

Abstract
Reactive surface lysine groups on the chimeric monoclonal antibody (3G4) and on human serum albumin (HSA) were labeled with two different PARACEST chelates. Between 7.4 – 10.1 chelates were added per 3G4 molecule and between 5.6 – 5.9 chelates per molecule of HSA, depending upon which conjugation chemistry was used. The immunoreactivity of 3G4 as measured by ELISA assays was highly dependent upon the number of attached chelates: 88% immunoreactivity with 7.4 chelates per antibody versus only 17% immunoreactivity with 10.1 chelates per antibody. Upon conjugation to 3G4, the bound water lifetime of Eu-1 increased only marginally, up from 53 μs for the non-conjugated chelate to 65–77 μs for conjugated chelates. Conjugation of a chelate Eu-2 to HSA via a single side-chain group also resulted in little or no change in bound water lifetime (73–75 μs for both the conjugated and non-conjugated forms). These data indicate that exchange of water molecules protons between the inner-sphere site on covalently attached PARACEST agent and bulk water is largely unaffected by the mode of attachment of the agent to the protein and likely its chemical surroundings on the surface of the protein.
Keywords: Eu3+ chelates, bifunctional PARACEST agents, 3G4 monoclonal antibody, HSA, bound water lifetimes
INTRODUCTION
Recently a novel class of MRI contrast agents were introduced called chemical exchange saturation transfer (CEST) agents1 that function by an entirely different mechanism compared to more traditional paramagnetic relaxation agents. This mechanism is based upon the ability to selectively pre-saturate an exchanging proton or water molecule resonance on an agent that subsequently exchanges into the bulk water pool and thereby causes a decrease in intensity of the bulk water signal. A number of different paramagnetic CEST agents have now been reported and numerous applications are being explored.2–6 The CEST efficiency of a molecule is conveniently evaluated by measuring the decrease in intensity of the bulk water signal after applying a long selective irradiation pulse at various frequencies across the entire proton spectrum. Any decrease in water intensity (Mz/M0) after application of a presaturation pulse is related to the concentration of CEST agent (C), the number of exchanging protons or water molecules (q), the T1 of bulk water, and water proton lifetime (τMH).
| (1) |
Of the parameters that contribute to equation 1, only the lifetime of the exchanging water proton molecule on the agent is readily modified by chemistry so this has become the key parameter of interest in the development of newer agents. Water proton exchange in aqueous lanthanide chelates depends upon the ionic radius of the Ln3+ cation, the nature of ligand side-chains that contribute electrons to the central Ln3+, the coordination geometry of the chelate, and the extent of prototropic and whole water exchange (the rate of water proton exchange is the sum of the rates of these last two effects). A more detailed discussion of the impact of these chemical features on the bound water lifetime can be found in other review articles.7–9
It has been shown that the rate of water proton exchange in T1-based low molecular weight Gd3+ chelates is often altered by binding of the chelate to proteins.10–14 This has an important impact in the design of high relaxivity agents because water exchange can ultimately limit the relaxivity of such chelates when they are covalently attached to macromolecules. Even non-covalent binding of a Gd3+ chelate to albumin (typically HSA or BSA) via either electrostatic or hydrophobic interactions can affect water exchange.15 Binding of a chelate to a hydrophobic site seems to have less impact on water exchange (typically slows about 2-fold) than binding via electrostatic interactions. This may reflect restricted access of molecules in the second hydration sphere whenever a Gd3+ chelate is bound at a hydrophobic site on a protein.10, 13 Strong electrostatic or hydrogen bonding interactions can impact water exchange even further. For example, a dramatic lengthening of the bound water lifetime from ~8 ns to 290 ns (36-fold) was reported for a gadolinium chelate with highly charged phosphonate pedant arms when bound to HSA.16 This was ascribed mainly to the electrostatic forces between negatively charged pendant arms and positively charged residues on the protein.16
The efficiency of PARACEST contrast agents can also be dramatically influenced by molecular interactions that alter the bound water lifetime. For this reason it is important to have a good understanding of the environmental parameters that may affect their overall performance. This report describes the changes that occur in the residence lifetime of Eu3+-bound water molecules before and after conjugation of the PARACEST chelate, Eu-1, to a chimeric 3G4 monoclonal antibody (Tarvacin™) as a model targeting vector for diagnostic and therapeutic applications. 3G4 has a high affinity for phosphatidylserine (PS) which is normally found on the inner leaflet of the plasma membrane of normal cells,17 maintained in this position by an ATP-dependent transporter and aminophospholipid translocase. Loss in PS symmetry caused by inhibition of aminophospholipid translocase or activation of scramblase18 is found in high abundance in the vascular endothelium that lines tumor blood vessels in a variety of tumors.19, 20 PS externalization is typically not observed in healthy cells so PS has become an appealing target for molecular imaging of cancer. Prior to attachment of PARACEST agents to 3G4, human serum albumin (HSA) was first used as a model protein to establish the best conditions for conjugation of the bifunctional CEST ligands developed here. Measurement of the bound water lifetimes for Eu-1–HSA and Eu-2–HSA conjugates and a comparison with the value found for the Eu-1–3G4 conjugates allowed us to evaluate how protein surface groups influence water exchange in systems with different points of chemical attachment. HSA decorated with Eu-1 or Eu-2 (Chart 1) also served as models for evaluating the detection limit of these systems for molecular imaging by CEST.
Chart 1.

The chemical structures of ligands 1 and 2.
RESULTS AND DISCUSSION
Tumor imaging relies heavily on the highest possible signal-to-noise ratio at the site of interest so targeting of multiple PARACEST agents so a single localized region by use of antibodies or peptides will be important. Given our prior experience with Gd3+ chelates, one would anticipate that the local chemical environment around an attached agent would indeed influence proton exchange and ultimately affect CEST efficiency. In order to examine the nature of these interactions, we selected 3G4 to serve as the targeting antibody model for conjugation to well characterized PARACEST systems, particularly those that are considered kinetically inert.21, 22 Given that ligands derived from macrocyclic structures are most favorable for this application, one could consider conjugating such ligands to proteins either via a carbon backbone functionalized macrocyclic structure (ligand 1) or via attachment through one pendant arm (ligand 2). Given that isothiocyanates and N-hydroxysuccinimide esters as the most widely used functional groups with controlled chemistries for conjugation of bifunctional agents, dyes, haptens to protein surfaces primarily via ε-amine lysine residues,23, 24 two different versions of ligands appropriate for PARACEST were chosen for this study.
Preparation of agents that conjugate via the macrocyclic backbone
p-NO2-benzyl-cyclen, synthesized by literature methods,25, 26 was chosen as the standard building block for the backbone C-functionalized ligand system. Introduction of the N-ligating substituents was achieved by exhaustive alkylation with excess 2-bromo-N-methylacetamide under standard conditions (K2CO3/MeCN) (Scheme 1). 2-Bromo-N-methylacetamide was synthesized using a method adapted for preparation of chloroacetoamide27 with significantly better yields (35% higher) compared to published values.28 Excess 2-bromo-N-methylacetamide was separated from the tetrasubstituted p-NO2 benzyl cyclen 4 by column chromatography. Reduction of the nitro aryl group was conducted under typical hydrogenation conditions using 10% palladium on carbon and H2 (45 psi) to yield the aniline derivative 5. The final chelate was obtained using two different approaches: 1) Eu-5 was first prepared by addition of a stoichiometric amount of europium chloride to the aqueous solution of 5 at pH 5.5 followed by conversion of Eu-6 to Eu-1 using thiophosgene and a two phase solvent system (H2O and CHCl3); or 2) 6 was first converted to 1 followed by chelate formation (as shown in Scheme 1). Eu-1 was characterized by high resolution 1H NMR and by IR spectroscopy.
