

Abstract
Therapy for hepatitis C virus (HCV) infection is on the cusp of a new era. Until now, standard of care (SOC) therapy has involved interferon (IFN) and ribavirin. With the first successful phase 3 trials of specific targeted antiviral therapy for HCV (STAT-C) compounds, as well as three trials in progress giving the first glimpse of IFN-free combinations of STAT-C agents, this review looks ahead to the new classes of anti-HCV agents currently in clinical development. Successful pharmacologic control of HIV and TB frames the discussion, as well as consideration of the mutation frequency of HCV replication. Maximizing synergy between agents and minimizing cumulative toxicity will be critical to the design of future IFN-free STAT-C regimens.
Hepatitis C: the status of the epidemic and the standard of care
An estimated 140–170 million people worldwide have hepatitis C, between 2–3% of the global population.[1] Chronic hepatitis C virus (HCV) infection carries a 25% lifetime risk of cirrhosis and a smaller but significant risk of developing life-threatening hepatocellular cancer.[2] In the U.S., HCV is the leading indication for liver transplantation[3] and the most common cause of hepatocellular carcinoma.[4] HCV is a member of the family Flaviviridae with a 9.6-kb positive-sense, single-stranded RNA genome.[5] The HCV genome encodes a single polyprotein (~3,000 amino acids), which is processed by host and viral proteases into at least 10 mature viral proteins: core, E1 and E2 envelope proteins, the p7 ion channel protein, and the non-structural (NS) proteins NS2, NS3, NS4A, NS4B, NS5A, and NS5B (Figure 1).[6, 7] The viral life cycle has been treated in depth in multiple recent reviews[8, 9] and is outlined in Box 1 and Figure 2.
Figure 1.

Current and future targets for anti-HCV therapies, organized by target location in the HCV genome. The first-generation agents (IFN/RBV) are currently in use; second-generation agents (protease/polymerase inhibitors) are in advanced clinical development; third-generation agents and beyond may target a number of viral and/or host processes.
Box 1.
| Step | Most Relevant Molecules |
|---|---|
| 1: Attachment to cell surface. | E2, LDL-R/GAGs(?) |
| 2: Cell entry. | E1, E2, SR-BI, CD81, claudin-1, occludin |
| 3: Uncoating of single-stranded RNA genome. | |
| 4: Translation of HCV genome into polyprotein. | HCV RNA IRES (miR-122 target) |
| 5: Cleavage of polyprotein. | NS2, NS3/NS4A (proteases) |
| 6a: Assembly of membranous web-based replication complex. | NS4B, NS5A(?), cyclophilin, PI4KIII-α(?) |
| 6b: Replication of genome. | NS5B (polymerase), NS3 (helicase) |
| 7: Assembly of virions and maturation in the ER lumen and at the cell surface. | Core, p7(?), NS2, ApoB, MTP |
| 8: Release of virions into the circulation and further post-release maturation. | NS5A(?), ApoB, ApoE |
| Interference with innate immune system | NS3 (proteolysis of TRIF and MAVS), core (interference with JAK/STAT), NS5A (repression of IFN-γ, PKR binding), E2 (PKR binding) |
| Interference with adaptive immune system | E2 (mutation of hypervariable region), NS5A |
Figure 2.

HCV life cycle.[8, 9, 19, 99] 1: Attachment to cell surface. 2: Cell entry. 3: Uncoating of single-stranded RNA genome. 4: Translation of HCV genome into polyprotein. 5: Cleavage of polyprotein by NS3/NS4A protease. 6: Assembly of membranous web-based replication complex and replication of genome by NS5B's polymerase activity. 7: Assembly of virions and maturation in the ER lumen and transport to the cell surface. 8: Release of virions into the circulation and further post-release maturation.
The current standard of care (SOC) for HCV is a combination of pegylated interferon (PegIFN) and ribavirin (RBV),[10] agents that are not specific for HCV. Efficacy of this therapy ranges from 6–84%, depending on viral genotype, severity of liver disease, viral load and age at start of treatment, as well as host genetics. Indeed, recent genome wide association studies (GWAS) have revealed that single nucleotide polymorphisms (SNPs) in the promoter region of IL28B (the gene encoding interferon lambda-3) are strongly associated with response to SOC therapy (although the negative predictive value of the current IL12B region SNPs alone is still only about 20–30%) [11–13]. Side effects of standard therapy may include fevers, chills, sweats, edema, neutropenia, thrombocytopenia, and depression. For many patients, these side effects represent relative or absolute contraindications to even starting treatment, an extremely challenging barrier to completing treatment for those who start therapy, and at the very least a major burden for those able to tolerate a full year of standard treatment. In fact, a multicenter study of US veterans with chronic HCV infection found that only 32–41% were considered eligible to receive SOC[14]. Moreover, a 48-week course of standard therapy costs over $20,000,[15] not including hematopoetic growth factors used to counter side effects of SOC and nonpharmaceutical costs.
Multiple new classes of anti-HCV drugs are in various stages of development. These include NS3/4A protease inhibitors, NS5B polymerase inhibitors, inhibitors of the binding of the NS4B protein to RNA, and inhibitors of the multifunctional viral protein NS5A.[16] This article will not describe novel IFNs, which are the subject of a recent review.[17] This article will also not discuss agents that have failed or been halted or abandoned at preclinical or clinical stages of development, or novel algorithms for determining duration of SOC therapy[18]—all of which have been well reviewed elsewhere. In addition, for a more detailed treatment of virus-host interactions, the reader is referred to a recent review in this journal.[19] For classes of compounds with one or more candidates in clinical development, compounds at the preclinical stage will be omitted or treated only briefly, as will classes such as inhibitors of entry or assembly, for which relatively sparse published data exist. Instead, the discussion will focus on the present state of, and future prospects for, specific anti-HCV therapy, with an emphasis on new classes of small-molecule agents that either are the subject of recent clinical trials or are under preclinical evaluation. Since the field is rapidly evolving, the reader is referred to clinicaltrials.gov for additional information on trials that may have been registered after the date of this writing.
Resistance and other challenges to the success of therapy
A mathematical model has been developed that describes the emergence of resistant virus during therapy with STAT-C (Box 2, Figure 3).[20] Because of the high turnover rate of HCV (1012 virions per day), the high error rate of the NS5B polymerase (μ=10−5), and the size of the HCV genome (~104 nt), the circulating pool of virus is expected to contain every possible single and double mutant even in the absence of therapy. Mutation at yet another position is expected to emerge within the first few days of therapy, as the most resistant pre-existing virus expands to become the dominant quasispecies under selective pressure.[20] As a result, a successful combination regimen consisting entirely of STAT-C agents may need to pose a barrier to resistance of at least four separate concurrent mutations.
Figure 3.

Model of emergence of resistance to an HCV therapeutic regimen (reprinted with permission from Rong et al.[20]). T: susceptible (uninfected target) cells; Is: cells infected with drug-sensitive virus; Ir: cells infected with resistant virus; Vs: population of drug-sensitive virus; Vr: population of drug-resistant virus; s: rate of generation of new target cells from precursors; ρT: rate of proliferation of target cells; d: rate of death of uninfected cells; δ: rate of death of infected cells; ps: rate of production of new drug-sensitive virus by infected cells; pr: rate of production of new drug-resistant virus by infected cells; μ: rate of mutation from drug-sensitive to drug-resistant state; c: rate of clearance of circulating virus; β: rate of initial infection of susceptible cells; εs: success rate of infections with drug-susceptible virus (rate of progression to release of new virus); εr: success rate of infections with drug-resistant virus. Red x's represent the effect of drug on εs and εr.
Given the above considerations, there clearly is an urgent need for new anti-HCV agents. Fortunately, because of the development of invaluable tools – such as the HCV replicon and infectious clone[21–25] – enabling HCV molecular genetics, an exciting pipeline of very interesting potential drugs is emerging. Unfortunately, there is no convenient and fully faithful animal model of hepatitis C, although a variety of immunodeficient mouse models harboring transplanted human hepatocytes have been developed.[e.g., 26]. The benefits and drawbacks of these diverse systems have recently been reviewed in this journal.[19]
There are several ways in which the probability of emergence of resistance can be decreased: i) the use of at least four STAT-C agents, each acting by a different mechanism or at least exhibiting no cross-resistance, such that one active agent would always be available to “mop up” mutants resistant to the others. (This is the strategy most analogous to highly active antiretroviral therapy for HIV or to multidrug therapy for tuberculosis.) In order to be able to select a four-drug regimen for any given patient, even in the absence of transmitted resistant virus, a significantly larger number of agents will need to make it through clinical development, as some candidates may have side effect or drug-drug interaction profiles that may preclude their use in certain patient populations. ii) Targeting host functions upon which the virus depends (because the targeted genetic locus is not under the control of the virus, this could raise the barrier to developing resistance because evolving an effective “work around” may involve more than a simple mutation). iii) Using agents that exert selective pressure to decrease viral “fitness” (as reflected by replication rate) in order to reduce the number of potentially resistant mutants generated. (This strategy is also sometimes used in the selection of regimens for HIV.[27] iv) Addition of novel agents to SOC may also accelerate the impairment of replication, as could enhancement/restoration of endogenous immune mechanisms. v) Another strategy, complementary to the strategies above, would be to use synergistic combinations of agents that work together to decrease the residual HCV replication rate below the critical threshold for emergence of newly resistant mutations; criteria for predicting the in vivo significance of synergy have been described[28] and applied.[29]
The need to overcome resistance places significant constraints on the choice of drugs for an anti-HCV regimen. Just as important is the consideration of toxicity, and, as a corollary, the need to promote adherence to therapy. Although any individual anti-HCV drug may have a tolerable side-effect profile when administered alone, the combination of two or more drugs with overlapping side effects may make the combination either too toxic to be given safely or so poorly tolerated that adherence diminishes, allowing resistance to emerge. By analogy, the adherence-resistance (A-R) curve for HIV therapy has an inverse U-shape, such that failure to take even 15–20% of prescribed doses, e.g., due to gastrointestinal side effects, can lead to maximal selection for resistant mutants[30]. Therefore, agents with additive toxicities should usually not be included in the same regimen, and combinations with toxicities that potentially limit complete adherence or tolerability will be less attractive than more benign combinations. Synergy, in such a case, can become extremely valuable; the excess “potency capital” provided by synergy could be used to decrease the dose of the most toxic member of a regimen while maintaining sufficient antiviral potency. Finally, in light of host factors, such as IL28B region genotype, that may affect response to (or toxicity of) a particular agent or combination, some cocktails may become preferred or contraindicated in certain individuals.
