

Abstract
There are very few fungal polyketide synthases that have been characterized by mass spectrometry. In this paper we describe the in vitro reconstitution and FT-ICR-MS verification of the full activity of an intact 277 kDa fungal polyketide synthase LovF of the lovastatin biosynthetic pathway. We report here both the verification of the reconstitution of fully functional holo-LovF by using 13C-labelled malonyl-CoA to form α-methylbutyrate functionality, and also detection of five predicted intermediates covalently bound to the 4′-phosphopantetheine at the acyl carrier protein (ACP) active site utilizing the phosphopantetheine ejection assay and high resolution mass spectrometry. Under in vitro conditions, the diketide acetoacetyl-intermediate did not accumulate on the ACP active site of holo-LovF following incubation with malonyl-CoA substrate. We found that incubation of holo-LovF with acetoacetyl-CoA served as an effective means of loading the diketide intermediate onto the ACP active site of LovF. Our results, demonstrate that subsequent α-methylation of the acetoacetyl intermediate stabilizes the intermediate onto the ACP active site, and facilitates the formation and mass spectrometric detection of additional intermediates en route to the formation of α-methylbutyrate.
Keywords: α-methylbutyrate, fourier transform ion cyclotron resonance, lovastatin, LovF, mass spectrometry, natural products, phosphopantetheine ejection assay, polyketide synthase
Polyketide natural products have served as a valuable source of useful therapeutics, due in large part to their broad structural variability and resulting functional diversity (1). Numerous compounds of polyketide origin, such as erythromycin (antibiotic) (2), rapamycin (immunosuppressant) (3), doxorubicin (antitumor) (4), and lovastatin (antihypercholesterolemic) (5), are commercially-available drugs and widely-used. The profound impact of this class of natural products on pharmaceutical development has lead to extensive efforts to characterize the enzymes responsible for their biosynthesis. The enzymes, polyketide synthases (PKSs), follow a general biosynthetic scheme in which simple acyl-coenzyme A (CoA) substrates are utilized to generate larger, more complex polyketide products by an array of catalytic domains (6). Polyketide biosynthesis progresses in a step-wise manner in which mulitple acyl substrates are selected by the enzyme, and through repeated cycles of Claisen-like condensation reactions, the individual building blocks are linked together via C-C bonds to form an elongated intermediate. Each round (iteration) of condensation may be followed by various combinations of tailoring reactions, each reaction occurs at the active sites of the various catalytic domains. The variation in structure of each polyketide is ultimately derived from the combination of different catalytic domains that make up the PKS and the acyl-substrate specificity of the acyl transferase (AT) domain(s).
Bacterial type I PKSs are composed of several modules, each of which contains a minimal set of catalytic domains and an array of additional tailoring domains that modify the polyketide intermediate after each iteration of elongation. Each module has, at minimum, a ketosynthase (KS) domain that catalyzes the Claisen-like condensation between the growing intermediate and “extender” acyl-units, an AT domain that selects and loads the extender acyl-unit from the CoA substrate (the initial module selects/loads the “starter” acyl subunit), and an acyl-carrier protein (ACP) domain that covalently tethers the growing intermediate to the enzyme and transfers it from one module to the next downstream module. The conserved serine at the active site of the ACP domain is post-translationally modified with a phosphopantetheine (PPant) moiety (7). PPant acts as a flexible/mobile arm, bearing a thiol group to which the growing polyketide intermediate remains covalently bound as a thioester, thereby facilitating the movement of the intermediate to the active sites of the various domains and between each module (This will be 8).
In contrast to bacterial type I PKSs, in which each domain is used only once per every molecule of product synthesized, the catalytic domains of fungal type I PKSs are re-used multiple times as successive rounds (iterations) of elongation and tailoring occur to yield a polyketide product. Essentially, a fungal type I PKSs resembles a single bacterial type I PKS module in terms of domain composition. However, fungal PKS possesses a smaller number of catalytic tailoring domains, but may use them iteratively in different combinations to generate a polyketide. A subgroup of fungal PKSs of particular interest is highly-reducing polyketide synthases (HR-PKSs) from filamentous fungi (9). HR-PKSs are capable of varying combinations of β-keto-reduction, dehydration, and/or enoyl-reduction after each iteration of elongation, but little is known about how these enzymes are programmed to do so.
The polyketide lovastatin is a potent HMG-CoA inhibitor (statin) (10, 11, 12) and was the first compound approved by the FDA to be used specifically for lowering serum cholesterol levels. Lovastatin was widely used and is the progenitor to modern statin analogues, such as the blockbuster drug simvastatin (13). The biosynthesis of lovastatin is accomplished in the filamentous fungus Aspergillus terreus by two HR-PKSs: the 335kDa nonaketide synthase (NKS) LovB and the 277kDa diketide synthase (DKS) LovF (14, 15, 16). LovB, in conjunction with a separate enoyl reductase LovC, synthesizes dihydromonacolin L acid (17, 18), which is subsequently oxidized by CYP450 enzymes to yield monacolin J acid (Figure 1A), the nonaketide core of lovastatin. LovF synthesizes the α-methylbutyrate side chain that is transferred by the dissociated acyltransferase LovD (19) to the C8-hydroxyl of monacolin J acid to yield the complete natural product. Acetyl-CoA, malonyl-CoA, NADPH, and SAM are utilized by LovF to yield α-methylbutyryl-S-LovF; a process involving a single condensation reaction followed by α-methylation, β-ketoreduction, dehydration, and α,β-enoyl reduction reactions at the active sites of the various catalytic domains (Figure 1B). The starting unit acetate and extender unit of malonate, originating from acetyl-CoA and malonyl-CoA respectively, are received by the malonyl-CoA:ACP acyl-transferase (MAT) domain and transferred to the free thiol of the PPant arm at the ACP active site. Under in vitro conditions, malonyl-CoA is the source of both the starter and extender units, therefore the formation of the malonyl-S-ACP intermediate is followed by decarboxylation to acetyl-S-ACP and the acetyl-intermediate is transferred to the KS domain, leaving the ACP unoccupied and available to receive the malonyl extender unit. Subsequent condensation occurs between the acetyl-intermediate residing at the KS active site and the decarboxylated malonate extender unit. The resulting acetoacetyl-S-ACP undergoes MT catalyzed C-methylation of the α-carbon. The methylated diketide undergoes β-ketoreduction, dehydration (forms an α,β-double bond), and enoyl reduction in the presence of NADPH. These reactions are catalyzed by the KR, DH, and ER domains respectively (Figure 1C).
Figure 1.
