). Interestingly, an artery that had been pretreated with apyrase and was perfused with apyrase showed an increase in cytosolic calcium, but with a very different calcium profile than observed in the control group. A smaller but similar calcium profile than in the control group was observed in the artery transfected with Pkd2 siRNA. Because, at a higher microscopic magnification, we observed that the freely placed artery was moved as a result of the motion from the luminal fluid perfusate, we predicted that the movement would result in stretching-like motion on the arterial wall. Consistent with this idea, we hypothesize that the luminal wall stretching would result in sustained cytosolic calcium increase, a mechanism that would involve ATP release.17,18 Furthermore, it is worth mentioning that the calcium profiles in apyrase-treated arteries and in isolated endothelial cells are very similar (Figures 2 through 6), indicating that apyrase might have diminished the stretch-induced calcium response in a freely placed artery.
Figure 7.

Integration of polycystin-2 and purinergic signaling in isolated artery. To differentiate mechanical forces generated by perfusate, an isolated artery was freely placed to provide unrestrained movement, or the artery was capillary-enclosed to limit stretching motion. Artery transfected with scrambled siRNA is denoted as control (ctr), whereas artery transfected with scrambled siRNA and pre-treated and perfused with apyrase is designated as apyrase (apy). Isolated artery was also transfected with Pkd2 siRNA (Pkd2). a, In the freely placed artery, as illustrated by the top image, calcium imaging studies show different cytosolic calcium profiles among control, apyrase, and Pkd2-treated groups. b, In the capillary-enclosed artery, as illustrated by the top image, flow-induced cytosolic calcium increase was diminished in Pkd2, compared to control or apyrase-treated arteries. Changes in cytosolic calcium was pseudocolored, white/green represents a low level, and yellow/red denotes a higher level of cytosolic calcium. In all cases, a similar fluid-flow rate of 164 μL/sec was initiated after time 0 second. c, Five responsive areas within the artery, if any were present, were randomly selected and analyzed for changes in cytosolic calcium levels. d, Both Pkd2+/− and Pkd2−/− endothelial cells were able to respond to 10 μmol/L ATP in the presence or absence of 1 mmol/L EGTA. N=3 arteries for each group and treatment, and N>2 for cells with passage 2, 3, or 4.
To further confirm this possibility, we carefully inserted an artery into a glass capillary tube (Figure 7b). The aorta inside the capillary tube had very limited room for perfusate pressure-induced arterial stretching or expending. In this capillary-enclosed setting, neither control nor treated arteries showed a sustained increase in cytosolic calcium in response to a similar flow rate of 164 μL/sec (Figure 7c). Most important is that the Pkd2 siRNA artery did not show a significant increase in cytosolic calcium, although it still responded to ATP (not shown). To verify these findings, we challenged both Pkd2+/− and Pkd2−/− endothelial cells with ATP in the presence and absence of EGTA (Figure 7d). Because Pkd2−/− endothelial cells were able to respond to ATP and because Pkd2-depleted arteries could respond to mechanical fluid flow in freely placed but not in capillary-enclosed settings, we propose that polycystin-2 functions as a mechanical channel and has a specific role in fluid shear sensing. We, therefore, propose that ciliary polycystins are only few examples of a large family of sensory proteins that a cell may have. Thus, depending on its sensory proteins, an endothelial cell could have different mechanisms to detect a range of mechanical stimuli.
Discussion
Dysfunction of many ciliary proteins has been linked to a list of human diseases, from cystic kidney and obesity to blindness and mental retardation. Although many ciliary functions have been proposed,19 their mechanical function as microsensory compartments has been the most described.20–22 In our present study, we suggest that polycystin-2 is a ciliary calcium channel that functions as one of the sensory machineries in endothelial cells. Our study also indicates that abnormality in polycystin-2 expression, localization and/or function is related to the inability of endothelial cells to generate NO in response to fluid shear stress. We further propose that failure to produce NO in response to shear stress is clinically relevant to the development of hypertension, particularly in PKD patients.
In the present study, we show, for the first time, that polycystin-2 is localized to endothelial cilia in cell culture and in vivo. We studied polycystin-2 extensively, using an siRNA approach and genetic model in mouse and human vascular endothelial cells. Although our siRNA approach using mouse endothelial cells did not provide similar inhibition levels of polycystin-2 expression, the transcript and expression levels were well correlated with the overall endothelial cell response to fluid shear. To confirm that polycystin-2 function is clinically relevant, we isolated inter-lobar endothelial cells from ADPKD kidneys. For each diseased kidney, however, we observed a mixed response from different arterial segments. This result is consistent with our previous findings whereby not all ADPKD kidney epithelial cells are irresponsive to fluid shear stress.23 We and others have found that only epithelial cells isolated from cyst-linings that do not show polycystin-1 or -2 localization to cilia are abnormal in flow sensing.23,24 In agreement with this notion, our data suggest that ciliary localization of polycystin-2 is required in fluid shear sensing. Moreover, we have also shown that ciliary localization of polycystin-2 could depend on functional polycystin-1 to cilia in human and mouse cells.10,23 Therefore, mutation(s) in PKD1 may alter subcellular ciliary localization of polycystin-2. Nonetheless, proper ciliary localization and function of polycystin-2 are requirements for fluid sensing in the endothelial cells.