Potential cocktail components
Protease (NS3/NS4A) inhibitors (Figure 2, step 5)
Multiple inhibitors of the NS3/NS4A protease are at various stages of preclinical and clinical development (Table 1a–c), and recent reviews have been published.[31, 32] The two compounds in phase 3 evaluation, telaprevir[33, 34] and boceprevir,[35, 36] have each recently been reviewed on their own, and have similar resistance profiles consistent with their similar mechanisms, albeit different major toxicities. Both drugs appear to be slated for thrice-daily dosing. Compounds currently being tested as part of IFN-free regimens in phase 2 trials include BMS-650032[37] and ITMN-191.[38]
Table 1a.
Virological data for protease inhibitors in clinical development.
| Name | Company | Dosing (mg) | Locus of Resistance | GT | Log drop in HCV-VL | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phase | IC50 (nM) | 155 | 156 | 168 | 36 | 54 | Other | mono | combo | ||||
| telaprevir (VX-950, MP-424) | Vertex | 750q8, 1125q12 | 3 | 7 | Y | Y | Y | Y | 1, 2, 4 | 4.0 (GT2) | 5.5 (GT2) | ||
| boceprevir (SCH 503034) | Schering | 800 tid | 3 | 14 | Y | Y | Y | Y | 170 | 1 | 2.06 | ||
| narlaprevir (SCH 900518) | Schering | 100–600 daily/r | 2 | 7 | Y | Y | 1, 2, 3 | 4.5 | |||||
| ABT-450 | Abbott/Enanta | 50–300 daily/r | 2 | 1 | |||||||||
| BI 201335 | Boehringer Ingelheim | 240 daily | 2 | Y | Y | 170 | 1 | 3 | 5.3 | ||||
| BMS-650032 | BristolMyers Squibb | 600q12 | 2 | 0.2–0.4 | Y | Y | 1, 4 | ||||||
| GS 9256 | Gilead | 300 daily, 75–200 bid | 2 | Y | Y | Y | 1 | 2.9 | |||||
| ITMN-191 (R7227, RG7227, RO5190591) | Intermune/Roche | 100–400 daily/r | 2 | Y | Y | 3.9 | 5.1 | ||||||
| TMC435 (TMC435350) | Tibotec | 200 daily | 2 | 13 | Y | Y | 80 | 1 | 3.9 | 5.3 | |||
| vaniprevir (MK-7009) | Merck | 300–600 bid | 2 | 0.06–1.4 | Y | Y | Y | 1, 2 | 4.6 | ||||
| ACH-1625 | Achillion | 300–500 bid | 1 | 4.2 | |||||||||
| IDX320 | Idenix | 1 | 0.8–23 | Y | Y | Y | 80 | ||||||
IC50: 50% inhibitory concentration in biochemical assays of protease; HCV-VL: hepatitis C virus viral load; mono: when used as monotherapy; combo: when used as one component of combination therapy, usually with SOC; GT: genotype; /r: with coadministration of 100mg daily of oral ritonavir to boost serum levels of the drug by altering hepatic metabolism; Y: mutation at locus detected in resistant virus
Table 1c.
Virological data for protease inhibitors in preclinical development.
| Name | Company | Locus of Resistance | GT | ||||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 (nM) | 155 | 156 | 168 | 36 | 54 | Other | |||
| IDX136 | Idenix | 1–3 | 1, 4 | ||||||
| IDX316 | Idenix | 1–3 | 1, 4 | ||||||
| ACH-2684 | Achillion | ||||||||
| AVL-181 | Avila | 0.4 | Y | Y | 43 | ||||
| AVL-192 | Avila | ||||||||
| EA-058 | Enanta | 0.04–16 | 1–4 | ||||||
| EA-063 | Enanta | 0.03–0.7 | 1–4 | ||||||
| Pheophytin a | 890 | Y | |||||||
IC50: 50% inhibitory concentration in biochemical assays of protease; GT: genotype; Y: mutation at locus detected in resistant virus.
Briefly, telaprevir, which is believed to be closer to approval than boceprevir, demonstrated a number needed to treat (NNT) of 4 to 5, when added to SOC in treatment-naïve patients (PROVE1 [NCT00336479] and PROVE2 [NCT00372385]), and an NNT of 3 when added to SOC in treatment-experienced patients (PROVE3 [NCT00420784]). The predominant adverse event was rash, which led to discontinuation of therapy in 7% of study participants in the PROVE1 and PROVE2 studies. Results from the Phase 3 studies ADVANCE (NCT00627926), ILLUMINATE (NCT00758043), and REALIZE (NCT00703118) are expected to be presented shortly.[34]
Boceprevir demonstrated an NNT of 3 when added to SOC in treatment-naïve patients (SPRINT-1 [NCT00423670]). Adverse events have included anemia and dysgeusia, as well as headache.[35] Data from the Phase 3 SPRINT-2 (NCT00705432) and RESPOND-2 (NCT00708500) trials are expected to be presented shortly, and a third Phase 3 trial (NCT00795431) has been registered.[36]
Although the NS3 protein also has a helicase activity, exploitation of this target has lagged behind development of protease inhibitors. Rapid emergence of virus resistant to protease inhibitors and significant side effects such as severe rash and anemia remain important hurdles for the most advanced members of this class, which were among the first to demonstrate in vivo the impossibility of using such agents as monotherapies.
Polymerase (NS5B) inhibitors: nucleoside and non-nucleoside (Figure 2, step 6)
Inhibitors of NS5B, the catalytic subunit of the HCV RNA-dependent RNA polymerase, are also at various stages of preclinical and clinical development (Table 2a–b). As for the NS3 protease, the availability of a crystal structure of NS5B and the precedence of protease and polymerase inhibitors being successfully developed against other viruses, has led to quite a number of candidate drugs. Several of these compounds have also recently been reviewed.[39] Polymerase inhibitors are further subclassified as either nucleoside analogues or non-nucleosides, with the former targeting the enzyme's active site and the latter targeting one of at least five allosteric binding sites on the polymerase and inducing a conformational change that inhibits polymerase activity.[39] Compounds tested as part of IFN-free regimens in phase 2 trials include the nucleoside analogue RG7128[38] and the non-nucleoside VCH-222 (renamed VX-222 with the purchase of Virochem by Vertex).[40]
Table 2a.
Virological data for NS5B (polymerase) inhibitors.
| Locus of Resistance | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name | Class | Company | Dosing (mg) | Phase | replicon EC50(nM) | 414 | 419 | 423 | 448 | 482 | 494 | 556 | Other | GT |
| ABT-072 | NNI | Abbott | 160 daily | 2 | 0.3–5.3 | Y | Y | Y | 316 | 1 | ||||
| ABT-333 | NNI | Abbott | 2 | 2–7 | Y | Y | Y | 316 | 1 | |||||
| ANA598 | NNI | Anadys | 200–400 bid | 2 | 0.5–52 | Y | 554 | 1 | ||||||
| filibuvir (PF-00868554) | NNI | Pfizer | 300–600 bid | 2 | 59 | Y | 422 | 1 | ||||||
| GS 9190 | NNI | Gilead | 40–240 bid | 2 | 0.6–2.5 | 1 | ||||||||
| IDX184 | Nuc | Idenix | 50–100 daily | 2 | ||||||||||
| PSI-7851 (PSI-7977) | Nuc | Pharmasset | 50–200 daily | 2 | 30 | 1, 4 | ||||||||
| RG7128 (RO5024048, prodrug of PSI-6130) | Nuc | Roche/Pharmasset | 1500 bid | 2 | 610 | 282 | 1–4 | |||||||
| silibinin (silybin) | NNI | Rottapharm-Madaus | 1400 iv daily ×2 or 20 mg/kg/d ×14 | 2 | 600 | 110, 285, 442, 498 | 1 | |||||||
| VX-222 (VCH-222) | NNI | ViroChem/Vertex | 750 bid | 2 | 12–23 | 1 | ||||||||
| VCH-759 | NNI | ViroChem/Vertex | 400–800 tid | 2 | 300 | Y | Y | Y | ||||||
| VCH-916 | NNI | ViroChem/Vertex | 200 tid / 300–400 bid | 2 | Y | Y | Y | Y | 1 | |||||
| BI 207127 | NNI | Boehringer Ingelheim | 400–800 tid | 1 | 1 | |||||||||
| IDX375 | NNI | Idenix | 1 | |||||||||||
| MK-3281 | NNI | Merck | 100–400q12 | 1 | 1b | |||||||||
Table 2b.