Scheme 1.

Synthesis of Eu-1. Reagents and conditions: i. BrCH2CONHCH3 (3) / K2CO3 / MeCN (71 %); ii. H2 / 10% Pd on C / EtOH (99 %); iii. SCCl2 / H2O / CHCl3 (99 %); iv. EuCl3 / H2O.
With the development of monoclonal antibody-radioisotope conjugates, two general synthetic methods are typically used: 1) pre-labeling involves preparation of the chelate followed by attachment of the chelate to the protein in a second step, and 2) post-labeling generates the antibody-ligand conjugate first followed by addition of the radionuclide in the second step.29 In the current study, the pre-labeling method was chosen because it eliminates non-specific binding of metal ions to the protein. In addition, this method maintains a ligand concentration that favors rapid kinetics of chelation which is not the case for post-chelation conditions where concentrations of the protein-ligand conjugate are typically no higher than 10−4–10−5 M. Conjugations of ML chelates to proteins were conducted as previously reported for conjugation of DTPA (diethylenetriaminepentaacetic acid)-isothiocyanate to human IgG.30 Given that 3G4 has twelve solvent accessible lysine residues, our initial conjugation experiments used a 100-fold excess of Eu-1 per antibody (8.3-fold excess per lysine) to achieve maximum modification while preserving the immunoreactivity of the antibody (Scheme 2). Although this gave an average of ten Eu-1 chelates per antibody (Table 1), the immunoreactivity of 3G4 was severely reduced to ~17% as determined by ELISA. This loss of immunoreactivity was unexpected given that no lysine residues are thought to be near the binding sites of the antibody. In a second experiment, a 50-fold excess of Eu-1 gave a product that contained 7.4 europium chelates per the antibody (Table 1). This product retained 88% immunoreactivity as shown by ELISA. In the ELISA assay, the monoclonal antibody is recognized by horseradish peroxidase (HRP) as a secondary antibody and one possible explanations for reduced immunoreactivity encountered at the higher loading value is that site recognition between HRP and 3G4 was disrupted by excess modification of lysine groups. Integrity of the antibody after the conjugation reaction was analyzed by gel electrophoresis (Figure 4). As can be seen for the 3G4 conjugate with 7.4 Eu-1 chelates result is similar to that of unmodified antibody, while for the 3G4 conjugate with 10.1 Eu-1 chelates some decomposition was observed. Using identical chemical conditions, a 20-fold excess of Eu-1 over HSA resulted in a product with an average loading value of 5.6 chelates per protein molecule (Table 1).
Scheme 2.
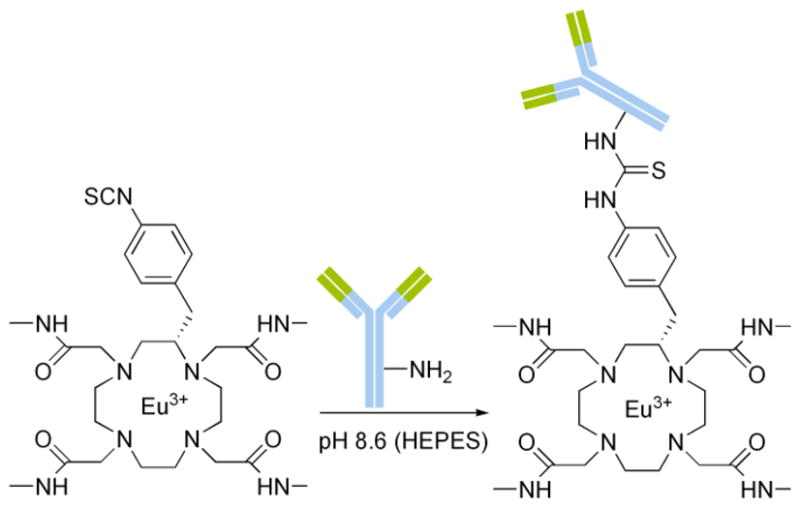
Schematic for the conjugation of Eu-1 chelates to 3G4.
Table 1.
Initial and final ratios of reactants in the conjugation reactions and immunoreactivity of the antibody
| Initial (EuL/P)i | Final (EuL/P)f | (R)f/(R)i | Immunoreactivitya of 3G4,% | |
|---|---|---|---|---|
| Eu-1 – 3G4 (1) | 100 | 10.1 | 0.10 | 17 ± 2 |
| Eu-1 – 3G4 (2) | 50 | 7.4 | 0.15 | 88 ± 2 |
| Eu-1 – HSA | 20 | 5.6 | 0.28 | - |
| Eu-2 – HSA | 100 | 5.9 | 0.06 | - |
average of duplicate experiments (ELISA assay)
Figure 4.

Determination of the detection limit of the CEST effect for Eu-1-HSA conjugate using two power of irradiation (B1 = 1370 Hz and B1 = 1075 Hz), irradiation time 2 s, 8 scans, 25 °C, pH 7.4 (5 mM HEPES/HCl, 140 mM NaCl), B0 = 270 MHz. Dilutions were made using HEPES buffer.
Preparation of agents that conjugate via a pendant arm
Eu-2 was synthesized as outlined in Scheme 3. Mono-CBz-cyclen 7 was alkylated with 2-bromo-N-methylacetamide in the presence of K2CO3 to produce 8 in high yield. Following column chromatography to remove excess 2-bromo-N-methylacetamide and deprotection of benzyloxycarbomate with H2 and 10% palladium on carbon, the tris-substituted amide 9 was obtained. N-(2-bromoacetyl)glycine benzyl ester was prepared by alkylation of the O-benzyl protected ester of glycine with bromoacetobromide in dichloromethane with excess of K2CO3 as a base. 9 was alkylated with N-(2-bromoacetyl) glycine benzyl ester 10 and the excess alkylating agent removed by column chromatography to obtain 11. The benzyl protecting groups were subsequently removed by hydrogenolysis (H2 and 10% palladium on carbon) and the europium chelate was prepared by mixing equimolar quantities of the ligand 2 and europium chloride at pH 5.5 in water. The N-hydroxysuccinimide ester of Eu-2 was generated in situ according to the literature procedure31 and a 100-fold excess of this activated ester was added to HSA (Scheme 4). After purification by dialysis and gel permeation chromatography, ICP analysis showed that ~5.9 Eu-2 chelates were attached to each HSA. Even through the conjugating efficiency (Table 1) was somewhat lower than desired; the simplified purification procedure eliminated many of the purification problems commonly encountered with other procedures.
Scheme 3.