We further hypothesize that vascular endothelia also require a “second-hit” in ADPKD in a similar manner to renal epithelia.14,15 This implies that a germ-line mutation (heterozygous) may not be sufficient to cause any clinical symptoms such as hypertension, but another random somatic mutation (homozygous) is required. To examine this possibility, we used a Pkd2 mouse model to compare Pkd2+/− and Pkd2−/− endothelial cells in response to fluid flow. In fact, there was only 1 study that assessed sensory polycystin-2 function using Pkd2 mouse model.11 Regardless, the results support our hypothesis that unlike Pkd2−/− cells, Pkd2+/− endothelial cells maintain responsiveness to fluid flow. More importantly, our studies confirm that polycystin-2 is an important shear-sensitive calcium channel in endothelial cells.
Although polycystin-1 and -2 have been shown to interact at the COOH termini,9,13 there is no study in vascular endothelial cells examining polycystin-1 and -2 interaction. Through coimmunoprecipitation studies, we confirmed that in endothelial cells, both polycystins interact to one another reciprocally. There were no apparent changes in polycystin-1 level between Pkd2+/− and Pkd2−/− endothelial cells. In lieu of these results, we propose that polycystin-1 mechanosensor interacts with polycystin-2 calcium channel, and this polycystin complex localizes in the microsensory compartment, cilium. An abrupt increase in blood pressure would result in fluid shear increase, followed by activation of cilia and polycystin complex to generate NO.
Throughout our studies, we used 2 different readouts to confirm the fluid shear sensing ability of the endothelial cells. Whereas the calcium readout is biophysically pertinent to basic science, NO is biochemically more relevant to the etiology of hypertension. Interestingly, we observed that if a Pkd2 knockdown or knockout cell shows a negative calcium readout, the NO readout is also negative and vice versa. To test the hypothesis that increases in cytosolic calcium are a prerequisite signaling event for NO biosynthesis, we used EGTA to chelate extracellular calcium. In the absence of extracellular calcium, the cytosolic calcium and NO increases were abolished, indicating that fluid shear sensing involves extracellular calcium influx, which in turn is required for NO production. To further verify our flow assay on the signaling event for NO biosynthesis, we used L-NAME to inhibit eNOS. As expected, L-NAME inhibited NO production but not calcium signaling in response to fluid flow. Because eNOS has a specific phosphorylation site for PKC,16 whose activity depends on calcium, we used calphostin C to demonstrate that PKC is required for shear-induced eNOS activation.
Because eNOS activation depends biochemically on calmodulin as a cofactor, we used W7 to inhibit calmodulin function. Our data shows that similar to L-NAME, W7 inhibited NO production but not calcium signaling. Not only was calmodulin a cofactor for eNOS, calcium–calmodulin complex has also been shown to activate Akt/PKB activity.16 To investigate whether Akt/PKB is involved in eNOS activity, we applied Akt inhibitor II in our system. Our data indicate that Akt/PKB is also involved in regulation of eNOS activation in response to fluid shear. In addition to calmodulin, Akt/PKB is also regulated by PI3K, which has been shown to be involved in shear stress–induced NO release.16 However, PI3K did not seem to play a major role in shear-induced eNOS activation, at least in our system. Collectively, our study suggests that endothelial cells require functional mechanosensory cilia and a list of intermediate machineries to produce NO in response to fluid shear stress. Upon sensing this mechanical signal, polycystin-2 promotes extracellular calcium influx that, in turn, activates PKC and binds to calmodulin; the calcium–calmodulin complex then increases Akt/PKB activity. Activation of eNOS by calmodulin, PKC, and Akt/PKB initiates an immediate NO synthesis.