Clinical data for NS5B polymerase inhibitors.
| Adverse Events | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Name | mono | combo | SVR active | SVR SOC | Derm | HA | Const | GI | Psych | Renal | Other |
| ABT-072 | |||||||||||
| ABT-333 | 4.0 | Y | Y | Y | |||||||
| ANA598 | 2.4 | Y | |||||||||
| filibuvir (PF-00868554) | 2.1 | 4.6 | Y | Y | Y | Y | Y | ||||
| GS 9190 | Y | QT | |||||||||
| IDX184 | 0.74 | Y | Y | Y | |||||||
| PSI-7851 (single enantiomer is PSI-7977) | 1.0 | Y | |||||||||
| RG7128 (R7128, RO5024048, prodrug of PSI-6130) | 5.0 | 65% (13/20) | 60% (6/10) | ||||||||
| silibinin (silybin) | Y | ||||||||||
| VCH-222 | 3.7 | ||||||||||
| VCH-759 | 2.5 | Y | Y | Y | |||||||
| VCH-916 | 1.5 | Y | Y | ||||||||
| BI 207127 | 5.6 | Y | |||||||||
| IDX375 | |||||||||||
| MK-3281 | 3.8 | myoclonus | |||||||||
SVR active/SOC: percentage of participants in experimental therapy/SOC group achieving sustained virologic response; Derm: dermatologic adverse events; HA: headache; Const: constitutional (e.g., fever or fatigue); GI: gastrointestinal; Psych: psychiatric; Renal: elevation in kidney function markers; QT: prolongation of the QT interval on electrocardiography, potentially fatal in a subset of patients receiving other QT-prolonging drugs or with the congenital long QT syndrome; Y: adverse event reported in experimental therapy group.
Both RG7128 and VX-222 have been evaluated in twice-daily dosing regimens, although several other compounds are being studied as once-daily agents (Table 2a). Adverse effects of RG7128 have not been reported, but reports of studies of VX-222 describe mild-to-moderate adverse effects.[41] Further evaluation of RG7128 (e.g., INFORM-1 [NCT00801255]) and VX-222 (NCT01080222) in combination regimens is ongoing.
The silibinins, natural products first isolated from the milk thistle extract silymarin also deserve mention as milk thistle products in over-the-counter preparations are not infrequently used by patients with hepatitis [42]. Hypothesized to inhibit NS5B polymerase [43], its precise mechanism of action remains uncertain [44] but clinical evaluation is ongoing. A silibinin extract is also available in many countries (and as an investigational agent in the US) as an intravenous antidote to Amanita phalloides mushroom ingestion, and its successful use has been described at the case report level in an HIV/HCV coinfected patient.[45]
NS4B inhibitors (Figure 2, step 6)
The NS4B protein has multiple functionalities, including formation of the “membranous web” structure [46] believed to represent the viral replication platform, interaction with other NS proteins in the replication complex, GTP hydrolysis, and RNA binding.[47] A microfluidic assay has been used to demonstrate that the NS4B protein uses an arginine-rich-like motif to bind specifically to the 3' terminus of the negative strand of HCV RNA, and to conduct high-throughput screening for inhibitors of this interaction.[16] One of the compounds identified by this screen, clemizole hydrochloride, is a clinically well-tolerated H1 histamine receptor antagonist that has seen extensive use as an antipruritic. Clemizole has demonstrated dramatic in vitro synergy with the protease inhibitors boceprevir and telaprevir.[29] This is a marked contrast to most combinations of anti-HCV agents to date, which typically display simple additivity (and even in some cases antagonism). Indeed, clemizole displayed additivity with IFN, RBV, and polymerase inhibitors, highlighting the specificity and uniqueness of the strong clemizoleprotease inhibitor synergy.[29] Such synergy usually has an underlying biologic basis, and in this case might reflect the genetic evidence supporting important interactions between NS4B and NS3.[29, 48] In vitro cross-resistance does not occur between clemizole and boceprevir.[29] Several phase 1b studies of clemizole are currently underway, (CLEAN studies, typified by NCT00945880 [CLEAN-1]), evaluating clemizole in populations with different GT distributions; while tolerability of therapy is the primary endpoint of these phase 1 studies, data are also being collected on the efficacy of clemizole as lead-in monotherapy and in combination with SOC or with other novel agents.
Anguizole, a compound first identified as an NS4B binder with anti-HCV replication activity, targets the second amino (N)-terminal amphipathic helix (AH2) of NS4B and alters its cellular distribution, impairing formation of the “membranous web” and interfering with replication.[47] Other small molecules against the NS4B AH2 target [49] and their derivatives are in preclinical development.
NS5A inhibitors (Figure 2, step 6, as well as other roles)
From a lead identified in an unbiased high-throughput screening effort of a million-compound corporate library, BMS-790052 was identified as an inhibitor of HCV replication in HCV replicon and cell-culture systems at picomolar concentrations, with a resistance profile mapping to mutations in NS5A.[50] Additive to synergistic effects were documented between BMS-790052 and multiple other drug classes, including IFN, NS3 inhibitors, and nucleoside and non-nucleoside NS5B inhibitors. In a dose-escalation trial in individuals with genotype (GT) 1a/1b infection, a single 100 mg dose achieved mean maximal reduction in HCV titers of 3.6 log10, with duration of effect greater than 144 h.[50] The addition of BMS-790052 to SOC raised rapid virologic response (RVR) rates from 1/12 (SOC alone) to 5/12 (3 mg daily), 11/12 (10 mg daily), or 10/12 (60 mg daily) and raised complete early virologic response (cEVR) rates from 5/12 (SOC alone) to 7/12 (3 mg daily) or 10/12 (10 mg daily and 60 mg daily).[51] Studies of the combination of BMS-790052 with SOC (NCT00874770, NCT01016912, NCT01017575, and NCT01125189 [HEPCAT]), as well as a study of the combination of BMS-790052 and BMS-650032 with or without SOC agents (NCT01012895) are underway.[50]
PPI-461 is another NS5A inhibitor currently in preclinical development. It shares with BMS-790052 a high therapeutic index in vitro and an apparent high mutational barrier to resistance. Reports from animal absorption, distribution, metabolism, excretion and toxicity (ADME/Tox) testing suggest once-daily dosing in humans may be possible.[52]
Inhibitors of p7 (Figure 2, possibly step 7)
Antiviral strategies targeting the 63-amino acid residue protein p7, composed largely of two amphipathic α-helices, have recently been reviewed.[53] Briefly, the p7 protein forms a hexameric barrel-shaped cation channel, and small-molecule inhibition of either oligomerization or ion flow blocks production of HCV virions in the infectious cell culture system, producing up to one log reduction in titer in a round of infection.[54] Small-molecule inhibitors of p7 include compound classes developed as antivirals against other viruses: adamantanes, n-nonyl nojirimycin derivatives, and amiloride derivatives. Amantadine, one of the adamantanes, in a meta-analysis appeared to increase SVR rates for nonresponders to SOC, but not for treatment-naïve patients or relapsers, when combined with SOC.[55] A 12-week trial of monotherapy with UT-231B, an imino sugar with anti-p7 activity (NCT00069511), failed to show antiviral activity. On the other hand, a phase 1b/2a trial of the amiloride analogue BIT-225 showed no serious adverse effects and a modest reduction in HCV-VL.[53]
Cyclophilin inhibitors (Figure 2, step 6)
Significant interest has arisen in analogues of cyclosporine A (CsA), believed to act by inhibiting cyclophilins (Cyps), as potential HCV therapeutics, and these have been reviewed recently.[56] Several models have been proposed for the mechanism of action of Cyp inhibitors:[56] they may block an interaction between Cyps and NS5A; they may block an interaction between Cyps and NS5B; they may block recruitment of NS5B to the replication complex; or they may interfere with proteolytic cleavage of the NS5A-NS5B junction within the HCV polyprotein precursor. A recent mammalian two-hybrid study demonstrated interaction between the peptidyl-prolyl isomerase pocket of CypA and NS5A.[57]
Although inhibition of HCV replication was first observed with CsA itself, the immunosuppressive side effects of CsA precluded its use in HCV therapy, and analogues lacking immunosuppressive effect but retaining anti-HCV effect (possibly by blocking Cyp's chaperoning or peptidyl-prolyl isomerase activity) are in clinical development.[56]
Among the cyclophilin inhibitors, alisporivir, formerly known as Debio-025, is the subject of a recent review.[58] Preclinical studies on replicon cell lines showed additive to slightly synergistic effects with IFN, RBV, or STAT-C drugs, with resistance mutations clustering in the NS5A gene.[59] In a dose-ranging study combining PegIFN with 200–1000 mg doses of alisporivir (loading with twice-daily dosing for one week, then daily dosing to complete a four-week course), alisporivir was additive with PegIFN for GT 1/4 but did not appear to confer additional benefit for GT 2/3, meeting criteria for RVR in 8/12 patients with GT 1/4 and in 5/6 patients with GT 2/3 at the 1000 mg dosing level.[60] Adverse effects considered treatment-related include hypertension, hyperbilirubinemia (believed due to inhibition of a biliary canalicular transporter), platelet reduction, nausea, headache, and fatigue.[60] An ongoing phase 2a trial of alisporivir combined with PegIFN±RBV for the first four weeks of therapy in prior nonresponders to PegIFN+RBV (NCT00537407) has demonstrated smaller HCV viral load (VL) reductions in treatment-experienced patients than in treatment-naïve patients at four weeks.[61] A phase IIB trial of alisporivir + PegIFN+RBV is also ongoing (NCT00854802).[58]
Another Cyp inhibitor, NIM811, has demonstrated tolerability in a two-week proof-of-concept study in healthy volunteers and HCV-infected patients.[62] In patients with relapse after SOC, NIM811+PegIFN achieved an HCV-VL decrease of 2.78 log after 14 days, as compared to a decrease of 0.58 log with PegIFN monotherapy.[63] Decrease in platelets was more pronounced in the NIM811+PegIFN arm than in the PegIFN arm.