Synthesis of Eu-2. Reagents and conditions: i. BrCH2CONHCH3 (3) / K2CO3 / MeCN (86 %); ii. H2 / 10% Pd on C / EtOH (99 %); iii. BrCH2CONHCH2COOBn (10) / K2CO3 / MeCN (79 %); iv. H2 / 10% Pd on C / EtOH; v. EuCl3 / H2O.
Scheme 4.

Schematic of the conjugation of Eu-2 to HSA through an active ester coupling. The image of HSA highlights solvent accessible lysine residues (nitrogen - blue, aliphatic chains - green). Hydrogen atoms were omitted for clarity.
NMR and CEST measurements
The Eu-1 and Eu-2 protein conjugates were analyzed by high resolution 1H NMR to confirm the integrity of the chelates once attached to the protein surface. The paramagnetic Eu3+ ion produces characteristic hyperfine shifts in ligand protons and, in particular, the characteristic H4 macrocyclic protons that appear between 23 and 31 ppm for both Eu-1 and Eu-2 are particularly useful. The presence of these diagnostic resonances in the isolated Eu-1/Eu-2 –protein/3G4 conjugates confirmed that the Eu3+ ion remains sequestered by the ligand even after conjugation (Figure 2).
Figure 2.

1H NMR spectra of PARACEST conjugates. 1H NMR spectrum of 5.5 mM Eu-1 in the HSA conjugate (top, 270 MHz) and 0.95 mM Eu-2 in the HSA conjugate (bottom, 500 MHz) pH 7.4 25 °C, pH 7.4 (5 mM HEPES/HCl, 140 mM NaCl). The water peak was partially suppressed.
Figure 3 shows typical CEST spectra of the Eu-1–HSA conjugate measured at different irradiation powers. The CEST exchange peak at ~57 ppm corresponds to the inner-sphere water molecules associated with the ~5–6 covalently attached Eu-1 chelates on the surface of HSA. The symmetry of this exchange peak indicates that these 5–6 chelates experience similar chemical environments on the surface of the protein. These combined data were fitted to a three-pool exchange model (bulk solvent water, Eu3+ coordinated water, and a third pool representing all other exchangeable protons associated with the protein; the 5–6 Eu-1 chelates on the protein surface were all assumed to be identical) to obtain a water proton exchange rate of ~15 × 103 s−1 (corresponding to a Eu3+-bound water lifetime of 65 ± 6 μs). Experimentally, a 27% signal reduction was observed for 5.5 mM Eu-1–HSA (0.98 mM HSA) using a presaturation field (B1) of 1370 Hz. Further dilutions of this sample with HEPES buffer to 0.35 mM Eu-1-HSA (Figure 4) resulted in a 3% reduction in water intensity using the same B1. Assuming that a 3% change in signal is the lower limit for reliable detection, then 0.35 mM could be considered the detection limit for the Eu-1–HSA system. However, it has been reported that CEST efficiency is optimal when the exchange rate, Cb, equals 2πB1.9 Thus, water proton exchange in this system is may be considered somewhat faster than optimal for a B1 of 1370 Hz.
Figure 3.

CEST spectra of a 5.5 mM solution of Eu-1–HSA conjugate recorded at B0 = 270 MHz, 25 °C, pH 7.4 (5 mM HEPES/HCl, 140 mM NaCl) with B1 = 1370 Hz, 1075 Hz and 833 Hz using an irradiation time of 2 s.
Only small amounts of antibody was available for experimentation so a final sample of Eu-1-3G4 (1 attempt, 10.1 chelates) prepared for CEST measurements contained only 0.32 mM Eu-1, near the estimated detection limit based upon the HSA results. Nevertheless, the same CEST spectrum was observed for this sample using a B1 of 1370 Hz (data not shown) as observed for Eu-1-HSA. The Eu3+-bound water proton lifetime determined from this single CEST spectrum was 71 μs, a value similar to that found for Eu-1 conjugated to HSA. During the second preparation of Eu-1-3G4 (7.4 chelates), a larger amount of material was obtained allowing preparation of a sample with a higher concentration of antibody (~170 μM) and Eu-1 (1.26 mM) for CEST measurements. CEST spectra acquired at 1370 and 1075 Hz were fitted to a similar three-pool model to obtain a Eu3+-bound water proton lifetime of 77 ± 2 μs. This suggests that water proton exchange in Eu-1 is relatively insensitive to protein identity, although more systems must be studied to substantiate this conclusion. Even though the concentration of 3G4 antibody in the first sample was only ~32 μM, this is still much higher than one would estimate for a local antibody concentration in a biological sample. Thus, we must tentatively conclude that CEST imaging using a Eu-1 protein conjugate may not be sensitive enough for a biological detection. However, it was recently shown that thulium chelates are substantially more sensitive than europium chelates for CEST imaging,32 so the detection limit may be well below that estimated here for Eu-1-HSA and Eu-1-3G4.
Although proton exchange rates were similar in Eu-1-HSA and Eu-1-3G4, it was of interest to compare these exchange rates with that of an appropriate model compound. Eu-6 was examined in place of Eu-1 as discrete chelate analog to eliminate possible sample degradation through hydrolysis of the isothiocyanate group of Eu-1 during the measurements. Interestingly, a fitting of CEST spectra of Eu-6 collected using different B1 values yielded a slightly shorter bound water proton lifetime (53 ± 2 μs) than those measured for the protein conjugates (see summary in Table 2). This suggests that proton exchange may be slowed slightly upon conjugation of Eu-1 to a protein. However, this increase in the bound water proton lifetime is modest compared to the previously reported changes10 but, in most cases, the previous studies have involved hydrophobic interactions of Gd3+ chelates with albumin binding pockets where the bound water molecule is likely significantly closer to the protein surface than in this study. Here, Eu-1 likely reacts with the most solvent accessible lysine residues presumably positioning the Eu3+-bound water molecule somewhat farther away from the protein surface.
Table 2.
Experimental and fitted parameters for calculation of bound water lifetimes
| T1, s | 100 -MS/M0, % | [EuL], mM | τMH, μs | |
|---|---|---|---|---|
| Eu-1 – 3G4 (1) a | 2.34 ± 0.09 | 3 | 0.32 | 71 |
| Eu-1 – 3G4 (2) a | 2.25 ± 0.09 | 8 | 1.26 | 77 ± 2 |
| Eu-1 – HSA a | 1.95 ± 0.07 | 27 | 5.5 | 65 ± 6 |
| Eu-6 a | 2.91 ± 0.12 | 44 | 12.5 | 53 ± 2 |
| Eu-2 – HSA b | 2.67 ± 0.12 | 12 | 0.95 | 75 ± 2 |
| EuDTMA b | 8 | 0.95 | 80 ± 2 | |
| Eu-2 b | 42 | 9.0 | 73 ± 3 |
Data was obtained at B0 = 270 MHz, B1= 1370, 1075, 833, 641 Hz (samples 3 and 4), B1 = 1370 Hz (sample 1), B1 = 1370 and 1075 Hz (samples 2), irradiation time 2 s, 8 scans, pH = 7.4 (samples 1–3 in 5 mM HEPES/HCl buffer, containing 140 mM NaCl) and 25 °C, Eu-6 in water pH = 7.4;
Data was measured at B0 = 500 MHz, B1 = 1412, 1075, 770, 540 Hz, irradiation time 2 s, 8 scans, pH = 7.4 (sample 5 in 5 mM HEPES/HCl buffer, containing 140 mM NaCl) and 25 °C.