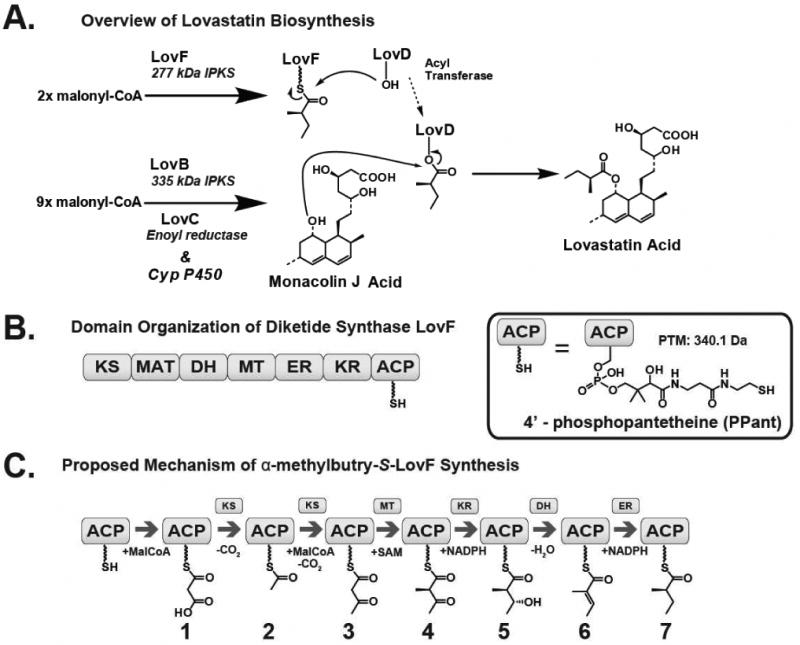
Biosynthesis of lovastatin. (A) Overview of the biosynthesis of lovastatin by LovB and LovF. LovD, a broadly acting acyl transferase, mediates the transfer of α-methylbutryate from LovF to the C8 hydroxyl position of monacolin J acid to form lovastatin acid. (B) LovF consists of multiple catalytic domains: β-ketoacyl synthase (KS), malonyl-CoA:ACP acyltransferase (MAT), dehydratase (DH), methyltransferase (MT), enoylreductase (ER), ketoreductase (KR), and acyl carrier protein (ACP). The 4′-phosphopantetheinylated ACP serves as a covalent tether for the acyl-intermediate as it is processed by the other catalytic domains of LovF (C) Based upon the catalytic domain composition of LovF, the enzyme is predicted to produce multiple covalent intermediates before biosynthesis of α-methylbutyrate.
Protein mass spectrometry (MS) has emerged as a valuable tool for the investigation of the biosynthesis done by both PKSs and the related non-ribosomal peptide synthetases (NRPSs). The value of mass spectrometry as a tool for dissecting these biosynthetic pathways can be largely attributed to the fact that intermediates are covalently bound to active site residues thus causing stable mass-shifts (20, 21, 22, 23). Additionally, the phosphopantetheinyl arm present on carrier domains of these enzymes is prone to be ejected when the protein (or protease digested peptide) bearing PPant is subjected to thermal activation by collision induced dissociation (CID) or infrared multi-photon dissociation (IRMPD) (24, 25, 26). The resulting ejected PPant arm gives rise to two characteristic 1+ ions at 261.1267 and 359.1036 m/z that will be shifted corresponding to the mass of a covalently-bound intermediate. Taking advantage of the aforementioned features of PKSs, and coupled with the use of high-resolution/high-mass-accuracy instruments, an unparalleled level of analysis of these massive enzymatic machines can be achieved.
To date there are only a handful of studies which utilize mass spectrometry to analyze iterative fungal PKSs, largely due to the difficulty in obtaining sufficient quantities of these enzymes. Recent efforts to engineer a yeast expression system from which to obtain large quantities of fully-functional holo-LovF have been successful (27), thereby providing sufficient quantities of the enzyme for thorough MS analysis. Still, many challenges exist which hinder the analysis of relatively small intermediates (<600Da) bound to large multifunctional PKSs, the most obvious of which is their massive size (>250kDa). The best studied fungal pathway is PksA, which assembles the precursor to aflatoxin B1 (28). In a recent investigation, PksA was extensively dissected into discrete domains prior to in vitro reconstitution in order for the protein to be more amenable to mass spectrometric investigations.
The aim for this study was to use mass spectrometry to detect the covalent intermediates of α-methylbutyrate biosynthesis formed by structurally intact holo-LovF. Our goals were to carry out in vitro reactions using the intact megasynthase first, followed by limited proteolysis of the PKS to generate smaller, active-site-containing peptide fragments that are more amenable to mass spectrometry analysis. We anticipated that this would provide insight into the active site occupancy of the intermediates during the biosynthesis of α-methylbutyrate of the lovastatin pathway. We report here the detection, by FT-ICR-MS, of multiple transient intermediates bound to the ACP active-site-containing peptide fragments from the intact LovF. The use of high mass-accuracy instrumentation allowed us to directly detect the results of incubation of LovF with its substrates and to determine suitable reaction conditions to generate there various intermediates.
Experimental Procedures
LovF ACP Active Site Identification for MS Analysis
The cloning, expression, and purification of C-terminal hexahistidine tagged LovF, post-translationally modified by inclusion of a copy of the npgA gene from A. nidulans to yield 4′-phosphopantetheinylated holo-LovF, were described previously (27). For the purposes of generating and identifying the LovF ACP active site, as well as all subsequent activity assays, purified holo-LovF was diluted to a concentration of 14.4 μM (4μg/μL) in 100mM of Tris-HCl buffer, pH 7.8, with 1mM TCEP in 50μL final volume (200μg total LovF). LovF was prepared as described above, and then incubated with 1mM malonyl-CoA (Sigma-Aldrich) for 10 minutes. After acyl-CoA incubation, the sample was digested using 1μg of proteomics grade trypsin (1:200 w/w) in 50 μL of 20mM NH4HCO3 reaction buffer, pH 8.2 (Trypsin Singles™ - Sigma), at 37°C. The tryptic digestion was quenched following 10 minutes using an equal volume (100μL) of 10% formic acid. Following proteolysis, the resulting mixtures of tryptic peptides from each holo-LovF reaction were fractioned using an Agilent 1200 HPLC equipped with a Jupiter reverse phase C4 5μ, 300A, 150 × 4.6 mm column (Phenomenex) using the water/acetonitrile gradient listed in the table below. The absorbance of the digestion mixture was monitored multiple wavelengths (e.g. 205nm and 280nm) by photo diode array detector. Fractions containing active site peptides were identified by comparing HPLC chromatographs of holo-LovF to those of acyl-CoA substrate-incubated LovF. All fractions analyzed, including those containing active site peptides, were collected and immediately frozen with dry ice, then lyophilized.