Biomechanical forces in the blood vessel can be observed in the many forms, including stretch resulting from muscle distention caused by blood pressure and shear stress resulting from drag force generated by blood flow. To differentiate these mechanical forces, we designed a capillary-enclosed system that would allow an isolated artery to experience shear stress only. In a step increase in fluid flow, the capillary-enclosed artery showed a short burst increase in cytosolic calcium, similar to those seen in perfused cultured cells. On the other hand, the conventional freely placed artery, which induced stretch and increased in arterial diameter, showed a sustained increase in cytosolic calcium in response to fluid flow. Stretch-induced ATP release has been shown in many systems,17 including in endothelial cells.18 To investigate this purinergic involvement in our system, the artery was first treated with apyrase to hydrolyze any nucleoside triphosphates or diphosphates. Our data show that apyrase-treated artery has a very different calcium profile, indicating that ATP may play a role in stretch-induced calcium increase in freely placed artery.
To verify that Pkd2−/− endothelial cells did not have abnormal response to ATP, we challenged both Pkd2+/− and Pkd2−/− cells with ATP in the absence and presence of extracellular calcium chelator, EGTA. Cells from both genotypes demonstrated similar calcium profiles in response to ATP, with or without EGTA. It is worth noting that EGTA abolished flow-induced, but not in ATP-induced, calcium changes, demonstrating the complexity of mechanosignal transduction systems in vasculature. Consistent with this idea, shear stress has been shown to potentiate ATP-induced cytosolic calcium increase.25 To further establish the mechanosignaling complexity, we have previously demonstrated that although Pkd1−/− endothelial cells failed to respond to shear stress, they were able to respond to other mechanical and pharmacological stimuli.12 Similarly, Pkd2−/− endothelial cells lost their responsiveness to shear stress but not to ATP. This confirms our data from mouse and human endothelial cells that polycystin-2 has a specific shear-sensing role in vascular endothelial cilia.
All in all, our present study helps to explain the hypertensive phenotype seen in patients with ADPKD. We show that polycystin-2 in cilia plays crucial roles in the mediation of fluid shear-sensing, as well as the transduction of these mechanical signals into changes in calcium signaling and NO synthesis in endothelial cells. Hence, ciliary polycystin-2 may play crucial roles in the regulation of cardiovascular homeostasis. In view of the data presented here, we propose that abnormal ciliary polycystin-2 functions can lead to compromised fluid sensing which will further impair synthesis of NO, a mediator for other downstream signaling pathways in smooth-muscle relaxation.
Supplementary Material
Acknowledgments
We thank Charisse Montgomery for editorial review of the manuscript. The authors are also grateful to Wendy Boone and Drs Keith Crist, Michael Rees, Matthew Rutter, and Robert Booth for assistance in human tissue collection. We extend our gratitude to the Yale Polycystic Kidney Disease Research Center (DK57328) for providing the Pkd2 mice.
Sources of Funding
This work was supported by American Heart Association Grant 0630257N; NIH grants HL084451 and DK080640; and, in part, by the University of Toledo research programs, including the deArce Memorial Endowment Fund and the University Research Awards Fellowships Mini-Grants Program.
Footnotes
This work was presented in part at the 2008 Federation of American Societies for Experimental Biology Summer Research Conference on Polycystic Kidney Disease, July 27 to August 1, 2008, Snowmass Village, Colo.
Disclosures
None.
References
- 1.Boucher C, Sandford R. Autosomal dominant polycystic kidney disease (ADPKD, MIM 173900, PKD1 and PKD2 genes, protein products known as polycystin-1 and polycystin-2) Eur J Hum Genet. 2004;12:347–354. doi: 10.1038/sj.ejhg.5201162. [DOI] [PubMed] [Google Scholar]
- 2.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 3.Hateboer N, Veldhuisen B, Peters D, Breuning MH, San-Millan JL, Bogdanova N, Coto E, van Dijk MA, Afzal AR, Jeffery S, Saggar-Malik AK, Torra R, Dimitrakov D, Martinez I, de Castro SS, Krawczak M, Ravine D. Location of mutations within the PKD2 gene influences clinical outcome. Kidney Int. 2000;57:1444–1451. doi: 10.1046/j.1523-1755.2000.00989.x. [DOI] [PubMed] [Google Scholar]
- 4.Hateboer N, v Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, Torra R, Breuning M, Ravine D. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet. 1999;353:103–107. doi: 10.1016/s0140-6736(98)03495-3. [DOI] [PubMed] [Google Scholar]