Similar fittings were performed on CEST spectra of the Eu-2–HSA conjugate to estimate a value for τM. In this case, both Eu-2 and EuDTMA (1,4,7,10–tetra(methylcarbonylamide)-1,4,7,10-tetraazacyclododecane) were compared as model chelates based on their structural similarities. From Table 2 it can be seen that bound water lifetime in Eu-2 and EuDTMA are the same as the average value found for the Eu-2 – HSA conjugate. This indicates that the protein surface has little or no influence on water proton exchange when Eu-2 is covalently attached to the protein via its sole carboxyl side-arm. This is in contrast to the small decrease in proton exchange rates reported for other similar systems.10–13 While Eu-1 and Eu-2 should have nearly identical inner-sphere Eu3+-bound water structures, the primary difference between these two chelates when covalently attached to a protein is their point of the attachment to the protein surface. Although one might anticipate a priori that the Eu3+-bound water site in a covalently attached Eu-1 species would be situated further from the protein surface via the rigid benzyl thiourea linkage, it is this structure that seems to be impacted more by its attachment to the surface that Eu-2. In the later case where Eu-2 is attached to a protein surface via its more flexible aliphatic side-chain, this appears to have less affect on water or proton exchange. Clearly, further experiments with different proteins will need to be done to see if this trend holds for other systems as well. These studies do illustrate that covalent attachment of a lanthanide complex to a surface has much less impact on water access to the lanthanide coordination sphere compared to small lanthanide chelates that bind to albumin non-covalently. This perhaps is not surprising since non-covalent binding of small molecules to albumin occur at sites buried further into the protein structure where interactions with other protein side chains is much more likely than for a lanthanide chelate attached to surface moiety.
EXPERIMENTAL
General remarks
All organic precursors and solvents were obtained from commercial sources and used as received unless otherwise stated. Human serum albumin (HSA, 66.7 kDa) was purchased from Fluka. The monoclonal antibody (3G4) was generously provided by Paragrine Pharmaceuticals. 1H and 13C NMR spectra were acquired on a JEOL Eclipse 270 spectrometer operating at 270.17 and 67.93 MHz, respectively. Chemical shifts (δ) are reported in parts per million. CEST spectra and T1 values were measured using either a JEOL Eclipse 270 or a Varian INOVA 500 spectrometer (operating at 499.99 MHz). Prior to data acquisition, samples were warmed to room temperature and equilibrated in the probe at 25 °C for at least 10 min before measurement. T1 was measured by using an inversion-recovery sequence with 15 different delay times and 8 averages. CEST spectra were recorded by application of a long presaturation pulse at selected frequencies across the spectrum followed by a single observe pulse to measure the residual water signal. Infrared spectra were recorded on a Nicolet Avatar 360 FT-IR spectrometer as either KBr pellets or as neat liquids. UV-vis absorption spectra were recorded using a Shimadsu UV-1601 double beam diode array spectrophotometer. Hydrogenations were performed using a Parr hydrogenation apparatus. [Eu3+] was determined analytically using an Elan 6100 DRC (PE Scien) ICP Mass Spectrometer. Electrospray ionization mass spectrometry (ESI-MS) was performed by HT Laboratories, San Diego, California. Fast atom bombardment mass spectrometry (FAB-MS) was performed by the Mass Spectrometry Facility at University of Alabama at Tuscaloosa. Elemental analyses were obtained from Galbraith Labs, Knoxville, Tennessee. Gel electrophoresis was performed on Pharmacia LKB (Phast System) using phast gel (Gradient 4–15 Amersham Bioscience). 1-Benzyloxycarbonyl-1,4,7,10-tetraazacyclododecane trihydrochloride salt (referred to as mono-protected CBz-cyclen)33, 2-p-nitrobenzyl-1,4,7,10-tetraazacyclododecane26, and 1,4,7,10–tetra(methylcarbonylamide)-1,4,7,10-tetraaza cyclododecane (DTMA)28 were prepared using published methods.
2-Bromo-N-methylacetamide (3)
K2CO3 (55.3 g, 400 mmol) and methylamine hydrochloride (13.5 g, 200 mmol) were added to dichloromethane (700 mL) and the suspension was cooled to 0 °C. Bromoacetyl bromide (39.75 g, 200 mmol) was then added dropwise with vigorous stirring, and the reaction mixture was stirred at 0 °C for an additional 1 h followed by 2 h at room temperature. Water (70 mL) was then added and the organic layer that separated was dried overnight over Na2SO4. The solvent were removed under reduced pressure and the residue was crystallized from diethylether to afford a colorless compound (23.4 g, 77% yield). 1H NMR (270 MHz, CDCl3): δ = 2.8 (3H, d, CH3), 3.9 (2H, s, CH2), 6.8 (1H, s br, NH); 13C NMR (67.5 MHz, CDCl3): δ = 27.0 (CH3), 29.2 (CH2), 166.1 (CONH). NMR data is consistent with published data 28.
2-p-Nitrobenzyl-1,4,7,10-tetraazacyclododecane-1,4,7,10 -tetra(methylcarbonylamide) (5)
K2CO3 (1.25 g, 9 mmol) was added to a solution of p-NO2 benzyl cyclen 4 (0.6 g, 1.95 mmol) in acetonitrile (10 mL) and the suspension was heated at 70 °C for 30 min. 2-bromo-N-methylacetamide (1.25 g, 8.21 mmol) was then added in one portion. The reaction mixture was heated for 7 days at 65 – 70 °C, allowed to cool and filtered. The solvents were then removed under reduced pressure. The resulting residue was purified by column chromatography over silica gel. The column was first eluted with dichloromethane to remove impurities followed by elution of the title compound with methanol. The residue obtained from the column was dissolved in chloroform, filtered and solvent was removed by vacuum to afford the title compound as a yellow solid (0.68 g, 71 % yield). Rf = 0.4 (SiO2, MeOH). 1H NMR (270 MHz, D2O, pD 14): δ = 2.5 (4H, m br, ring CH2), 2.75–2.8 (12H, m, CH3), 3.01–3.12 (4H, m br, ring CH2), 3.2–3.4 (7H, m, ring CH2), 3.7–4.14 (10H, m, CH2Ar and CH2CO), 7.5 (2H, d, Ar), 8.2 (2H, d, Ar). 13C NMR (67.5 MHz, D2O pD 14): δ = 25.7–26.1 (CH3), 32.0 (CH2Ar), 51.01–52.5 (CH2 ring br), 56.1–56.4 (CH2CO) 124.1 (Ar), 130.5 (Ar), 146.4 (Ar), 147.8 (Ar), 171.0–173.7(CONH, br). FTIR (KBr): 1181, 1203, 1348, 1382, 1518, 1561, 1673, 2850, 2971, 3083, 3250. m/z (ESI+) 614 ([M+Na]+), 630 ([M+K]+). Anal. Found C = 51.7%, H = 7.8%, N =19.8% C27H45N9O6.2H2O requires C = 51.7%, H = 7.9%, N = 20.1%.