A third Cyp inhibitor, SCY-635, was reported to have some synergy with IFN and additivity with RBV in vitro.[64] A phase 1b study in GT1 patients (11/20 treatment-naive) found no safety issues and demonstrated a decrease in HCV-VL only at the highest dose, 300 mg thrice daily (tid) (median nadir, 2.20 log decrease).[65] Follow-up genotype analysis of patients receiving 300 mg tid demonstrated single point mutations in NS5B in two patients, unassociated with virologic rebound.[66]
Modulators of the innate immune response
In order for HCV infection to become chronic, it must evade elimination by the innate and adaptive immune responses.[9, 19] The virus has developed multiple immune evasive mechanisms, several of which are targets for therapeutic strategies under development.
Nitazoxanide
The thiazolide drug nitazoxanide (NTZ), which is approved in the U.S. to treat specific parasitic gastroenteritides, was serendipitously observed to decrease aminotransferase levels in HCV/HIV coinfected patients. This observation led to evaluation of NTZ and analogues as an anti-HCV agent,44 and a recent review of this line of research has been written [67]. The antiviral mechanism of NTZ involves stimulation of the host response via activation of the double-stranded RNA activated protein kinase (PKR),[68] a classical antiviral effector of IFN, and in vitro replicon studies showed that NTZ pretreatment sensitized the virus to IFN.[69] A subsequent randomized placebo-controlled trial of NTZ monotherapy in GT 4 infected individuals demonstrated sustained virologic response (SVR) in 4/23 (17%) with only virological responders showing a significant decrease in HCV-VL.[70]
Nitazoxanide was then further evaluated at 500 mg twice daily (bid) in GT 4 infected individuals in combination with PegIFN±RBV in a three-arm randomized controlled trial (RCT) in Egypt (STEALTHC-1, NCT00421434).[71] Individuals not in the SOC comparator arm received a 12-week lead-in with NTZ, to which PegIFN±RBV was added for the remaining 36 weeks of therapy. Among treatment-naïve individuals, the triple therapy arm achieved SVR in 79% of patients, as compared to 50% for SOC (p=0.023) and 61% for PegIFN+NTZ (p>0.05 vs. SOC). In a separate stratum of treatment-experienced individuals, who were excluded from the SOC arm, 1/12 achieved SVR with PegIFN+NTZ and 3/12 achieved SVR with triple therapy.[72] Adverse events were similar across groups, with the exception of an increased rate of anemia in the arms receiving RBV.[71] A group of 44 patients with similar characteristics, recruited later and given a four-week lead-in with NTZ followed by 36 weeks of PegIFN+NTZ, showed similar rates of RVR, early virologic response (EVR), and ETR to the groups given 12-week NTZ lead-in in STEALTHC-1.[73] A double-blinded RCT of 64 treatment-experienced individuals with GT 1 (STEALTHC-2, NCT00495391) administered NTZ or placebo for a four-week lead-in period, followed by 48 weeks of triple therapy or SOC, reported an SVR rate of 7% (3/42) in the NTZ group and none in the placebo group.[74] A double-blinded RCT of the same regimen in 110 treatment-naïve individuals with GT 1 (STEALTHC-3, NCT00637923) reported preliminary SVR12 (sustained virologic response measured at 12, as opposed to the standard 24, weeks after conclusion of therapy) rates of 44% (NTZ) versus 32% (placebo).[75]. An extended-release NTZ formulation has been developed, and second-generation thiazolides in pre-clinical development have been alluded to in the literature.[67]
Toll-like receptor (TLR) agonists
The rationale for, and early history of, the development of TLR agonists in chronic liver disease has been recently reviewed.[76, 77] Briefly, downstream signals proceeding from the recognition of dsRNA by TLR3 and RIG-I, ssRNA by TLR7, and non-methylated CpG-containing DNA by TLR9 appear to be blocked by HCV nonstructural proteins before they can activate the innate immune system and upregulate IFN production. Specifically, it has been demonstrated in vitro that TLR3 signaling is blocked by NS3/NS4A proteolysis of its downstream effector TRIF, and RIG-I signaling is blocked by NS3/NS4A proteolysis of its downstream effector MAVS (also called IPS-1).[9] The TLR7 and TLR9 signals are blocked by NS5A binding to their common downstream effector MyD88, also as demonstrated in vitro.[76] Plasmacytoid dendritic cells, which were recently shown to respond to HCV-infected hepatocytes by producing interferon in a TLR7-mediated process, may therefore be an important activator of the innate immune response to HCV.[78]
Since the demonstration that isatoribine, an agonist of TLR7, induced a decrease in HCV-VL, presumably through immune activation,[79] several candidate TLR7 and TLR9 agonists have been nominated for clinical development. Agonists of TLR7 have been administered as oral prodrugs both to improve bioavailability and to reduce side effects from activation of gastrointestinal immune tissue.[77]
One TLR7 agonist currently in clinical development is the oral prodrug ANA773, which was recently the subject of a phase 1 double-blind placebo-controlled dose-escalation monotherapy study.[80] Patients of any genotype were enrolled regardless of prior receipt of SOC and received every-other-day dosing of ANA773 or placebo for 28 days. Adverse events were dose-dependent and included mild to moderate flu-like symptoms, and the median maximum decline in HCV-VL was 0.79 log in the highest-dose group for which data were available (1600 mg), compared to 0.30 log in the placebo group (p=0.04). Data from the 2000 mg dose group are pending.
An injectable TLR9 agonist, IMO-2125, is the subject of two phase 1 placebo controlled studies: IMO-2125-001 (NCT00728936), a monotherapy dose-escalation study in prior nonresponders to SOC,[81] and IMO-2125-201 (NCT00990938), a dose-escalation study in combination with RBV in therapy-naïve GT 1 patients. Results from several dose cohorts of the monotherapy study were recently presented.[81] Adverse events included brief mild to moderate flu-like symptoms, headache, and injection site reactions. Post-dose serum IFN-α increased in a dose-dependent fashion and with repeated doses, and the median nadir HCV-VL in the highest dose group (0.32 mg/kg/wk) represented a roughly 1.4 log decline from baseline. Further planned development includes a 12-week combination trial with RBV and an investigation of twice-weekly dosing.[81]
Modulators of the adaptive immune response
Failure of the adaptive immune system to adequately control HCV infection is the hallmark of chronic hepatitis C, characterized by immune cell dysfunction and inadequate responses at multiple levels. For example, evasion of the adaptive immune response by HCV can be achieved by simple mutation of epitopes allowing for escape.[82] CD8 T-cell exhaustion is also observed, associated with upregulation of the programmed death 1 (PD-1)/PD-L1 regulatory pathway; blockade of this pathway in blood-derived CD8 T cells using anti-PD-L1 antibodies results in reversal of the exhaustion phenotype in vitro.[83] However, such blockade using anti-PD-1 antibodies alone was ineffective in liver-derived HCV-specific CD8 T cells; the exhaustion phenotype was only reversed with the simultaneous blockade of either CTLA-4 [84] or CD137 [85]. While the therapeutic benefits of anti-PD-1 or anti-PD-L1 antibodies may need to be balanced against the potential risk of autoimmunity, a recently completed clinical trial of MDX-1106, a human monoclonal anti-PD-1 antibody (NCT00703469), may shed light on this question.[82]
Some other promising approaches in early development
Virus particle lysis (Figure 2, step 8)
A peptide derived from the N-terminal domain of NS5A, the NS5A AH peptide, is capable of rupturing HCV virions as well as other viruses [86], a property that appears to depend on the target particle size.[87] Clinical studies of AH peptide have not yet been reported, but a more interesting application may be in extracorporeal clearance of viremia at the time of liver transplantation.
Host lipid metabolism interference (Figure 2, steps 1, 2, 7, and 8)
The microenvironment in which HCV replication occurs is believed to be enriched for specific lipids, including cholesterol, sphingomyelin, phosphatidylinositol-4-phosphate (PI4P) and phosphatidylinositol-4,5-bisphosphate (PI4,5P2) lipids.[88–90] The statins, inhibitors of de novo cholesterol synthesis, have demonstrated anti-HCV activity in cell culture[91], but this may be due at least in part to inhibition of protein geranylgeranylation rather than cholesterol synthesis.[92]
The synthetic machinery for sphingomyelin and PI4P lipids is important to HCV replication.[88, 91, 93–97] as is the ability of HCV proteins to specifically bind to a key metabolite of PI4P, PI4,5P2.[90] Sphingomyelin synthesis can be blocked by inhibiting serine palmitoyltransferase using, for example, the natural product myriocin in vitro, which was reported to attenuate HCV replication in a synergistic fashion with IFN or simvastatin.[88] A more recently described serine palmitoyltransferase inhibitor, NA 808, reduced HCV-VL in a humanized chimeric mouse model and appeared to enhance the effect of PegIFN in this model.[98] Compounds targeting the PI4P synthesis pathway have not yet been developed but represent an attractive potential approach.