HPLC Gradient for Active Site Purification
Solvent A is HPLC grade water (J.T. Baker) with 0.1% TFA (Sigma-Aldrich) and solvent B is HPLC grade acetonitrile (J.T. Baker) with 0.1% TFA. Gradient was run at a flow-rate of 1 ml/min.
| min | 0.0 | 10.0 | 15.0 | 55.0 | 60.0 | 60.1 | 60.2 | 62.6 | 63.0 | 65.0 | 66.0 | 71.0 |
| % A | 90 | 90 | 70 | 30 | 10 | 10 | 95 | 95 | 5 | 5 | 90 | 90 |
| % B | 10 | 10 | 30 | 70 | 90 | 90 | 5 | 5 | 95 | 95 | 10 | 10 |
Generation of LovF-Bound Acyl-Intermediates
For all assays, holo-LovF was diluted to 4μg/μL as described above. Samples of LovF were incubated with either 0.25, 0.5, 1.0, 2.0, 4.0, 8.0, 34.7, 69.4 molar equivalents (3.6, 7.2, 14.4, 28.8, 57.6 and 115.2 μM, 0.5mM, 1mM) of malonyl-CoA with & without 1mM nicotinamide adenine dinucleotide phosphate (NADPH) and 1 mM S-adenosylmethionine (SAM) for prior to tryptic digestion. 1mM [1,2,3-13C3] malonyl-CoA (Sigma-Aldrich) was incubated with the holo-LovF, 1 mM SAM, and 1 mM NADPH to generate 13C-labelled intermediates. In order to generate acetoacetyl-S-LovF by direct acylation holo-LovF was incubated with 1mM of acetoacetyl-CoA (Sigma-Aldrich) at room temperature. For the detection of additional intermediates, holo-LovF was co-incubated first with 1mM acetoacetyl-CoA and 1mM SAM first, then subsequently incubated with 1mM NADPH.
LovF Mass Spectrometric Analysis
Lyophilized HPLC fractions were dissolved in 40-80 μL of an ESI solution containing 50% LC-MS grade methanol (J.T. Baker), 49% LC-MS grade water (J.T. Baker), and 1% formic acid (Acros Organics), then gently vortexed for 30 seconds and centrifuged for 2 minutes at 14,000 rpm. Analysis was performed using a hybrid 6.4T LTQ-FT (Thermo Electron - North America) mass spectrometer. The instrument first tuned to 816.3 m/z using the bovine cytochrome C (Sigma). Samples were introduced by electrospray ionization (1.30 - 1.5 kV; 0.3 psi) using a Biversa Nanomate 100 (Advion Biosciences) nanospray device in positive ion mode. Detection using the low-resolution LTQ was performed using 1000 millisecond (ms) maximum ion accumulation time. An 8000 ms maximum ion accumulation time and a resolution of 100,000 were used for FT-ICR-MS detection. MS2 and MS3 utilized normalized collision energy of 30%, an activation Q of 0.200, and activation times between 100-300 ms depending on intensity of the ions being isolated. Lowering the activation Q was critical for the detection of low m/z fragment ions, in particular PPant ejection ions, that were less than 1/3 the mass of the parent peptide. Although multiple charge states of the ACP active site peptides were detected the highly-charge peptides with a mass below 800 m/z were found to have very low signal intensity. In the initial rounds of MS analysis, fractions were quickly surveyed, using the ion trap detector, for the presence of phosphopantetheine ejection ion signature by subjecting individual peptide ions to collision induced dissociation (CID) and looking for the presence of a pantetheine ejection ion at (calculated mass 261.1 m/z) and the phosphopantetheine ejection ion at (calculated mass 359.1 m/z, as well as the charge-loss post-ejection parent peptide. This was accomplished using data-dependent data acquisition method to quickly survey fractions (see below) prior to more thorough analysis of individual ions by FT-ICR-MS.
For the majority in depth FT-ICR-MS analysis of the ACP occupancy, the 17+ charge state of the larger LovF ACP active-site containing peptide (Y2420-H2538 at 803.9991 m/z) was isolated and fragmented in order to detect substrates and/or intermediate occupancy of the ACP active site. Specifically, ions in the range of 800-820 m/z were isolated and fragmented. This mass range contained mostly peptide ions that resulted from the 17+ charge ACP active site peptide or mass shifted forms, shifts that were due to covalently bound intermediates, oxidation, or non-covalent adducts such as sodium and/or potassium. There were few contaminating ions. All ions between 800-820 m/z were collectively trapped and fragmented by CID to efficiently generate ejection ions being a range of intermediates depending on the reaction conditions used. Following the in vitro incubation reactions between holo-LovF and the various substrates, mass-shifted ACP peptides bearing covalently bound intermediates still fit well within the 800-820 m/z range. The identity of intermediates of α-methylbutyrate covalently bound to the pantetheine ejection ion were confirmed by both the exact mass as determined by FT-MS2 analysis, as well as IT-MS3 detection of diagnostic MS3 fragments. Pantetheine ejection ions subjected to MS3 were fragmented using a normalized collision energy of 30%, an activation Q of 0.200, and activation time of 100 ms at each stage of MSn. Typically, a minimum of 250-500 individual FT-ICR-MS spectra were acquired for each at each stage of MS1, MS2, or MS3. The spectra were averaged to post-acquisition data analysis using Qualbrowser (Thermo Electron Corp).
Data-Dependent PPant Ejection Screening Method
In order to rapidly screen mixtures of tryptic peptides in each HPLC fraction for ACP active site containing peptides, a data-dependent tandem-MS method was employed. As each lyophilized HPLC fraction was initially infused into the mass spectrometer using the Nanomate nanospray source, tandem MS data was gathered using a preprogrammed data-acquisition method that was generated with the instruments controlling software - Xcalibur 1.0 (Thermo Electron Corp). This data acquisition method involved repeatedly acquiring a single low-resolution MS1 scans followed by data-dependent CID (MS2) of the top 5 most abundant ions from the MS1 scan. Each ion was trapped and fragmented twice before being added to an exclusion list. The exclusion list size was set to 500 ions (maximum allowable by the software), with an exclusion duration that lasted the full 5 minute duration of the acquisition method. The m/z isolation width was set to +/-1.5, activation Q at 0.200, the activation time set to 1000 ms, and the activation energy set to 35%. The use of the low-resolution, high-sensitivity LTQ for the data-dependent data acquisition permitted the rapid detection of low-intensity PPant ejection ions that may have been below the detection limits of the FT-ICR-MS (even when employing in-source fragmentation) without incurring significantly longer data acquisition times. As the data-dependent method continued to collect spectra from each fraction, the previously acquired MS2 spectra were continuously refreshed and visually screened for the presence of PPant ejection ions. After the conclusion of the data-dependent method, any candidate parent ions that appeared to yield pantetheine ejection ions were subsequently trapped and fragmented. All parent ions identified as being PPant modified were subjected to more thorough FT-ICR-MS analysis.