2-p-Aminobenzyl-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetra(methylcarbonylamide) (6)
10% palladium (0.4 g) on carbon was added to a solution of 5 (0.41 g, 0.69 mmol) in ethanol (10 mL). The reaction mixture was placed in a hydrogenation vessel for 24 h with the H2 pressure set to 45 psi. The sample was mixed twice during this 24 h period for 10 – 15 min each time. The reaction mixture was filtered through Celite®545 (Aldrich) and the filtrate concentrated under reduced pressure to afford the title compund as a pale solid (0.39 g, 99 % yield). 1H NMR (270 MHz, D2O pD 14): δ = 2.60–3.67 (37H, m), 6.78 (2H, d, Ar), 7.02 (2H, d, Ar). 13C NMR (67.5 MHz, D2O pD 14): δ = 26.8-26.02 (CH3), 30.9 (CH2Ar), 51.3–52.6 (CH2 ring, br), 57.0–57.4 (CH2CO, br), 116.9 (Ar), 130.3 (Ar), 145.5 (Ar), 146.4 (Ar), 173.7–174.6 (CONH, br). FTIR (KBr): 1156, 1247, 1357, 1410, 1464, 1557, 1676, 2827, 2968, 3076, 3450 cm−1. m/z (ESI+) 562 ([M+H]+), 584 ([M+Na]+).
2-p-Isothiocyanatobenzyl-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetra(methylcarbonylamide) (1)
A solution of thiophosgene (0.051 g, 446 μmol) in chloroform (2 mL) was added to a solution of 6 (0.065 g, 116 μmol) in water (1 mL) and the mixture was stirred at room temperature for 2 hours. The water fraction was separated and the organic layer extracted with water (2 × 1 mL). The water fractions were combined and freeze dried to give the title compound as a pale solid (0.07 g, 99% yield). 1H NMR (270 MHz, D2O pD = 2) δ = 2.62–2.77 (12H, m, CH3), 3.05–4.11 (25H, m, CH2Ar, CH2CO and ring CH2), 7.31 (4H, m, Ar). FTIR (KBr): 1158, 1236, 1362, 1410, 1471, 1571, 1689, 2116 (N=C=S), 2947, 2975, 3099, 3267. m/z (ESI+) 604 ([M+H]+), 626 ([M+Na]+).
1-Benzyloxycarbonyl-1,4,7,10-tetraazacyclododecane-4,7,10-tri(methylcarbonylamide) (8)
K2CO3 (3.37 g, 24.4 mmol) was added to a solution of mono-protected CBz-cyclen 7 (1.1 g, 2.44 mmol) in acetonitrile (20 mL), the suspension was heated warmed to 70 °C, and 2-bromo-N-methylacetamide (1.15 g, 24.9 mmol) was added in one portion. The reaction mixture was stirred at 65 – 70 °C for 12 hours, filtered, then concentrated under vacuum. The resulting residue was dissolved in water, extracted with dichloromethane (3 × 50 mL), and the combined organic fractions dried over Na2SO4 overnight. After filtering, the solvent was removed under reduced pressure and the residue purified by silica gel column chromatography, eluting with 10% methanol in chloroform to afford the title compound as a colorless gum (1.08 g, 85 % yield). Rf = 0.3 (SiO2, MeOH/CHCl3, 1:9). 1H NMR (270 MHz, CD3CN): δ = 2.82 (8H, s br, ring CH2), 2.89 (6H, s, CH3), 2.93 (3H, s, CH3), 3.14 (4H, s br, ring CH2), 3.30 (4H, s br, ring CH2), 3.53 (2H, s, CH2CO), 3.74 (4H, s, CH2CO), 5.3 (2H, s, OCH2Ph), 7.52 (2H, m br, NH), 7.58 (5H, m, Ph), 7.7 (1H, m br, NH). 13C NMR (67.5 MHz, CD3CN) δ = 25.4 (CH3), 25.6 (CH3), 47.0 (CH2 ring), 47.4 (CH2 ring), 53.4 (CH2 ring br), 58.3 (NCH2CO), 67.0 (OCH2Ph), 128.0 (Ph), 128.3 (Ph), 129.9 (Ph), 137.6 (Ph), 156.7 (CONH), 171.7 (CONH), 171.9.9 (COO). FTIR (NaCl pallet, CHCl3): 1049, 1119, 1157, 1219, 1417, 1533, 1673, 2827, 3013, 3056, 3352 cm−1. m/z (ESI+) 558 (100% [M+K]+).
1,4,7,10-Tetraazacyclododecane-1,4,7-tri(methylcarbonylamide) (9)
10% palladium on carbon (0.2 g) was added to a solution of 8 (0.93 g, 1.5 mmol) in ethanol (30 mL). The reaction mixture was placed in a hydrogenation vessel for 72 h with the H2 pressure set to 50 psi. The sample was mixed for 10 – 15 min twice per day during this 72 h period. The catalyst was filtered through Celite®545 (Aldrich), the solvents were removed under reduced pressure to afford the title compound colorless oil (0.57 g, 99 % yield). 1H NMR (270 MHz, D2O pD 9): δ = 2.54 (4H, s br, ring CH2), 2.63 (4H, s br, ring CH2), 2.69–2.70 (9H, m, NHCH3), 2.77 (4H, s br, ring CH2), 3.06–3.10 (6H, m, CH2CONH), 3.23–3.24 (4H, s br, ring CH2). 13C NMR (67.5 MHz, D2O pD 9) δ = 25.8 (CH3), 26.9 (CH3), 44.9 (CH2 ring), 49.5 (CH2 ring), 51.1 (CH2 ring), 53.4 (CH2 ring), 56.3 (CH2CONH), 58.3 (CH2CONH), 174.1 (CONH), 174.5 (CONH). FTIR (NaCl pallet, CHCl3): 1111, 1212, 1305, 1417, 1546, 1647, 2842, 2924, 3091, 3317 cm−1. m/z (ESI+) 386 ([M+H]+), 408 ([M+Na]+). Anal. Found C = 45.3%, H = 9.0%, N = 21.5% C17H35N7O3 × 3.5H2O requires C = 45.5%, H = 9.4%, N = 21.8%.
N-(2-Bromoacetyl) glycine benzyl ester (10)
K2CO3 (13.83 g, 10 mmol) was added to a solution of glycine benzyl ester, p-toluenesulfonate salt (1.68 g, 5 mmol) in dichloromethane (50 mL). The suspension was cooled to 0 °C and, with vigorous stirring, bromoacetyl bromide (0.99 g, 5 mmol) was added drop wise over ~5 min. After the addition was complete, the reaction mixture was stirred at 0 °C for 1 h and at room temperature for an additional 2 h. Water (10 mL) was then added and the organic layer was separated and dried over Na2SO4 overnight. The drying agent was then filtered and the solvent removed under reduced pressure. The resulting residue was crystallized from hexane to afford the title compound as a colorless solid (1.04 g, 73% yield). 1H NMR (270 MHz, CDCl3): δ = 3.9 (2H, s, CH2CO), 4.11 (2H, d, CH2CO2), 5.2 (2H, s, CH2Ph), 6.97 (1H, s br, NH), 7.36 (5H, m, Ph). 13C NMR (67.5 MHz, CDCl3): δ = 28.7 (CH2CONH), 42.0 (CH2CO2), 67.6 (CH2Ph), 128.6 (Ph), 128.8 (Ph), 134.9 (Ph), 165.8 (CONH), 169.1 (CO2). FTIR (KBr): 940, 1041, 1188, 1262, 1382, 1444, 1538, 1662, 1763, 2877, 2966, 3064, 3282 cm−1. m/z (ESI−) 284 (100% [M-H]−).