The metabolic machinery for lipoprotein particles is also believed to play a crucial role in entry, assembly, and maturation of HCV virions.[99] Briefly, HCV particles may initially associate with the host low density lipoprotein (LDL) receptor on target hepatocytes and then with the lipoprotein receptor SR-BI. Assembly of core around the newly replicated genomic RNA also takes place in a lipid-rich environment at the ER, and proteins important in the synthesis of very low density lipoprotein (VLDL) particles, including microsomal triacylglycerol transfer protein (MTP), ApoB, and ApoE, appear to be essential for the assembly, release, and maturation of HCV particles.[99] Interference with these processes may be a fruitful area for development of new classes of anti-HCV agents.
Inhibition of the initiation of translation (Figure 2, step 4)
Translational initiation for the HCV polyprotein takes place at an internal ribosome entry site (IRES). The microRNA element miRNA-122, preferentially expressed in hepatocytes and in the HCV-susceptible Huh7 cell line, appears to play a role in the stimulation of translation (as well as other aspects of the viral life cycle), and the HCV IRES contains at least two miRNA-122 binding sites.[100] Sequestration of miRNA-122 either by protected antisense oligonucleotides in vitro[100] or by the antisense locked nucleic acid (LNA) analogue SPC3649 in primates[101] suppresses viral replication and may provide a novel mechanism for future therapy in humans.
Therapeutic vaccination
Several immunotherapeutic approaches to HCV are also being pursued. The peptide vaccine IC41 had low immunogenicity and produced at best transient 1–2 log reductions in HCV RNA levels when used as monotherapy,[102] and results of a phase 2 trial adding IC41 to SOC for the last 20 weeks of a 48-week course were inconclusive.[103] Discouraging efficacy results of a recent phase I trial of an autologous monocyte therapy using T-cell epitopes aimed at core, NS3, and NS4B suggest that immunologic response to current vaccination approaches may not, by itself, be sufficient to alter the course of HCV infection,[104] but the role of therapeutic vaccines, either alone or in combination with other agents, remains to be fully explored.
Projected directions of future development
In the short term, the greatest therapeutic benefit may be gained from increasing the efficacy of SOC regimens. In this first phase, the challenge is to maximize cure rates across genotypes. The addition of novel agents to SOC, such as protease or polymerase inhibitors or other agents of new classes, represents the most obvious route. Even this may be difficult to achieve if incremental increases in efficacy are offset by significant toxicities of new agents. Moreover, until a collection of such new agents is available, extreme caution will need to be exercised. On the one hand, we now have,[11–13] and are soon to have more, powerful genetic tests that can help select the patients most appropriate to receive so-called triple therapy (SOC plus a direct acting antiviral such as a protease inhibitor). The predictive power of these genetic tests can be further augmented by supplementing with levels of certain serum markers (e.g. IP-10)[105] and clinical factors already known to influence successful response to SOC. On the other hand, administering triple therapy to patients who are destined to be null-responders to SOC may be akin to functional monotherapy with the new agent, putting the patients at high risk for selecting resistant virus and treatment failure. The latter could be compounded by these patients now being precluded from receiving the full benefits of a future drug cocktail dependent upon that new agent. To avoid such pitfalls (and potential liability) physicians and drug manufacturers will need to appropriately inform patients who have so patiently waited for improved therapies and avoid premature treatment of patients with regimens that are not suited to their individual cases.
Another critical consideration is that many of the new agents have suboptimal or no effect on genotypes predominant outside the industrialized world (i.e. nonGT 1), where the greatest burden of HCV lies. The ultimate goal, however, to be met by the second phase of advances in anti-HCV therapy, is to replace the most toxic or onerous components of SOC (e.g. IFN) with less toxic, all-oral regimens. As multiple classes of agents reach clinical maturity, it appears likely that we will soon have the cumulative raw antiviral suppressive power to attempt to make this goal a reality.
In order for a future STAT-C cocktail to effectively block replication and tip the balance away from the development of resistance and toward clearance of infection, several conditions are likely to be required. First, given that cross-resistance within a class is common, the components of a cocktail are likely to require complementary mechanisms of action, rather than combining multiple agents of the same class. Second, given the high rate of generation of mutant viral genomes, a premium might be placed on synergy between agents as well as the inclusion of agents that interact with host targets.
Finally, given that an effective STAT-C cocktail may be composed of at least 3–4 drugs, attention must be paid to cumulative toxicity. Pairs of agents with overlapping or even synergistic toxicities will be more difficult to combine in a single regimen, and the most useful agents will be those with minimal or no significant toxicities or adverse interactions. Indeed, in the context of a multi-component cocktail, a marginal increase in efficacy could be supplemented by adding another drug with a distinct mechanism of action, but toxicity that is additive with that of other drugs could doom the regimen altogether. Therefore, it becomes less important that any single component potently reduces the viral load than that it provides for a novel mechanism of action and minimal toxicity. Moreover, synergy between two components may enable the use of lower doses of the more toxic component, thus further reducing overall toxicity of the regimen. With potential toxicity a significant barrier to eligibility for SOC, and with a large proportion of discontinuations of SOC due to toxicity, a premium will be placed on agents that enable dose decreases for other agents in the regimen.
Fortunately, with multiple agents under development in a broad variety of classes, it is likely that suitable regimens composed entirely of novel oral agents will emerge in the coming years. While speculation on the future approval of pharmaceuticals is prone to significant uncertainties, some general projections may be made.
In the short term (2–3 years), physicians who treat chronic HCV infection will face encouragement from multiple sources to place their patients on an interferon-containing regimen supplemented by one or two new agents (Figure 4, center bar). As mentioned above, it is crucial to choose candidates for IFN-containing hybrid regimens both cautiously and strategically. That is, individuals fated to respond poorly to IFN-containing regimens, due either to predictable (i.e., viral genotype, host IL28B genotype) or unforeseen factors (i.e., toxicity requiring discontinuation of therapy or failure to achieve SVR), could experience functional monotherapy or two-drug therapy. Since protease and/or polymerase inhibitors are expected to remain as backbone elements of IFN-sparing regimens, this could make choosing a salvage regimen more difficult.[18]
Figure 4.
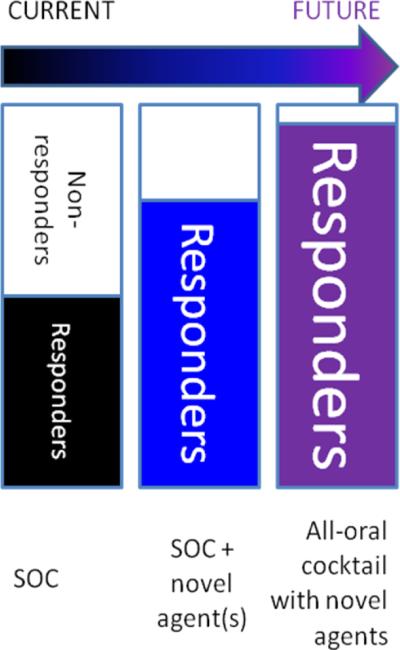
Projected evolution of anti-HCV regimen composition. As response rates increase with new therapeutic cocktails, the number of options is expected to increase as well, and as the most toxic components of SOC regimens (i.e. IFN) are eliminated, discontinuation due to toxicity should also decrease.
In the intermediate future (5–10 years; Figure 4, rightmost bar), we foresee the availability of multiple new classes of orally bioavailable agents, with the expected attendant increase in efficacy and decrease in toxicity.
Since specific anti-HCV therapies entered the development pipeline, there has been a growing trend to “warehouse” patients believed unlikely to respond to, or to tolerate, therapy with SOC pending the availability of new agents.[18] We feel, however, that attention should be paid not only to the “inventory” of patients awaiting therapy, but also to the inventory of novel drug classes available. We favor reserving the IFN-containing hybrid regimens for patients whose indication for therapy is urgent enough that awaiting the availability of an IFN-sparing regimen would pose an unacceptable risk of disease progression, and who are projected to tolerate and respond to an IFN-containing regimen. A similar strategy was recently employed for many individuals with highly resistant HIV, who were placed on a combination of multiple newly approved agents (darunavir, raltegravir, and maraviroc) rather than adding any of these agents singly to a failing background regimen. With such unprecedentedly favorable prospects for novel agents against HCV, the matching of patients to drug regimen cocktails will become a matter of strategy as well as a matter of timing.
Box 2. Model of HCV resistance to therapy[20].
a: deriving the frequency of mutations on a per-genome basis from the polymerase error rate
Pi-mutant: probability of i single-nucleotide substitutions occurring in a single replication event
L: length of the viral genome
μ: probability of a substitution occurring at the replication of a given nucleotide
Rong et al. calculate that P1=0.087, P2=0.0042, and P3=0.00013. Since 9.1×1011 virions per day are generated in the absence of therapy, 9.1×1011×P2 = 4.2×109, so each of the 4.1×108 possible double-substitution mutants is generated each day. With effective therapy (an initial 5-log decline in HCV VL), 9.1×106 new virions are generated daily, so each of the 2.9×104 possible single-substitution compensatory mutants is generated among the 9.1×106×P1 = 8.7×105 single-mutant virions produced daily. Assuming that a single substitution is sufficient to confer resistance to a single agent, mutants resistant to any dual therapy are likely present in the therapy-naïve viral pool, and further mutation can confer resistance to a third agent even with a 5-log suppression of replication. Therefore, a barrier of at least four nucleotide substitutions (translating, under this assumption, to a 4-drug regimen) is necessary in order to prevent emergence of resistance.
b: Differential equations describing the model of resistance emergence.