Results
HPLC-directed LovF active site detection
To analyze the biosynthetic intermediates covalently bound to LovF by FT-ICR-MS, the entire 277kDa holo-megasynthase was treated to brief proteolytic digestion following in vitro reactions. This approach was employed to yield a complex mixture of peptide (typically 5-35 kDa) fragments, many of which contained multiple missed trypsin-cleavage sites. Digested samples of LovF were first subjected to off-line fractionation prior to MS analysis by HPLC. In order to rapidly identify the peptide fragments containing active sites, two samples of LovF were prepared: one incubated with malonyl-CoA prior to proteolysis and the other not. Both reaction mixtures were incubated with trypsin then separated by HPLC using identical gradients. Great care was taken to prepare the two samples identically (incubation time, pH, duration of proteolysis) with the sole difference being the presence of the CoA substrate. It was anticipated that after incubation with malonyl-CoA the active-site-containing peptide fragments of LovF would have differences in HPLC retention times on account of the covalently linked substrate, resulting in identifiable peak shifts on the chromatograms when compared to a substrate-free sample. The resulting HPLC chromatograms (205 nm abs) of the two digested samples were aligned to reveal separate peaks that appeared shifted in the malonyl-CoA treated sample of LovF (Figure 2). Fractions were collected, flash frozen, and lyophilized, then re-dissolved in an ESI solution containing 50% MeOH and 1% formic acid and directly introduced into the mass spectrometer using a Biversa Nanomate nanospray ESI source; a sample introduction system that allows extensive time for multiple methods of data acquisition. Each fraction was subjected to an automated, data-dependent tandem-MS (MS2) method that fragmented the majority of peptide ions and MS2 spectra were quickly inspected, using just the linear ion trap detector, for the presence a characteristic ejection ions 261.1 m/z or 359.1 m/z which would indicating the presence of the ACP active site.
Figure 2.
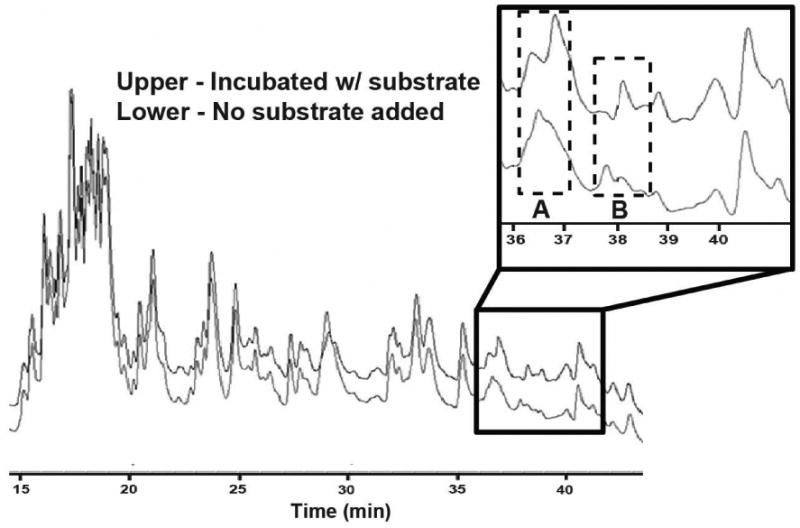
HPLC chromatogram comparison of LovF digests indicates active sites. Samples of holo-LovF and holo-LovF incubated with 1 mM malonyl-CoA were each subjected to limited proteolysis by trypsin and separated by HPLC. Comparing the chromatograms (abs 205nm) of these separate samples of LovF revealed two regions (A and B) where peak shifts occurred due to the presence of substrate covalently bound to active-site-containing tryptic peptides. Points A and B correlate to ACP active site containing peptides of two different sizes.
During the data-dependent spectra acquisition, several different parent peptides produced fragment ions with masses similar to PPant ejection ions. Therefore, to confirm the identity of these fragment ions as being PPant-related, the candidate parent ions were individually re-trapped in the ion trap portion of the instrument and fragmented for an additional round of fragmentation (MS3) (29). Fragmentation of the PPant ejection ions resulted in a series of diagnostic MS3 fragment. Parent peptide ions that were determined to yield PPant ejection ions were re-analyzed by FT-ICR-MS in order to unambiguously determine the exact mass of the peptide as well as confirming the exact mass of the PPant ejection ion. Using this overall data acquisition strategy, it was possible to map these parent peptides to the primary sequence of holo-LovF.
LovF ACP active site detection by FT-ICR-MS
When comparing the chromatograms of the trypsin digested LovF and that of LovF incubated with malonyl-CoA prior to digestion, two distinct differences noted (Figure 2 – Regions A & B) between 51.25% and 52.00% acetonitrile (36.25 to 37.00 min during the gradient) and 52.75% to 53.25% acetonitrile (37.75 to 38.25 min). FT-ICR-MS analysis of the fraction collected at point A revealed the presence of multiple charge states (11+ through 20+ observed) of a single peptide with a monoisotopic mass of 13650.86 Da (Figure 3A), within 0.9 ppm of the calculated exact mass (13650.85 Da) of the holo-ACP active-site-containing tryptic peptide Y2420-H2538 of LovF (Figure 3B). The HPLC fraction collected at point B was also found to contain multiple charge states (7+ thru 15+) of a peptide with an monoisotopic mass of 10440.26 Da (Figure 3C), which was within 1.4ppm of the theoretical mass (10440.25 Da) of the holo-ACP active-site-containing tryptic peptide A2448-H2538 of LovF (Figure 3D). Subjecting these ACP active site peptides to CID activation yielded phosphopantetheine ejection ions at 261.1267 m/z and 359.1036 m/z (Figure 4 A & B), serving as additional confirmation that these two tryptic peptides contained the phosphopantetheinylated active site residue (S2480). These two ACP peptides were consistently generated, in approximately equal quantites, following subsequent limited tryptic digestions of holo-LovF. All HPLC fractions were also inspected for peptides containing the KS and MAT active sites but these specific peptides were not identified following limited proteolysis.
Figure 3. FT-ICR-MS of holo-LovF ACP active site containing peptides.