1- N-Acetyl benzyl glycinate-1,4,7,10-tetraazacyclododecane-4,7,10-tri(methylcarbonyl amide) (11)
K2CO3 (0.21 g, 1.5 mmol) was added to a solution of 9 (0.5 g, 1.3 mmol) in acetonitrile (10 mL), the suspension was heated for 20 min at 70 °C, and N-(2-bromoacetyl) glycine benzyl ester 10 (0.43 g, 1.5 mmol) was added in one portion. The reaction mixture was stirred at 65 – 70 °C overnight, then filtered and solvent removed under reduced pressure. The resulting residue was purified by silica gel column chromatography. The column was first eluted with CHCl3/MeOH (9.5:0.5) to remove impurities, and the title compound was then eluted with CHCl3/MeOH/NH4OH (3:2:1) as the slowest moving fraction. The fractions were concentrated, dissolved in 2% methanol in chloroform, and filtered and the solvents were removed under vacuum to afford title compound (0.61 g, 79.5 % yield). Rf = 0.65 (SiO2, CHCl3/MeOH/NH4OH, 3:2:1). 1H NMR (270 MHz, CD3CN ): δ = 2.56–3.19 (33H, ring CH2, CH3, CH2CO), 3.95 (2H, d, NHCH2CO2), 5.13 (2H, s, OCH2Ph), 6.73 (4H, m br, CONH), 7.38 (5H, m, Ph). 13C NMR (67.5 MHz, CD3CN) δ = 25.3 (CH3), 25.5 (CH3), 41.0 (CH2COO), 50.1–50.9 (CH2 ring br), 56.9 (NCH2CO), 57.1 (NCH2CO), 66.5 (CH2Ph), 128.2 (Ph), 128.3 (Ph), 129.7 (Ph), 136.1 (Ph), 169.7 (CONH), 172.0 (CONH), 172.5 (CO2). FTIR (NaCl pallet, CHCl3): 672, 761, 1216, 1642, 1988, 3080, 3394 cm−1. m/z (ESI+) 591 ([M+H]+, 613 ([M+Na]+).
1-N-acetylglycine-1,4,7,10-tetraazacyclododecane-4,7,10-tri(methylcarbonylamide) (2)
10% palladium on carbon (0.1 g) was added to a solution of 11 (0.45 g, 0.76 mmol) in ethanol (10 mL). The suspension was placed on a hydrogenation apparatus operating at 45 psi and room temperature for 96 hours. The catalyst was filtered through Celite®545 (Aldrich) and the solvents removed under reduced pressure to afford the title compound as a colorless amorphous solid (0.37 g, 97 % yield). 1H NMR (270 MHz, D2O pD = 8): δ = 2.72–2.73 (9H, 2 s, CH3), 2.9 (4H, s br, ring CH2), 3.1–3.4 (12H, m br, ring CH2), 3.5 (4H, s, CH2CONH), 4.73–4.75 (6H, m, CH2CO). 13C NMR (67.5 MHz, D2O) δ = 25.9 (CH3), 26.0 (CH3), 43.5 (CH2CO2), 49.81 (CH2 ring, br), 50.2 (CH2 ring, br), 51.3 (CH2 ring, br), 51.9 (CH2CONH), 54.6 (CH2CONH), 56.1 (CH2CO2), 166.2 (CONH), 169.5 (CONH), 172.0 (CONH), 172.9 (CONH), 172.9 (CONH), 176.0 (CO2). FTIR (NaCl pallet, CHCl3): 769, 1212, 1642, 1992, 3060, 3414 cm−1. m/z (ESI−) 499 (100 % [M-H]−).
General procedure for preparation of lanthanide chelates
An aqueous solution of the appropriate ligand was adjusted to a pH of ~5.5 using 1 M NaOH/1 M HCl as necessary. A stoichometric quantity of standardized lanthanide chloride aqueous solution was added and the reaction mixture was stirred at room temperature with constant adjustment of pH to 5.0 – 5.5 using 1 M NaOH. The extent of the chelation reaction was monitored by following changes in the reaction pH, and were typically observed to be complete ~30 min. The absence of free lanthanide ions was established with xylenol orange (0.15 M acetate buffer, pH 5.5). The pH was then adjusted to 7 and the samples were filtered and freeze-dried to give the chelates. Eu-1: FTIR (KBr): 2116 (N=C=S) cm−1; m/z (FAB+) 754 and 756 [Eu3+1 – 2H]+; Eu-2: m/z (FAB+) 649 and 651 [Eu3+2− - H]+; Eu-6: m/z (FAB+) 710 and 712 [Eu3+6− - H]+ with an appropriate isotope pattern was observed.
Conjugation of Eu-1 to HAS
HSA (40 mg, 0.6 μmol) was dissolved in 1.5 mL of HEPES/HCl buffer (5 mM, 140 mM NaCl, pH 8.6). Eu-1 (11.3 mg, 12 μmol) was dissolved in 0.15 mL of water and added to the protein solution. The reaction mixture was incubated at room temperature for 3 hours, then purified by dialysis against HEPES/HCl buffer (5 mM, 140 mM NaCl, pH 7.4) for 48 h (4 × 2L) at room temperature and followed by gel permeation chromatography over Sephadex (G-25) equilibrated with HEPES/HCl buffer (5 mM, 140 mM NaCl, pH 7.4). The purified conjugate (~2.2 mL) was concentrated to ~0.2 mL using an Ultrafree-MC Microcentrifuge filter (NMWL 5000 Da). After the first concentration step, 0.3 mL of fresh buffer was added and the volume was reduced again to ~0.4 mL.
Conjugation of Eu-1 to 3G4
3G4 (23 mg/mL, 77 nmol) was dialyzed in a Tube-O-Dialyzer™ (2 mL, GBioscences) against HEPES/HCl buffer (5 mM, 140 mM NaCl, pH 8.6) for 24 h (2 × 2L) at 4 °C. The final concentration was obtained by measuring the absorbance at 280 nm (ε = 1.4 mL/(cm × mg)). Eu-1 (3.6 mg, 3.85 μmol) was dissolved in 0.1 mL of water and added to the 3G4 solution (22 mg/mL). The mixture was incubated at room temperature for 3 hours, followed by dialysis against HEPES/HCl buffer (5mM, 140 mM NaCl, pH 7.4) for 48 h (4×2L) at 4°C. The purified conjugate was concentrated using absorbent (Spectra/Gel Absorbent, Spectrum®, Spectrum Lab) for 6 h at 4 °C.
Europium 1-N-Hydroxysuccinimidylacetatoamido glycinate-1,4,7,10-tetraazacyclododecane- 4,7,10-tri(methylcarbonylamide) chloride was obtained by the method of Lewis et al. (29). Eu-2 (0.11 g, 0.12 mmol) was dissolved in water (0.86 mL, 4 °C), and the pH adjusted to 5.5, followed by addition of N-hydroxysuccinimide (13.7 mg, 0.12 mmol) in 0.1 mL of water. The reaction mixture was stirred for 5 min and 0.02 mL 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) (12.2 mg, 60 μmol) aqueous solution was added. The reaction mixture was stirred for 30 minutes at 0 °C followed by pH adjustment to 7.5 with 0.25 M Na2HPO4. The europium activated ester chelate 12 was used for conjugation without isolation.