T: susceptible (uninfected target) cells
Is: cells infected with drug-sensitive virus; Ir: cells infected with resistant virus
Vs: population of drug-sensitive virus; Vr: population of drug-resistant virus
s: rate of generation of new target cells from precursors
ρT: rate of proliferation of target cells
N: cells not prone to infection
Tmax: hepatocyte carrying capacity of the liver
d: rate of death of uninfected cells; δ: rate of death of infected cells
ps: rate of production of new drug-sensitive virus by infected cells; pr: rate of production of new drug-resistant virus by infected cells
μ: rate of mutation from drug-sensitive to drug-resistant state
c: rate of clearance of circulating virus
β: rate of initial infection of susceptible cells
εs: success rate of infections with drug-susceptible virus (rate of progression to release of new virus); εr: success rate of infections with drug-resistant virus
Adapted from Rong et al.[20] with permission.
Table 1b.
Clinical data for protease inhibitors in clinical development.
| Name | Treatment naïve | Treatment experienced | Adverse Events | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Trial | SVR active | SVR SOC | Trial | SVR active | SVR SOC | Derm | HA | Const | GI | Psych | Bili | Other | |
| telaprevir | PROVE1 | 61% | 41% | PROVE3 | 53% | 14% | Y | ||||||
| PROVE2 | 69% | 46% | |||||||||||
| boceprevir | SPRINT-1 | 75% | 38% | Y | Y | Y | dysgeusia | ||||||
| narlaprevir | 81% | 38% | 38% | 38% | Y | Y | Y | ||||||
| ABT-450 | |||||||||||||
| BI 201335 | Y | Y | |||||||||||
| BMS-650032 | |||||||||||||
| GS 9256 | Y | Y | |||||||||||
| ITMN-191 | Y | Y | Y | dysgeusia | |||||||||
| TMC435 | Y | Y | Y | ||||||||||
| vaniprevir | Y | Y | |||||||||||
SVR active/SOC: percentage of participants in experimental therapy/SOC group achieving sustained virologic response; Derm: dermatologic adverse events; HA: headache; Const: constitutional (e.g., fever or fatigue); GI: gastrointestinal; Psych: psychiatric; Bili: elevation in biliary markers; Y: adverse event reported in experimental therapy group.
Table 3.
Reported synergistic relationships between selected anti-HCV agents.
| IFN | RBV | telaprevir | IDX316 | PSI-7977 | PSI-6130 (R7128) | IDX375 | ANA773 | clemizole | |
|---|---|---|---|---|---|---|---|---|---|
| telaprevir | sa | X | + | S | |||||
| boceprevir | 0 | S | |||||||
| narlaprevir | s | ||||||||
| vaniprevir | + | s | |||||||
| ANA598 | s | s | s | s | |||||
| IDX184 | s | s | s | s | |||||
| clemizole | + | + |
S, strong synergy predictive of likely in vivo synergy; s, possible, or minor but significant, synergy;
+, additivity or insignificant synergy; X, antagonism. Blank entries represent combinations for which no published data could be located, although the field is evolving rapidly and more precise quantitative data may become available. Similarly, agents not reported to be in active clinical or preclinical development have been omitted from this table in the interest of providing a snapshot of the agents with greatest clinical utility.
Acknowledgments
Financial support: This work was supported by NIH Training Grant T32 DK007056 (to MAG), NIH RO1 AI087917, NIH 1R41AI088793, and a Burroughs Wellcome Fund Clinical Scientist Award in Translational Research (to JSG).
JSG is a consultant to, receives research funding from, or has an equity interest in: Eiger BioPharmaceuticals, Genentech, Roche, Romark Laboratories, and Merck.
Glossary Box
- Complete early virologic response (cEVR)
undetectable HCV-VL at week 12 of therapy
- Early virologic response (EVR)
HCV-VL that is either at least 2 log10 decreased from baseline or undetectable at week 12 of therapy; failure to achieve EVR is considered by many to be grounds for early cessation of therapy in GT1/4, as negative predictive value of EVR is better than its positive predictive value
- End-of-treatment response (ETR)
undetectable HCV-VL at conclusion of therapy
- Genotype (GT)
there are six major genotypes of HCV, of which GT1 is the most common worldwide; GT1/4 have lower rates of response to SOC therapy than do GT2/3.
- JFH-1
a patient-derived GT2a strain of HCV RNA that can be used to infect cultured human hepatoma (Huh7.5) cells; allowing for evaluation of entry and assembly inhibitors.
- Membranous web
an ultrastructural alteration, visible by electron microscopy in HCV-infected cells, characterized by “vesicles embedded in a membranous matrix of circular or very tightly undulating membranes”[46] that serves to support viral RNA synthesis.
- NS proteins
nonstructural proteins expressed as part of the HCV polyprotein that function as part of the intracellular portion of the HCV life cycle
- Rapid virologic response (RVR)
undetectable HCV-VL at week 4 of therapy; achievement of RVR is considered strongly predictive of eventual success of therapy, but failure to achieve RVR does not have strong negative predictive value
- Replicon
a cell-culture model system in which the HCV genome is stably expressed but can be cleared by effective therapy; available for multiple GTs
- SOC
standard of care for HCV infection, involving 24–48 weeks (depending on genotype) of subcutaneously injected PegIFN and oral RBV therapy
- Specifically targeted antiviral therapy for hepatitis C (STAT-C)
drugs that exert an anti-HCV effect by interacting with one or more viral molecules (protein or RNA)
- Sustained virologic response (SVR)
undetectable HCV-VL 24 weeks after the end of therapy, considered equivalent to cure
- TLR
Toll-like receptor, a member of a family of proteins that sense pathogen-associated molecular patterns and stimulate the innate immune system, including IFN production, through shared effector pathways
- Very rapid virologic response (VRVR)
undetectable HCV-VL at week 2 of therapy
- VL
viral load, determined by RT-PCR of serum for HCV RNA
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure: MAG has no commercial or other association that might pose a conflict of interest.
References
- 1.Lavanchy D. The global burden of hepatitis C. Liver International. 2009;29:74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- 2.The Global Burden of Hepatitis C Working Group Global Burden of Disease (GBD) for Hepatitis C. J. Clin. Pharmacol. 2004;44:20–29. doi: 10.1177/0091270003258669. [DOI] [PubMed] [Google Scholar]
- 3.Wiesner RH, et al. Report of the First International Liver Transplantation Society Expert Panel Consensus Conference on Liver Transplantation and Hepatitis C. Liver Transplantation. 2003;9:S1–S9. doi: 10.1053/jlts.2003.50268. [DOI] [PubMed] [Google Scholar]
- 4.Marrero JA. Viral Hepatitis and Hepatocellular Carcinoma. In: Shetty K, Wu GY, editors. Chronic viral hepatitis: diagnosis and therapeutics. 2nd edn Springer; 2009. pp. 431–447. [Google Scholar]
- 5.Miller RH, Purcell RH. Hepatitis C virus shares amino acid sequence similarity with pestiviruses and flaviviruses as well as members of two plant virus supergroups. Proc. Natl. Acad. Sci. U. S. A. 1990;87:2057–2061. doi: 10.1073/pnas.87.6.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tellinghuisen TL, et al. Studying hepatitis C virus: making the best of a bad virus. J. Virol. 2007;81:8853–8867. doi: 10.1128/JVI.00753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sklan EH, et al. Mechanisms of HCV survival in the host. Nat. Rev. Gastroenterol. Hepatol. 2009;6:217–227. doi: 10.1038/nrgastro.2009.32. [DOI] [PubMed] [Google Scholar]
- 8.Sharma SD. Hepatitis C virus: molecular biology & current therapeutic options. Indian J. Med. Res. 2010;131:17–34. [PubMed] [Google Scholar]
- 9.Rehermann B. Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J. Clin. Invest. 2009;119:1745–1754. doi: 10.1172/JCI39133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kronenberger B, Zeuzem S. Current and future treatment options for HCV. Ann. Hepatol. 2009;8:103–112. [PubMed] [Google Scholar]
- 11.Tanaka Y, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat. Genet. 2009;41:1105–1109. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 12.Suppiah V, et al. IL28B is associated with response to chronic hepatitis C interferon-[alpha] and ribavirin therapy. Nat. Genet. 2009;41:1100–1104. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- 13.McCarthy JJ, et al. Replicated Association Between an IL28B Gene Variant and a Sustained Response to Pegylated Interferon and Ribavirin. Gastroenterology. 2010;138:2307–2314. doi: 10.1053/j.gastro.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bini EJ, et al. Prospective multicenter study of eligibility for antiviral therapy among 4,084 U.S. veterans with chronic hepatitis C virus infection. Am. J. Gastroenterol. 2005;100:1772–1779. doi: 10.1111/j.1572-0241.2005.41860.x. [DOI] [PubMed] [Google Scholar]
- 15.Grishchenko M, et al. Cost-effectiveness of pegylated interferon and ribavirin for patients with chronic hepatitis C treated in routine clinical practice. Int. J. Technol. Assess. Health Care. 2009;25:171–180. doi: 10.1017/S0266462309090229. [DOI] [PubMed] [Google Scholar]