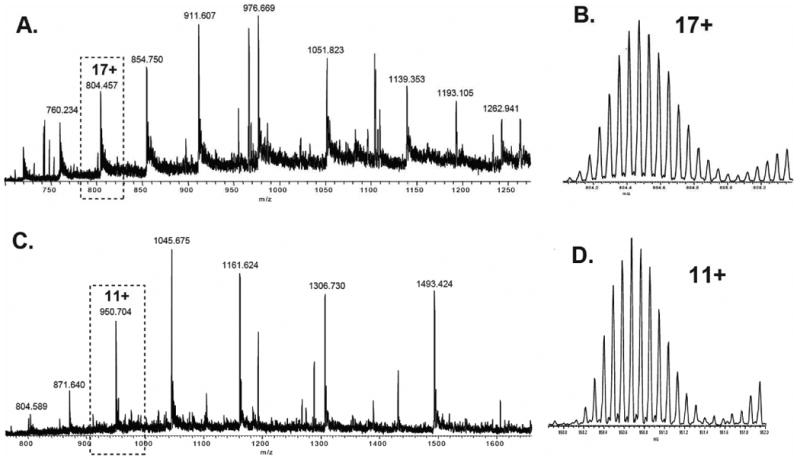
Analysis of individual HPLC fractions by FT-ICR-MS revealed the presence of two different ACP active-site-containing peptides in two separate HPLC fractions. Multiple charge states of each tryptic peptide were detected. (A) FT-ICR-MS of the HPLC fraction collected between 36-37 minutes resulted in detection of multiple charges of the ACP peptide Y2420-H2538. (B) Detail of the 17+ charge state of the ACP active site peptide Y2420-H2538. (C) FT-ICR-MS of the HPLC fraction collected between 37-38 minutes revealed multiple charges of the ACP peptide Y2448-H2538. (D) Detail of the 11+ charge state of the ACP active site peptide Y2448-H2538.
Figure 4. Phosphopantetheine elimination from holo-LovF ACP active site peptides.
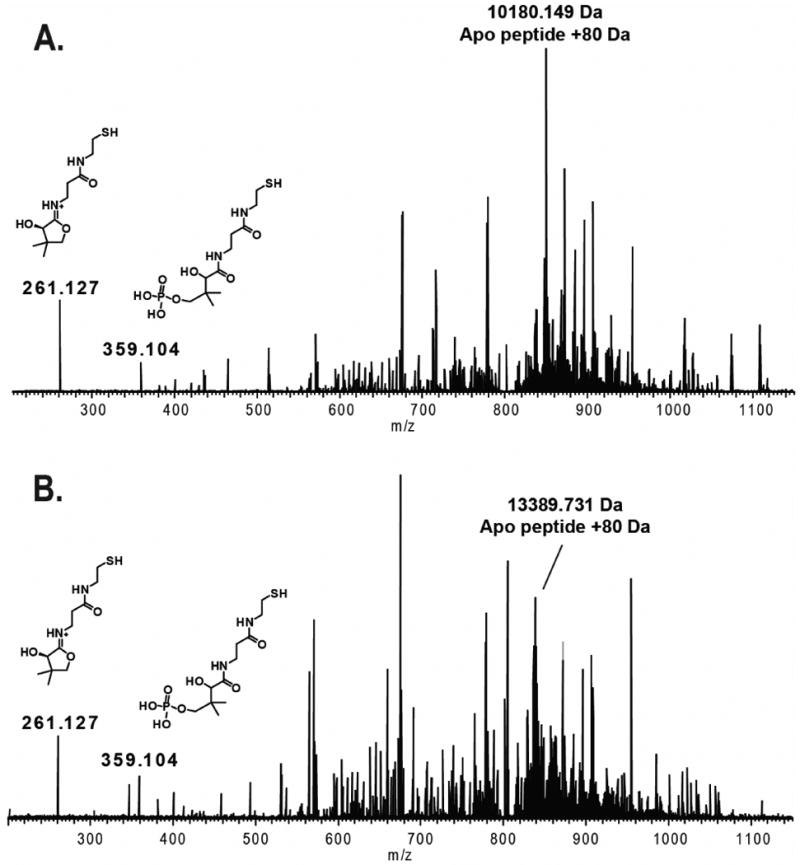
FT-ICR-MS2 of the two different sizes of ACP active-site-containing tryptic peptides from a sample of holo-LovF not incubated with malonyl-CoA revealed the presence of both a pantetheine ejection ion (Pant) at 261.127 m/z and a phosphopantetheinyl (PPant) ejection ion at 359.104 m/z. The resulting charge-loss parent phosphopeptide was clearly detectable among the fragment ions. (A) MS2 spectrum of the ACP active Y2448-H2538. (B) MS2 spectrum of the ACP active Y2420-H2538.
LovF and malonyl-CoA incubation
Having the ability to reproducibly generate ACP active site peptides from the intact holo-LovF PKS, we turned our attention towards monitoring this active site's occupancy following incubation of the megasynthase with substrate. We anticipated that incubation with malonyl-CoA would result in the formation of acetoacetyl-S-ACP intermediate. To this end, holo-LovF was initially incubated with only malonyl-CoA, digested, and then subjected to MS analysis. When analyzing the fraction that contained the ACP peptides, a wide range (e.g. 800-820 m/z) of ions were trapped, such that only a single charge-state (e.g. 17+) of the ACP active site peptide was isolated and fragmented. The use of this wide isolation window permitted the detection of mass additions to both the intact peptide and to the resulting PPant ejection ions. This allowed the determination of the relative quantities of different acyl-intermediates bound to the ACP active site at the time of proteolysis. Following incubation with malonyl-CoA, both of the holo-LovF ACP active-site-containing peptides had mass additions of 86.00 Da, corresponding to the formation of malonyl-S-LovF. Upon CID, these peptides yielded a pantetheine ejection ion with a mass of 347.128 m/z (calculated 347.1371 Da, 1.8ppm) (Figure 5). The identities of the pantetheine ejection ions were further verified by MS3 and analysis of the resulting fragmentation pattern of the pantetheine ejection ion (SI Figure 1). In addition, acetyl-S-ACP was detected in the same spectra as malonyl-S-ACP following incubation of holo-LovF with malonyl-CoA (Figure 5). The acetyl-intermediate at the ACP active site was the result of decarboxylation of the malonyl-S-ACP.
Figure 5. Detection of malonyl–S-LovF by PPant ejection.
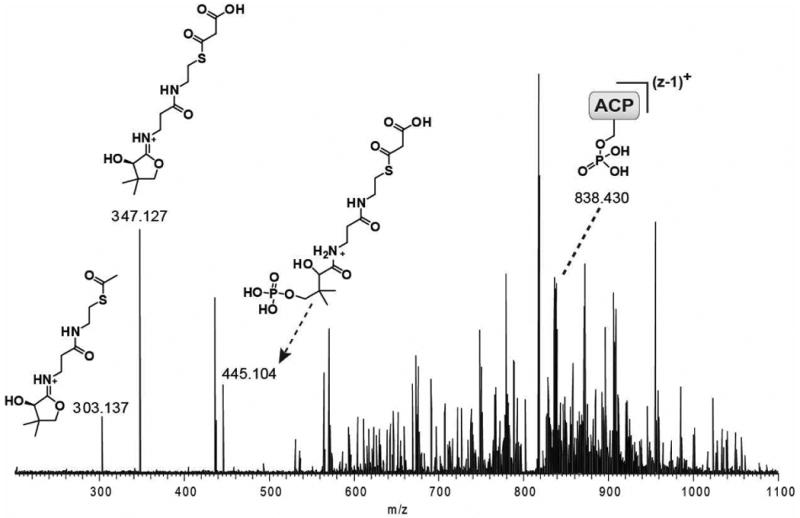
The FT-ICR-MS2 of ACP active site containing peptide following incubation of holo-LovF with 1 mM malonyl-CoA. Both pantetheine and phosphopantetheine ejection ion with a mass of 347.127 Da corresponding to the addition of malonyl was detected. The post-ejection charge-loss parent phosphopeptide (MS1 17+ to MS2 16+) was detected at 838.430 m/z (13,398.90 Da). FT-ICR-MS2 of the ACP active site containing peptide following incubation of holo-LovF with low concentrations (<0.1 mM) of malonyl-CoA resulted in the detection of a pantetheine ejection ion at 303.137 m/z corresponding the presence of an acetyl-intermediate.