- 5.Wu G, Markowitz GS, Li L, D’Agati VD, Factor SM, Geng L, Tibara S, Tuchman J, Cai Y, Park JH, van Adelsberg J, Hou H, Jr, Kucherlapati R, Edelmann W, Somlo S. Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nat Genet. 2000;24:75–78. doi: 10.1038/71724. [DOI] [PubMed] [Google Scholar]
- 6.Lu CJ, Du H, Wu J, Jansen DA, Jordan KL, Xu N, Sieck GC, Qian Q. Non-random distribution and sensory functions of primary cilia in vascular smooth muscle cells. Kidney Blood Press Res. 2008;31:171–184. doi: 10.1159/000132462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torres VE, Cai Y, Chen X, Wu GQ, Geng L, Cleghorn KA, Johnson CM, Somlo S. Vascular expression of polycystin-2. J Am Soc Nephrol. 2001;12:1–9. doi: 10.1681/ASN.V1211. [DOI] [PubMed] [Google Scholar]
- 8.Ong AC, Ward CJ, Butler RJ, Biddolph S, Bowker C, Torra R, Pei Y, Harris PC. Coordinate expression of the autosomal dominant polycystic kidney disease proteins, polycystin-2 and polycystin-1, in normal and cystic tissue. Am J Pathol. 1999;154:1721–1729. doi: 10.1016/S0002-9440(10)65428-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature. 2000;408:990–994. doi: 10.1038/35050128. [DOI] [PubMed] [Google Scholar]
- 10.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 11.McGrath J, Somlo S, Makova S, Tian X, Brueckner M. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell. 2003;114:61–73. doi: 10.1016/s0092-8674(03)00511-7. [DOI] [PubMed] [Google Scholar]
- 12.Nauli SM, Kawanabe Y, Kaminski JJ, Pearce WJ, Ingber DE, Zhou J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation. 2008;117:1161–1171. doi: 10.1161/CIRCULATIONAHA.107.710111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsiokas L, Kim E, Arnould T, Sukhatme VP, Walz G. Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc Natl Acad Sci U S A. 1997;94:6965–6970. doi: 10.1073/pnas.94.13.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pei Y, Watnick T, He N, Wang K, Liang Y, Parfrey P, Germino G, St George-Hyslop P. Somatic PKD2 mutations in individual kidney and liver cysts support a “two-hit” model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1999;10:1524–1529. doi: 10.1681/ASN.V1071524. [DOI] [PubMed] [Google Scholar]
- 15.Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87:979–987. doi: 10.1016/s0092-8674(00)81793-6. [DOI] [PubMed] [Google Scholar]
- 16.Boo YC, Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am J Physiol. 2003;285:C499–508. doi: 10.1152/ajpcell.00122.2003. [DOI] [PubMed] [Google Scholar]
- 17.Burnstock G. Release of vasoactive substances from endothelial cells by shear stress and purinergic mechanosensory transduction. J Anat. 1999;194(pt 3):335–342. doi: 10.1046/j.1469-7580.1999.19430335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomes P, Srinivas SP, Van Driessche W, Vereecke J, Himpens B. ATP release through connexin hemichannels in corneal endothelial cells. Invest Ophthalmol Vis Sci. 2005;46:1208–1218. doi: 10.1167/iovs.04-1181. [DOI] [PubMed] [Google Scholar]
- 19.Nauli SM, Zhou J. Polycystins and mechanosensation in renal and nodal cilia. Bioessays. 2004;26:844–856. doi: 10.1002/bies.20069. [DOI] [PubMed] [Google Scholar]
- 20.Kolb RJ, Nauli SM. Ciliary dysfunction in polycystic kidney disease: an emerging model with polarizing potential. Front Biosci. 2008;13:4451–4466. doi: 10.2741/3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Praetorius HA, Spring KR. A physiological view of the primary cilium. Annu Rev Physiol. 2005;67:515–529. doi: 10.1146/annurev.physiol.67.040403.101353. [DOI] [PubMed] [Google Scholar]
- 22.Hierck BP, Van der Heiden K, Alkemade FE, Van de Pas S, Van Thienen JV, Groenendijk BC, Bax WH, Van der Laarse A, Deruiter MC, Horrevoets AJ, Poelmann RE. Primary cilia sensitize endothelial cells for fluid shear stress. Dev Dyn. 2008;237:725–735. doi: 10.1002/dvdy.21472. [DOI] [PubMed] [Google Scholar]
- 23.Nauli SM, Rossetti S, Kolb RJ, Alenghat FJ, Consugar MB, Harris PC, Ingber DE, Loghman-Adham M, Zhou J. Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J Am Soc Nephrol. 2006;17:1015–1025. doi: 10.1681/ASN.2005080830. [DOI] [PubMed] [Google Scholar]
- 24.Xu C, Rossetti S, Jiang L, Harris PC, Brown-Glaberman U, Wandinger-Ness A, Bacallao R, Alper SL. Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin localization and loss of flow-induced Ca2+ signaling. Am J Physiol Renal Physiol. 2007;292:F930–945. doi: 10.1152/ajprenal.00285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.James NL, Harrison DG, Nerem RM. Effects of shear on endothelial cell calcium in the presence and absence of ATP. FASEB J. 1995;9:968–973. doi: 10.1096/fasebj.9.10.7615166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.