- 16.Einav S, et al. Discovery of a hepatitis C target and its pharmacological inhibitors by microfluidic affinity analysis. Nat. Biotechnol. 2008;26:1019–1027. doi: 10.1038/nbt.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clark V, Nelson DR. Novel Interferons for Treatment of Hepatitis C Virus. Clin. Liver Dis. 2009;13:351–363. doi: 10.1016/j.cld.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 18.Shiffman ML. Treatment of hepatitis C in 2011: what can we expect? Curr. Gastroenterol. Rep. 2010;12:70–75. doi: 10.1007/s11894-009-0085-4. [DOI] [PubMed] [Google Scholar]
- 19.Georgel P, et al. Virus-host interactions in hepatitis C virus infection: implications for molecular pathogenesis and antiviral strategies. Trends Mol. Med. 2010;16:277–286. doi: 10.1016/j.molmed.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Rong L, et al. Rapid emergence of protease inhibitor resistance in hepatitis C virus. Sci Transl Med. 2010;2:30ra32. doi: 10.1126/scitranslmed.3000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindenbach BD, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 22.Lohmann V, et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 23.Blight KJ, et al. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290:1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 24.Wakita T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhong J, et al. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meuleman P, Leroux-Roels G. The human liver-uPA-SCID mouse: A model for the evaluation of antiviral compounds against HBV and HCV. Antiviral Res. 2008;80:231–238. doi: 10.1016/j.antiviral.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 27.QuiñonesMateu ME, et al. Viral Drug Resistance and Fitness. In: Kuan-Teh J, editor. Adv. Pharmacol. Academic Press; 2008. pp. 257–296. [DOI] [PubMed] [Google Scholar]
- 28.Prichard MN, et al. MacSynergy II manual. University of Michigan; 1993. [Google Scholar]
- 29.Einav S, et al. The Hepatitis C Virus (HCV) NS4B RNA Binding Inhibitor Clemizole Is Highly Synergistic with HCV Protease Inhibitors. J. Infect. Dis. 2010 doi: 10.1086/653080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Braithwaite RS, et al. Explaining variability in the relationship between antiretroviral adherence and HIV mutation accumulation. J. Antimicrob. Chemother. 2006;58:1036–1043. doi: 10.1093/jac/dkl386. [DOI] [PubMed] [Google Scholar]
- 31.Chary A, Holodniy M. Recent Advances in Hepatitis C Virus Treatment: Review of HCV Protease Inhibitor Clinical Trials. Rev. Recent Clin. Trials. 2010 doi: 10.2174/157488710792007293. [DOI] [PubMed] [Google Scholar]
- 32.Chen KX, Njoroge FG. A review of HCV protease inhibitors. Curr. Opin. Investig. Drugs. 2009;10:821–837. [PubMed] [Google Scholar]
- 33.Weisberg IS, Jacobson IM. Telaprevir: Hope on the Horizon, Getting Closer. Clin. Liver Dis. 2009;13:441–452. doi: 10.1016/j.cld.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 34.Fowell A, Nash K. Telaprevir: a new hope in the treatment of chronic hepatitis C? Adv. Ther. 2010;27:512–522. doi: 10.1007/s12325-010-0047-0. [DOI] [PubMed] [Google Scholar]
- 35.Berman K, Paul YK. Boceprevir, an NS3 Protease Inhibitor of HCV. Clin. Liver Dis. 2009;13:429–439. doi: 10.1016/j.cld.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 36.Mederacke I, et al. Boceprevir, an NS3 serine protease inhibitor of hepatitis C virus, for the treatment of HCV infection. Curr. Opin. Investig. Drugs. 2009;10:181–189. [PubMed] [Google Scholar]
- 37.McPhee F. Identification and Preclinical Profile of the Novel HCV NS3 Protease Inhibitor BMS-650032. J. Hepatol. 2010;52:S296–S296. [Google Scholar]
- 38.Gane E, et al. Early On-Treatment Responses During Pegylated IFN Plus Ribavirin are Increased Following 13 Days of Combination Nucleoside Polymerase (RG7128) and Protease (RG7227) Inhibitor Therapy (INFORM-1) J. Hepatol. 2010;52:S291–S292. [Google Scholar]
- 39.Burton JR, Jr., Everson GT. HCV NS5B polymerase inhibitors. Clin. Liver Dis. 2009;13:453–465. doi: 10.1016/j.cld.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 40.Amorosa V. New Frontiers of HCV Therapy in HIV/HCV Co-infection. Current HIV/AIDS Reports. 2010;7:117–126. doi: 10.1007/s11904-010-0051-7. [DOI] [PubMed] [Google Scholar]
- 41.Cooper C, et al. Safety, Tolerability and Pharmacokinetics of the HCV Polymerase Inhibitor VCH-222 Following Single Dose Administration in Healthy Volunteers and Antiviral Activity in HCV-Infected Individuals. J. Hepatol. 2009;50:S342–S342. [Google Scholar]
- 42.White CP, et al. Complementary and alternative medicine use by patients chronically infected with hepatitis C virus. Can. J. Gastroenterol. 2007;21:589–595. doi: 10.1155/2007/231636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmed-Belkacem A, et al. Silibinin and related compounds are direct inhibitors of hepatitis C virus RNA-dependent RNA polymerase. Gastroenterology. 2010;138:1112–1122. doi: 10.1053/j.gastro.2009.11.053. [DOI] [PubMed] [Google Scholar]
- 44.Wagoner J, et al. Multiple effects of silymarin on the hepatitis C virus lifecycle. Hepatology. 2010;51:1912–1921. doi: 10.1002/hep.23587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Payer BA, et al. Successful HCV eradication and inhibition of HIV replication by intravenous silibinin in an HIV-HCV coinfected patient. Journal of Clinical Virology. doi: 10.1016/j.jcv.2010.07.006. In Press. [DOI] [PubMed] [Google Scholar]
- 46.Egger D, et al. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002;76:5974–5984. doi: 10.1128/JVI.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bryson PD, et al. A small molecule inhibits HCV replication and alters NS4B's subcellular distribution. Antiviral Res. 2010;87:1–8. doi: 10.1016/j.antiviral.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paredes AM, Blight KJ. A Genetic Interaction between Hepatitis C Virus NS4B and NS3 Is Important for RNA Replication. J. Virol. 2008;82:10671–10683. doi: 10.1128/JVI.00875-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cho N-J, et al. Identification of a Class of HCV Inhibitors Directed Against the Nonstructural Protein NS4B. Sci. Transl. Med. 2010;2:15ra16–15ra16. doi: 10.1126/scitranslmed.3000331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao M, et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465:96–100. doi: 10.1038/nature08960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pol S, et al. Once-Daily NS5A Inhibitor (BMS-790052) Plus Peginterferon-Alpha-2a And Ribavirin Produces High Rates of Extended Rapid Virologic Response in Treatment-Naive HCV-Genotype 1 Subjects: Phase 2a Trial. J. Hepatol. 2010;52:S462–S462. [Google Scholar]
- 52.Colonno R, et al. Identification and Characterization of PPI-461, a Potent and Selective HCV NS5A Inhibitor with Activity Against All HCV Genotypes. J. Hepatol. 2010;52:S14–S15. [Google Scholar]
- 53.Griffin S. Inhibition of HCV p7 as a therapeutic target. Curr. Opin. Investig. Drugs. 2010;11:175–181. [PubMed] [Google Scholar]
- 54.Griffin S, et al. Genotype-dependent sensitivity of hepatitis C virus to inhibitors of the p7 ion channel. Hepatology. 2008;48:1779–1790. doi: 10.1002/hep.22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deltenre P, et al. Evaluation of amantadine in chronic hepatitis C: a meta-analysis. J. Hepatol. 2004;41:462–473. doi: 10.1016/j.jhep.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 56.Gallay PA. Cyclophilin inhibitors. Clin. Liver Dis. 2009;13:403–417. doi: 10.1016/j.cld.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 57.Chatterji U, et al. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J. Hepatol. 2010;53:50–56. doi: 10.1016/j.jhep.2010.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watashi K. Alisporivir, a cyclosporin derivative that selectively inhibits cyclophilin, for the treatment of HCV infection. Curr. Opin. Investig. Drugs. 2010;11:213–224. [PubMed] [Google Scholar]
- 59.Coelmont L, et al. Particular In Vitro Anti-HCV Activities and Resistance Profile of the Cyclophilin Inhibitor Debio 025. J. Hepatol. 2009;50:S36–S36. [Google Scholar]
- 60.Flisiak R, et al. The cyclophilin inhibitor Debio 025 combined with PEG IFNalpha2a significantly reduces viral load in treatment-naive hepatitis C patients. Hepatology. 2009;49:1460–1468. doi: 10.1002/hep.22835. [DOI] [PubMed] [Google Scholar]
- 61.Nelson DR, et al. Efficacy and Safety of the Cyclophilin Inhibitor Debio 025 in Combination with Pegylated Interferon Alpha-2a and Ribavirin in Previously Null-Responder Genotype 1 HCV Patients. J. Hepatol. 2009;50:S40. [Google Scholar]
- 62.Ke J, et al. Safety and Tolerability of Nim811, a Novel Cyclophilin Inhibitor for HCV, Following Single and Multiple Ascending Doses in Healthy Volunteers and HCV-Infected Patients. J. Hepatol. 2009;50:S229–S229. [Google Scholar]
- 63.Lawitz E, et al. Safety and Antiviral Efficacy of 14 Days of the Cyclophilin Inhibitor Nim811 in Combination with Pegylated Interferon [Alpha]2a in Relapsed Genotype 1 HCV Infected Patients. J. Hepatol. 2009;50:S379–S379. [Google Scholar]