Most surprising was our inability to detect the acetoacetyl-intermediate, which was expected to form in the presence of holo-LovF and malonyl-CoA, a result of decarboxylation and condensation between a malonyl extender unit and the acetate that resulted from decarboxylation of the initial malonyl unit loaded to the ACP (30). Initially, holo-LovF was incubated with 1mM malonyl-CoA for 10 minutes prior to proteolysis. Different incubation durations (e.g. 1, 2, 4, 8, 15, 30 minutes) were tested, but did not yield different results. Inclusion of SAM and NADPH during incubation also failed to generate the acetoacetyl-S-ACP. Therefore, we hypothesized that at the 1mM malonyl-CoA concentration (∼70 molar equivalents relative to holo-LovF used) was excessively high and was overwhelming the ability of holo-LovF to shuttle the diketide from the KS active site back to the ACP active site by congesting the ACP with malonyl. To test this hypothesis, holo-LovF was incubated with lower concentrations of malonyl-CoA such that 0.5, 1, 2, 4, 8 molar equivalents of substrate were used, both with and without 1mM SAM and 1mM NADPH. Given that two moles of malonyl-CoA and one mole holo-LovF are needed to generate of one mole of acetoacetyl-S-LovF, we anticipated that the lowered malonyl-CoA concentration in the incubations would yield the sought-after intermediate(s). However, the lower substrate concentrations also failed more to yield detectable quantities of the acetoacetyl-S-ACP peptide (Figure 6), producing mostly malonyl-S-ACP and, to a lesser extent, acetyl-S-ACP. At this point in time the occupancy of the LovF KS active site cannot be determined.
Figure 6. Concentration Dependent Malonyl-S-LovF Accumulation.
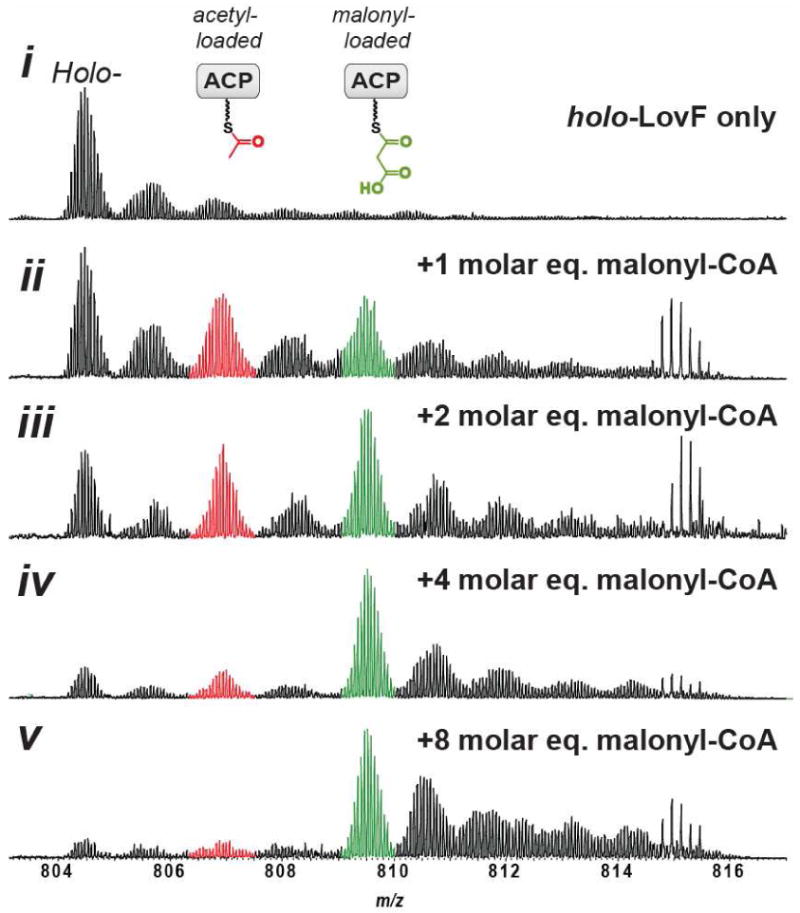
Holo-LovF was incubated with different molar equivalent concentrations of malonyl-CoA. In vitro reactions contained 14.4μM holo-LovF incubated with SAM, NADPH, and 0, 14.4, 28.8, 57.6 or 115.2 μM malonyl-CoA. The ACP active site peptide was predominately occupied with malonyl after incubation with 115.2μM malonyl-CoA. The relative intensity of acetyl-S-LovF was substantially higher at lower concentrations of malonyl-CoA.
During these sets of experiments, scaling up the quantity of enzyme being used led us detect, by FT-ICR-MS, that a small quantity (<5%) of α-methylbutyryl-S-LovF detectable after incubation with 0.5 equivalents of malonyl-CoA, as previously reported (27). PPant ejection and MS3 were used to verify this assignment. However, the α-methylbutyryl-product was found to be present even in the absence of malonyl-CoA incubation (SI - Fig 2). Therefore, we were faced with two unanticipated obstacles in our attempt to detect multiple LovF intermediates. First was the lack of detectable acetoacetyl-S-ACP and second was the presence of a small amount of α-methylbutyryl-S-ACP regardless of whether or not substrate was included. In light of these two observations, it was unclear whether LovF was capable of production of α-methylbutyrate in the absence of LovD under these particular in vitro reaction conditions. We address both of the aforementioned obstacles in the next section.
LovF incubation with 13C-labelled malonyl-CoA substrate
In order to determine whether a portion of the α-methylbutyryl-S-LovF observed, following incubation of holo-LovF with substrate, was due to de novo synthesis under in vitro reaction conditions it was necessary to further evaluate our assay using 13C-labeled substrate. Samples of holo-LovF were incubated with 1mM each of [1,2,3-13C3] malonyl-CoA, SAM, and NADPH for 10 minutes prior to trypsin digestion. Upon CID fragmentation of the ACP active site peptide, [1,2,3,4-13C4] α-methylbutyryl-loaded PPant ejection ion (349.198 m/z) was clearly detectable in the resulting tandem mass spectrum, along with the unlabelled α-methylbutyrate product that co-purified with holo-LovF (Figure 7). These results demonstrated that under our in vitro reaction conditions holo-LovF was a fully competent enzyme capable of de novo biosynthesis of [1,2,3,4-13C4] α-methylbutyrate using [1,2,3-13C3] malonyl-CoA provided during the in vitro reactions. Just as when holo-LovF was incubated with unlabelled malonyl-CoA and there was no detectable formation of acetoacetate-S-LovF, we did not detect the presence of a [1,2,3,4-13C4] acetoacetyl-loaded PPant ejection ion (calculated: 349.161 m/z) which would have been distinguishable by FT-ICR-MS from the [1,2,3,4-13C4] α-methylbutyryl-loaded PPant ejection ion.