Conjugation of activated chelate 12 to HAS
HSA (40.0 mg, 0.6 μmol) was dissolved in 0.78 mL of HEPES/HCl buffer (5 mM, 140 mM NaCl, pH 7.4) followed by addition of the europium activated ester chelate 12 (theoretical amount: 60 μmol). The reaction mixture was then incubated at room temperature for 2 hours. The reaction mixture was purified by dialysis against HEPES/HCl buffer (pH 7.4) for 48 h (4 × 2L) at room temperature followed by gel permeation chromatography over Sephadex (G-25) equilibrated with HEPES/HCl buffer (pH 7.4). The purified conjugate (~2.2 mL) was concentrated to ~0.2 mL using Ultrafree-MC Microcentrifuge filters (NMWL 5000 Da). After the first concentration step, 0.3 mL of fresh buffer was added and the once again the volume was reduced to ~0.4 mL.
Determination of Eu-Antibody/Protein Ratio
The number of europium chelates per antibody or HSA was measured by Inductively Coupled Plasma Mass Spectrometry (ICP MS). The uncertainty in the measurement reflects the deviation of three parallel measurements (n = 3). The peak intensities were fitted to a four point calibration curve to obtain a mean value. The antibody/protein concentrations were determined by standard Lowry assay. Three measurements were performed per each conjugate preparation.
ELISA assays
Eu-1 – 3G4 samples were diluted with 2% fetus bovine serum (FBS) in PBS buffer to a concentration of 2 mg/mL antibody. As a control, unmodified 3G4 was used at the same concentration. Phosphatidylserine (50 μL, 10 μg/mL) in chloroform was added to each well in the 96-well plate and chloroform was evaporated at room temperature. 0.2 mL of the blocking buffer (10% FBS in PBS) was added to each well and this was incubated at 37 °C for 1 hour. The blocking buffer was removed and the plate was washed three times with PBS buffer. Samples were prepared in duplicate using a two fold dilution of 2 mg/mL 3G4 with blocking buffer. After incubating the plate at 37°C for 2 hour, it was washed three times with PBS to remove unbound 3G4. 0.1 mL of 0.8 μg/mL HRP (Horseradish peroxidase) conjugated goat anti-human IgG was then added to each well and the samples were incubated at 37°C for 1 h. The plate was then washed three times with PBS buffer followed by addition 100 μL of a developing reagent (10 mL of 0.2 M Na2PO4, 10 mL of 0.1 M citric acid, 10 mg of 1,2 -diaminobenzene, 10 μL of H2O2) to detect the second antibody. The samples were incubated for 10 min, followed by addition of 0.1 mL of 0.18 M H2SO4 to stop the reaction. The optical density at 490 nm was read using a 96 well plate reader (7520-Microplate reader, Cambridge Technology, Inc).
Fitting of CEST spectra (Z-spectra) to a three pool exchange model
The water exchange kinetics of each chelate-conjugate were determined by fitting the acquired Z-spectra to the Bloch equations modified for exchange using a process described previously34. In general EuDOTA-tetraamide chelates can only be fitted by using a 3-pool exchange model that includes bulk water, the Eu3+-bound water molecule and the ligand amide protons (typically four per chelate). The amide protons in these Eu3+ chelates experience rather small paramagnetic shifts and are often not fully resolved from the bulk water peak itself. Nevertheless, inclusion of these known exchange sites in the fitting procedure is important in correctly fitting the shape of the bulk water peak and in obtaining the correct exchange rate constants for the more highly shifted Eu3+-bound water exchange peak.
For Eu3+ chelates conjugated to a protein, the CEST fitting procedure is further complicated by a large number of exchangeable protons and/or water molecules closely associated with the protein. In order to fit these spectra it is important to be mindful of the way in which various parameters affect the fitting routine. Each saturation peak must be fitted to an appropriate intensity and width (shape). The intensity of the peak is predominantly determined by two factors the concentration of the exchanging pool and the T1 of protons within that pool. In general it is preferable to experimentally determine both these parameters. However, the exchangeable protons associated with the protein represent a diverse range of protons and environments consisting of: amide, hydroxyl and amine protons of the protein as well as water molecule hydrogen bound to the protein structure. For this reason determining a precise value of concentration or T1 is not possible. And yet, it is possible to take advantage of the nature of the fitting routine’s reliance on these two parameters for determining peak intensity to produce a valid fitting without knowing either value exactly. The T1 value of this pool was allowed to float substantially and even take unrealistic values. In so doing the fitting routine itself effectively self corrects for any error in the chosen concentration value – if the concentration value is too high then the T1 value used in fitting will be shorter to compensate.
Equally this third pool is inhomogeneous in chemical shift and will necessarily be broad. But line-width (shape) in the model is predominantly determined by the exchange rate of each pool with the bulk (note, there is no exchange in this model between the bound water pool and the third exchanging pool) and T2. One may take advantage of this to overcome the shift problem by allowing the center of the peak to shift over a reasonable diamagnetic range (1–6 ppm) and allowing T2 to adopt unrealistically short values. In this way the peak is allowed to become wide enough to encompass spins of all chemical shifts. Since there is little or no shift difference between the bulk water pool and this third pool, exchange between these pools will have little effect upon the shape of either peak and the shape of the peak of the third pool will be predominantly determined by T2.
Since it is exchange between bound and bulk water pools that is being probed the values concerning the bulk water and Eu3+-bound water pools were then tightly constrained: T1 values were experimentally determined for the bulk and values previous determined for the bound pool in a related system employed.33 T2 values were constrained to be shorter than T1 and fall within the range of values previously determined for similar systems. In this way the MATLAB™ fitting routine was constrained to fit the shape of the bound pool predominantly by altering water proton exchange alone, and was thus unaffected by the apparent increase in the width of the direct saturation peak at 0 ppm. This approach to fitting accommodates the signal arising from the protein without affecting the line-widths and shapes of the two exchange pools of interest. However, the fitting does as a consequence become inherently more dependent upon the line-shape of the bound pool; as a result, as B1 is increased and the spectrum broadens the values of τMH obtained in the fitting shorten slightly because the shape of this peak is broader but less well defined and therefore provides a somewhat less reliable fit.
Figure 1.

Gel electrophoresis of modified antibody containing in average 7.4 Eu-1 (1) chelates and unmodified antibody (2) used in preparation of the conjugate, modified antibody with average 10.1 Eu-1 chelates (3) and unmodified antibody (4).