- 64.Hopkins S, et al. SCY-635, a novel nonimmunosuppressive analog of cyclosporine that exhibits potent inhibition of hepatitis C virus RNA replication in vitro. Antimicrob. Agents Chemother. 2010;54:660–672. doi: 10.1128/AAC.00660-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hopkins S, et al. Safety, Plasma Pharmacokinetics, and Anti-Viral Activity of SCY-635 in Adult Patients with Chronic Hepatitis C Virus Infection. J. Hepatol. 2009;50:S36–S36. [Google Scholar]
- 66.Hopkins S, et al. Resistance Selection Following 15 Days of Monotherapy With SCY-635, a Non-Immunosuppressive Cyclophilin Inhibitor with Potent Anti-HCV Activity. J. Hepatol. 2010;52:S15–S15. [Google Scholar]
- 67.Keeffe EB, Rossignol JF. Treatment of chronic viral hepatitis with nitazoxanide and second generation thiazolides. World J. Gastroenterol. 2009;15:1805–1808. doi: 10.3748/wjg.15.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Elazar M, et al. The anti-hepatitis C agent nitazoxanide induces phosphorylation of eukaryotic initiation factor 2alpha via protein kinase activated by double-stranded RNA activation. Gastroenterology. 2009;137:1827–1835. doi: 10.1053/j.gastro.2009.07.056. [DOI] [PubMed] [Google Scholar]
- 69.Korba BE, et al. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antiviral Res. 2008;77:56–63. doi: 10.1016/j.antiviral.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 70.Rossignol JF, et al. Clinical trial: randomized, double-blind, placebo-controlled study of nitazoxanide monotherapy for the treatment of patients with chronic hepatitis C genotype 4. Aliment. Pharmacol. Ther. 2008;28:574–580. doi: 10.1111/j.1365-2036.2008.03781.x. [DOI] [PubMed] [Google Scholar]
- 71.Rossignol JF, et al. Improved virologic response in chronic hepatitis C genotype 4 treated with nitazoxanide, peginterferon, and ribavirin. Gastroenterology. 2009;136:856–862. doi: 10.1053/j.gastro.2008.11.037. [DOI] [PubMed] [Google Scholar]
- 72.Rossignol JF, et al. Randomized Controlled Trial of Nitazoxanide-Peginterferon-Ribavirin, Nitazoxanide-Peginterferon and Peginterferon-Ribavirin in the Treatment of Patients with Chronic Hepatitis C Genotype 4. J. Hepatol. 2008;48:S30–S30. [Google Scholar]
- 73.Rossignol JF, et al. Evaluation of a 4 Week Lead-In Phase with Nitazoxanide Prior to Nitazoxanide+Peginterferon in Treating Chronic Hepatitis C. J. Hepatol. 2008;48:S312–S312. [Google Scholar]
- 74.Shiffman ML, et al. Phase 2 Randomized, Double-Blind, Placebo-Controlled Study of Nitazoxanide with Peginterferon Alfa-2a and Ribavirin in Nonresponders (NR) with Chronic Hepatitis C Genotype 1: Final Report. J. Hepatol. 2010;52:S461–S461. [Google Scholar]
- 75.Bacon BR, et al. Phase 2 Randomized, Double-Blind, Placebo-Controlled Study of Nitazoxanide Plus Peginterferon and Ribavirin in HCV Genotype 1 Naive Patients: Week 12 Sustained Virologic Response Rate. J. Hepatol. 2010;52:S463. [Google Scholar]
- 76.Mencin A, et al. Toll-like receptors as targets in chronic liver diseases. Gut. 2009;58:704–720. doi: 10.1136/gut.2008.156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fletcher S, et al. Masked oral prodrugs of toll-like receptor 7 agonists: a new approach for the treatment of infectious disease. Curr. Opin. Investig. Drugs. 2006;7:702–708. [PubMed] [Google Scholar]
- 78.Takahashi K, et al. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. U. S. A. 2010;107:7431–7436. doi: 10.1073/pnas.1002301107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Horsmans Y, et al. Isatoribine, an agonist of TLR7, reduces plasma virus concentration in chronic hepatitis C infection. Hepatology. 2005;42:724–731. doi: 10.1002/hep.20839. [DOI] [PubMed] [Google Scholar]
- 80.Janssen HL, et al. ANA773, an Oral Inducer of Endogenous Interferons that Acts Via TLR7, Reduced Serum Viral Load in Patients Chronically Infected with HCV. Hepatology. 2009;50:1022A. [Google Scholar]
- 81.Muir A, et al. A Phase 1, Multi-Center, Randomized, Placebo-Controlled, Dose-Escalation Study of IMO-2125, a TLR9 Agonist, in Hepatitis C-Nonresponders. J. Hepatol. 2010;52:S14–S14. [Google Scholar]
- 82.Burke K, Cox A. Hepatitis C virus evasion of adaptive immune responses: a model for viral persistence. Immunol. Res. 2010;47:216–227. doi: 10.1007/s12026-009-8152-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Urbani S, et al. Restoration of HCV-specific T cell functions by PD-1/PD-L1 blockade in HCV infection: effect of viremia levels and antiviral treatment. J. Hepatol. 2008;48:548–558. doi: 10.1016/j.jhep.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 84.Nakamoto N, et al. Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade. PLoS Pathog. 2009;5:e1000313. doi: 10.1371/journal.ppat.1000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fisicaro P, et al. Functional T Cell Restoration Induced by PD-1/PD-L1 Blockade in Chronic Hepatitis B and C is Improved by Simultaneous Modulation of the Co-Stimulatory CD137/CD137L Pathway. J. Hepatol. 2009;50:S49. [Google Scholar]
- 86.Cheng G, et al. A virocidal amphipathic {alpha}-helical peptide that inhibits hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 2008;105:3088–3093. doi: 10.1073/pnas.0712380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cho NJ, et al. Mechanism of an amphipathic alpha-helical peptide's antiviral activity involves size-dependent virus particle lysis. ACS Chem. Biol. 2009;4:1061–1067. doi: 10.1021/cb900149b. [DOI] [PubMed] [Google Scholar]
- 88.Amemiya F, et al. Targeting lipid metabolism in the treatment of hepatitis C virus infection. J. Infect. Dis. 2008;197:361–370. doi: 10.1086/525287. [DOI] [PubMed] [Google Scholar]
- 89.Hsu NY, et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010;141:799–811. doi: 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cho NJ, et al. Phosphatidylinositol 4,5-bisphosphate is a ligand of HCV NS5A and mediates viral genome replication. doi: 10.1053/j.gastro.2014.11.043. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ikeda M, Kato N. Life style-related diseases of the digestive system: cell culture system for the screening of anti-hepatitis C virus (HCV) reagents: suppression of HCV replication by statins and synergistic action with interferon. J. Pharmacol. Sci. 2007;105:145–150. doi: 10.1254/jphs.fm0070050. [DOI] [PubMed] [Google Scholar]
- 92.Kapadia SB, Chisari FV. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. U. S. A. 2005;102:2561–2566. doi: 10.1073/pnas.0409834102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berger KL, et al. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A. 2009;106:7577–7582. doi: 10.1073/pnas.0902693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Borawski J, et al. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J. Virol. 2009;83:10058–10074. doi: 10.1128/JVI.02418-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tai AW, et al. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe. 2009;5:298–307. doi: 10.1016/j.chom.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Trotard M, et al. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB J. 2009 doi: 10.1096/fj.09-131920. [DOI] [PubMed] [Google Scholar]
- 97.Vaillancourt FH, et al. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology. 2009;387:5–10. doi: 10.1016/j.virol.2009.02.039. [DOI] [PubMed] [Google Scholar]
- 98.Hirata Y, et al. A Newly Synthesized Serine Palmitoyltransferase Inhibitor, NA 808, Has the Strong Effect to Hepatitis C Virus In Vitro And In Vivo. J. Hepatol. 2009;50:S344–S344. [Google Scholar]
- 99.Popescu CI, Dubuisson J. Role of lipid metabolism in hepatitis C virus assembly and entry. Biol. Cell. 2010;102:63–74. doi: 10.1042/BC20090125. [DOI] [PubMed] [Google Scholar]
- 100.Niepmann M. Activation of hepatitis C virus translation by a liver-specific microRNA. Cell Cycle. 2009;8:1473–1477. doi: 10.4161/cc.8.10.8349. [DOI] [PubMed] [Google Scholar]
- 101.Lanford RE, et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010;327:198–201. doi: 10.1126/science.1178178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Klade CS, et al. Therapeutic Vaccination of Chronic Hepatitis C Nonresponder Patients With the Peptide Vaccine IC41. Gastroenterology. 2008;134:1385–1395.e1381. doi: 10.1053/j.gastro.2008.02.058. [DOI] [PubMed] [Google Scholar]
- 103.Wedemeyer H, et al. Therapeutic vaccine IC41 as late add-on to standard treatment in patients with chronic hepatitis C. Vaccine. 2009;27:5142–5151. doi: 10.1016/j.vaccine.2009.06.027. [DOI] [PubMed] [Google Scholar]
- 104.Gowans EJ, et al. A phase I clinical trial of dendritic cell immunotherapy in HCV-infected individuals. J. Hepatol. 2010;53:599–607. doi: 10.1016/j.jhep.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Diago M, et al. Association of pretreatment serum interferon gamma inducible protein 10 levels with sustained virological response to peginterferon plus ribavirin therapy in genotype 1 infected patients with chronic hepatitis C. Gut. 2006;55:374–379. doi: 10.1136/gut.2005.074062. [DOI] [PMC free article] [PubMed] [Google Scholar]