Figure 7. [1,2,3,4-13C4] α-methylbutyrate product detection.
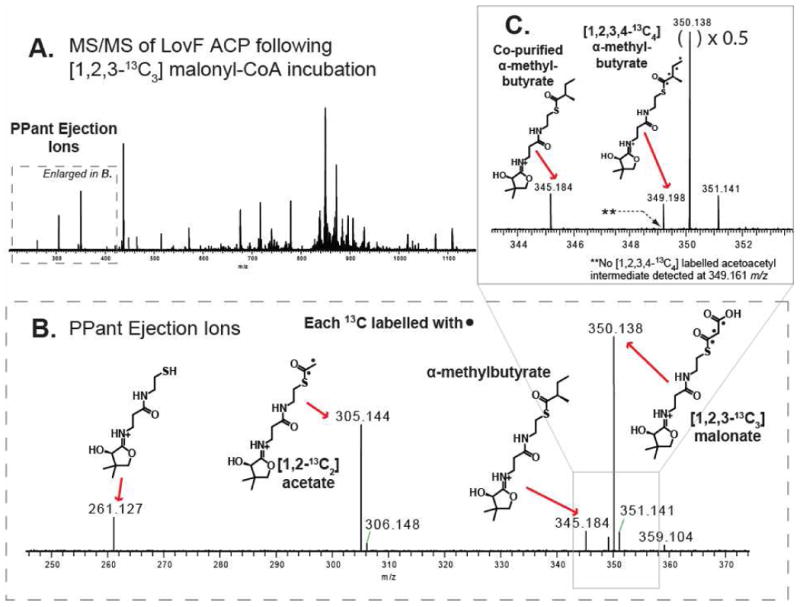
To confirm the de novo biosynthesis of α-methylbutyrate, holo-LovF was incubated with [1,2,3-13C3]-malonyl-CoA and analyzed by FT-ICR-MS (A) FT-ICR-MS2 of all parent ions between 800-820 m/z resulted in detection of multiple PPant ejection ions. (B) A more detailed view reveals the presence of a [1,2-13C2] acetyl-loaded and [1,2,3-13C3] malonyl-loaded PPant ejection ions. (C) The unlabelled α-methylbutyryl-loaded PPant ejection ion was present. However, the [1,2,3,4-13C4] α-methylbutyryl-loaded PPant ejection ion was detected in very low quantity, validating that the holo-LovF used in these assays was fully competent and capable of generating the product from malonyl-CoA added to the in vitro reactions.
Direct acylation with acetoacetyl-CoA
Because we did not detect the acetoacetyl intermediate on the ACP active site, we sought alternate solutions for evaluating the function of the individual catalytic domains by detecting the formation of the additional biosynthetic intermediates formed by LovF en route to the formation of α-methylbutyrate. In an attempt to load acetoacetate onto the ACP active site, apo-LovF was incubated with acetoacetyl-coenzyme A and Sfp (31, 32). At pHs between 7.0 – 8.0, Sfp-mediated phosphopantetheinylation of apo-LovF to acetoacetyl-S-ACP failed. The Sfp reaction was repeated in MES buffer at a lower pHs (6.0-6.5), but apo-LovF began precipitating as the pH was lowered below 7.0. Sfp reactions were reactions were typically incubated for 3 hours, so an overnight Sfp reaction at pH 7.5 was attempted. This reaction successfully yielded acetoacetyl-S-LovF, but only an extremely low conversion occurred. Incubation of apo-LovF with Sfp and CoA failed to produce holo-LovF, whereas a truncated protein comprised of just the apo-ACP domain, derived from LovF, was efficiently phosphopantheinylated by Sfp to yield either holo-ACP or acetoacetyl-S-LovF.
An alternate approach was employed which relied upon a direct acylation of acetoacetate from acetoacetyl-CoA to the free thiol of the phosphopantetheine at the ACP active site (33) of holo-LovF. Using this direct approach, holo-LovF was incubated with 1mM acetoacetyl-CoA for 15, 60, 90 and 120 minutes prior to tryptic digestion and MS analysis (Figure 8A). It was determined that maximal formation of acetoacetyl-S-LovF was achieved after 15 minutes (Figure 9B). Following direct acylation with acetoacetyl-CoA, a PPant ejection ion bearing an acetyl intermediate, was detected suggesting a retro-aldol cleavage of acetoacetyl-S-LovF to yield acetyl-S-LovF (Figure 8B). This reaction was not observed when we used the stand-alone construct in the formation of acetoacetyl-S-ACP.
Figure 8. Acetoacetyl-CoA Transthioesterification Reaction.
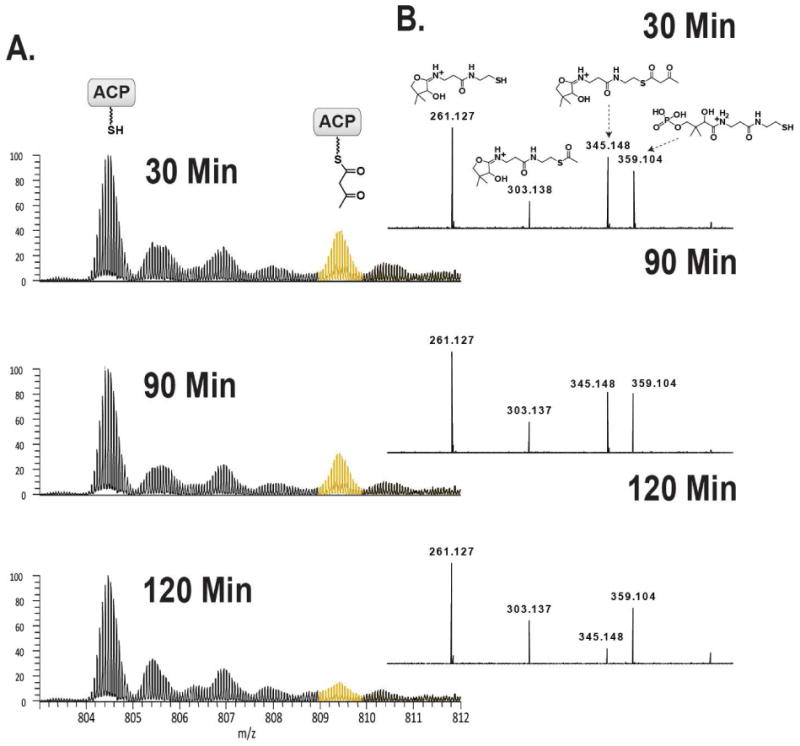
Samples of holo-LovF were incubated with 1mM acetoacetyl-CoA (acac-CoA) in order to directly transfer acetoacetate to the free thiol of the 4′-phosphopantetheine of the ACP active site. Using this strategy it was possible to circumvent the absence of this intermediate when the holo-LovF was incubated with malonyl-CoA. Holo-LovF was incubated with 1mM acetoacetyl-CoA for various lengths of time. (A) A mass shift of 84.0211 Da was observed on the ACP active site peptide corresponding to the formation of acetoacetyl-S-LovF. Longer incubation times were found to result in lower quantities of detectable acetoacetyl-S-LovF. The optimal incubation time was ultimately found to be 15 minutes. (B) FT-ICR-MS2 detection of the relative abundance of the acetoacetyl-loaded PPant ejection ion. In addition, acetyl-loaded PPant ejection ion was detected.