Acknowledgments
This work was supported in parts by grants from the National Institutes of Health (CA-115531 and RR-02584) and the Robert A. Welch Foundation (AT-584). We also thank Dr. Matthew Leybourne for his assistance with ICP MS measurements.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ward KM, Aletras AH, Balaban RS. J Magn Reson. 2000;143:79. doi: 10.1006/jmre.1999.1956. [DOI] [PubMed] [Google Scholar]
- 2.Aime S, Barge A, Castelli DD, Fedeli F, Mortillaro A, Nielsen FU, Terreno E. Magnetic Resonance in Medicine. 2002;47:639. doi: 10.1002/mrm.10106. [DOI] [PubMed] [Google Scholar]
- 3.Trokowski R, Zhang S, Sherry AD. Bioconjug Chem. 2004;15:1431. doi: 10.1021/bc0498976. [DOI] [PubMed] [Google Scholar]
- 4.Zhang S, Malloy CR, Sherry AD. J Am Chem Soc. 2005;127:17572. doi: 10.1021/ja053799t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ren JM, Trokowski R, Zhang SR, Malloy CR, Sherry AD. Magnetic Resonance in Medicine. 2008;60:1047. doi: 10.1002/mrm.21722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Terreno E, Castelli DD, Violante E, Sanders HMHF, Sommerdijk NAJM, Aime S. Chemistry-a European Journal. 2009;15:1440. doi: 10.1002/chem.200801766. [DOI] [PubMed] [Google Scholar]
- 7.Zhang S, Merritt M, Woessner DE, Lenkinski RE, Sherry AD. Acc Chem Res. 2003;36:783. doi: 10.1021/ar020228m. [DOI] [PubMed] [Google Scholar]
- 8.Sherry AD, Woods M. Annu Rev Biomed Eng. 2008;10:391. doi: 10.1146/annurev.bioeng.9.060906.151929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woods M, Woessner DE, Sherry AD. Chem Soc Rev. 2006;35:500. doi: 10.1039/b509907m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aime S, Chiaussa M, Digilio G, Gianolio E, Terreno E. J Biol Inorg Chem. 1999;4:766. doi: 10.1007/s007750050349. [DOI] [PubMed] [Google Scholar]
- 11.Aime S, Gianolio E, Longo D, Pagliarin R, Lovazzano C, Sisti M. Chembiochem. 2005;6:818. doi: 10.1002/cbic.200400364. [DOI] [PubMed] [Google Scholar]
- 12.Aime S, Gianolio E, Terreno E, Giovenzana GB, Pagliarin R, Sisti M, Palmisano G, Botta M, Lowe MP, Parker D. J Biol Inorg Chem. 2000;5:488. doi: 10.1007/pl00021449. [DOI] [PubMed] [Google Scholar]
- 13.Caravan P, Cloutier NJ, Greenfield MT, McDermid SA, Dunham SU, Bulte JWM, Amedio JC, Looby RJ, Supkowski RM, Horrocks WD, McMurry TJ, Lauffer RB. Journal of the American Chemical Society. 2002;124:3152. doi: 10.1021/ja017168k. [DOI] [PubMed] [Google Scholar]
- 14.Toth E, Connac F, Helm L, Adzamli K, Merbach AE. Journal of Biological Inorganic Chemistry. 1998;3:606. [Google Scholar]
- 15.Caravan P, Parigi G, Chasse JM, Cloutier NJ, Ellison JJ, Lauffer RB, Luchinat C, McDermid SA, Spiller M, McMurry TJ. Inorganic Chemistry. 2007;46:6632. doi: 10.1021/ic700686k. [DOI] [PubMed] [Google Scholar]
- 16.Aime S, Botta M, Crich SG, Giovenzana GB, Pagliarin R, Piccinini M, Sisti M, Terreno E. Journal of Biological Inorganic Chemistry. 1997;2:470. [Google Scholar]
- 17.Devaux PF. Annual Review of Biophysics and Biomolecular Structure. 1992;21:417. doi: 10.1146/annurev.bb.21.060192.002221. [DOI] [PubMed] [Google Scholar]
- 18.Comfurius P, Senden JM, Tilly RH, Schroit AJ, Bevers EM, Zwaal RF. Biochim Biophys Acta. 1990;1026:153. doi: 10.1016/0005-2736(90)90058-v. [DOI] [PubMed] [Google Scholar]
- 19.Ran S, Downes A, Thorpe PE. Cancer Res. 2002;62:6132. [PubMed] [Google Scholar]
- 20.Ran S, Thorpe PE. Int J Radiat Oncol Biol Phys. 2002;54:1479. doi: 10.1016/s0360-3016(02)03928-7. [DOI] [PubMed] [Google Scholar]
- 21.Pasha A, Tirsco G, Benyo ET, Brucher E, Sherry AD. Eur J Inorg Chem. 2007:4340. doi: 10.1002/ejic.200700354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sherry AD, Caravan P, Lenkinski RE. J Magn Reson Imag. 2009;30:1240. doi: 10.1002/jmri.21966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brinkley M. Bioconjug Chem. 1992;3:2. doi: 10.1021/bc00013a001. [DOI] [PubMed] [Google Scholar]
- 24.Liu S, Edwards DS. Bioconjug Chem. 2001;12:7. doi: 10.1021/bc000070v. [DOI] [PubMed] [Google Scholar]
- 25.McMurry TJ, Brechbiel M, Kumar K, Gansow OA. Bioconjugate Chemistry. 1992;3:108. doi: 10.1021/bc00014a004. [DOI] [PubMed] [Google Scholar]
- 26.Renn O, Meares CF. Bioconjug Chem. 1992;3:563. doi: 10.1021/bc00018a017. [DOI] [PubMed] [Google Scholar]
- 27.Woods M, Sherry AD. Inorganic Chemistry. 2003;42:4401. doi: 10.1021/ic0300823. [DOI] [PubMed] [Google Scholar]
- 28.Bianchi A, Calabi L, Giorgi C, Losi P, Mariani P, Paoli P, Rossi P, Valtancoli B, Virtuani M. Journal of the Chemical Society-Dalton Transactions. 2000:697. [Google Scholar]
- 29.Li M, Meares CF, Zhong GR, Miers L, Xiong CY, DeNardo SJ. Bioconjug Chem. 1994;5:101. doi: 10.1021/bc00026a001. [DOI] [PubMed] [Google Scholar]
- 30.Wu C, Gansow OA, Brechbiel MW. Nucl Med Biol. 1999;26:339. doi: 10.1016/s0969-8051(98)00112-7. [DOI] [PubMed] [Google Scholar]
- 31.Lewis MR, Kao JY, Anderson AL, Shively JE, Raubitschek A. Bioconjug Chem. 2001;12:320. doi: 10.1021/bc0000886. [DOI] [PubMed] [Google Scholar]
- 32.Vinogradov E, Zhang S, Lubag A, Balschi JA, Sherry AD, Lenkinski RE. J Magn Reson. 2005;176:54. doi: 10.1016/j.jmr.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 33.Woods M, Kiefer GE, Bott S, Castillo-Muzquiz A, Eshelbrenner C, Michaudet L, McMillan K, Mudigunda SD, Ogrin D, Tircso G, Zhang S, Zhao P, Sherry AD. J Am Chem Soc. 2004;126:9248. doi: 10.1021/ja048299z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woods M, Woessner DE, Zhao P, Pasha A, Yang MY, Huang CH, Vasalitiy O, Morrow JR, Sherry AD. J Am Chem Soc. 2006;128:10155. doi: 10.1021/ja061498t. [DOI] [PMC free article] [PubMed] [Google Scholar]