Figure 9. Detection of additional intermediates.
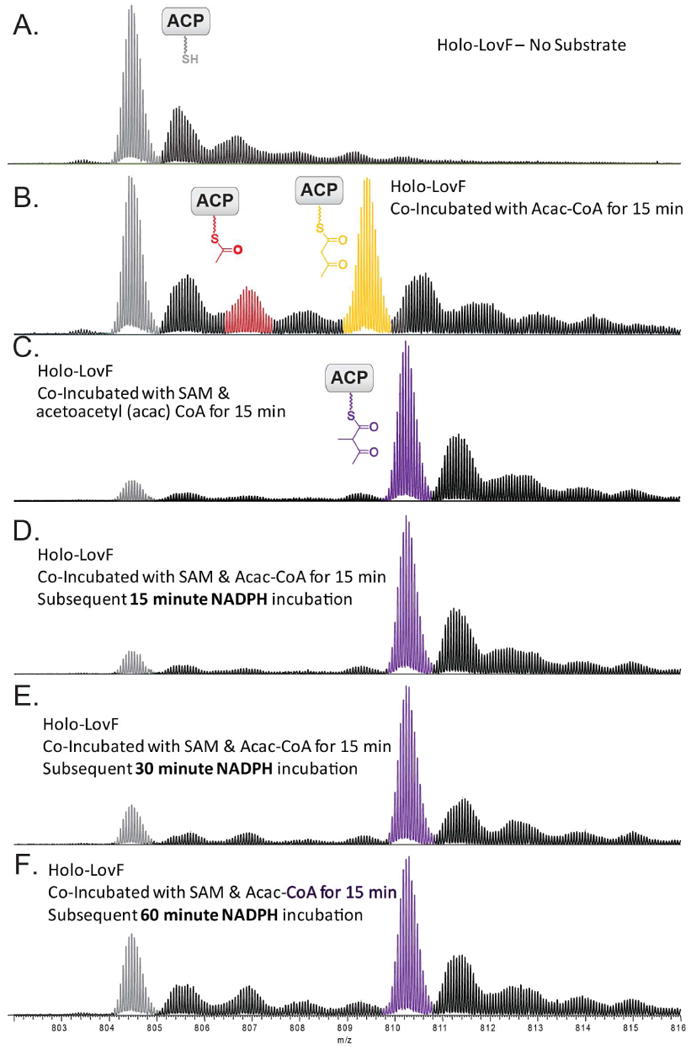
Following co-incubation of holo-LovF with acetoacetyl-CoA and SAM, NADPH was added to the solution and the reaction was allowed to incubate for 15, 30, and 60 minutes prior to tryptic digestion. (A) holo-LovF ACP active site for comparison. (B) holo-LovF with acetoacetyl-CoA and (C) holo-LovF with acetoacetyl-CoA and SAM. Incubation of holo-LovF plus SAM, followed by subsequent incubation of NADPH for (D) 15 minutes (E) 30 minutes (F) 60 minutes. From these three reactions, the formation of α-methylacetoacetyl-S-LovF was pronounced.
The direct observation of additional LovF intermediates
Direct acylation with acetoacetyl-CoA yielded sufficient quantities of acetoacetyl-S-LovF for probing the formation of additional intermediates in the presence of SAM and NADPH. In the biosynthetic pathway of α-methylbutyrate, methylation of the α-carbon is proposed to occur following formation of the diketide intermediate, which is catalyzed by the MT domain of LovF. Therefore, the logical next step following in vitro direct acylation was addition of SAM. holo-LovF and acetoacetyl-CoA were co-incubated with SAM for 15 minutes prior to limited trypsin digestion and the resulting mixture was separated by HPLC as done previously. The product of C-methylation at the α-carbon of the acetoacetyl-S-LovF was readily detected on the ACP active site peptide. During our assays, >95% of the holo-ACP peptides were found to be loaded with the α-methylated diketide following addition of SAM; the majority of the ACP active site peptide was detected as α-methyl-acetoacetyl-S-ACP (Figure 9C). In the absence of SAM, a different trend was observed. After a 15 min incubation of holo-LovF with acetoacetyl-CoA alone, at most 60% of the ACP active site peptide was occupied (Figure 9B) and detected as acetoacetyl-S-ACP.
Following methylation of the acetoacetyl intermediate, LovF is predicted to catalyze β-ketoreduction, dehydration of the β-hydroxyl group, and α,β-enoyl reduction of the intermediate to yield α-methylbutyrate (Figure 1C). holo-LovF was co-incubated with acetoacetyl-CoA and SAM, then NADPH was added followed by an additional 15, 30, or 60 minutes of co-incubation (Figure 10A iv-vi). This reaction yielded detectable, albeit small, amounts of the reduced β-hydroxy-α-methylbutyryl intermediate. The formation of this β-ketoreduced intermediate could only be detected via PPant ejection assay, yet it was still possible to observe a time-dependent build-up of this intermediate (Figure 10B). Under the in vitro incubation conditions attempted, there was no detection of the α-methylbut-α,β-enoyl intermediate, suggesting that further reduction of this intermediate to α-methylbutyrate occurs very rapidly when it is present on the ACP active site. Also, co-incubation of holo-LovF with acetoacetyl-CoA and NADPH, while excluding SAM, did not yield a detectable amount of shunt intermediates or butyryl-S-ACP. The utility of the high-mass accuracy capabilities of the FT-ICR-MS for detecting the formation of the different intermediates became exceptionally clear from this series of experiments, as several of the PPant ejection ions shared the same nominal mass, yet their exact masses differed by a mere <0.04 amu.
Figure 10. Intermediate detection by pantetheine ejection ions.
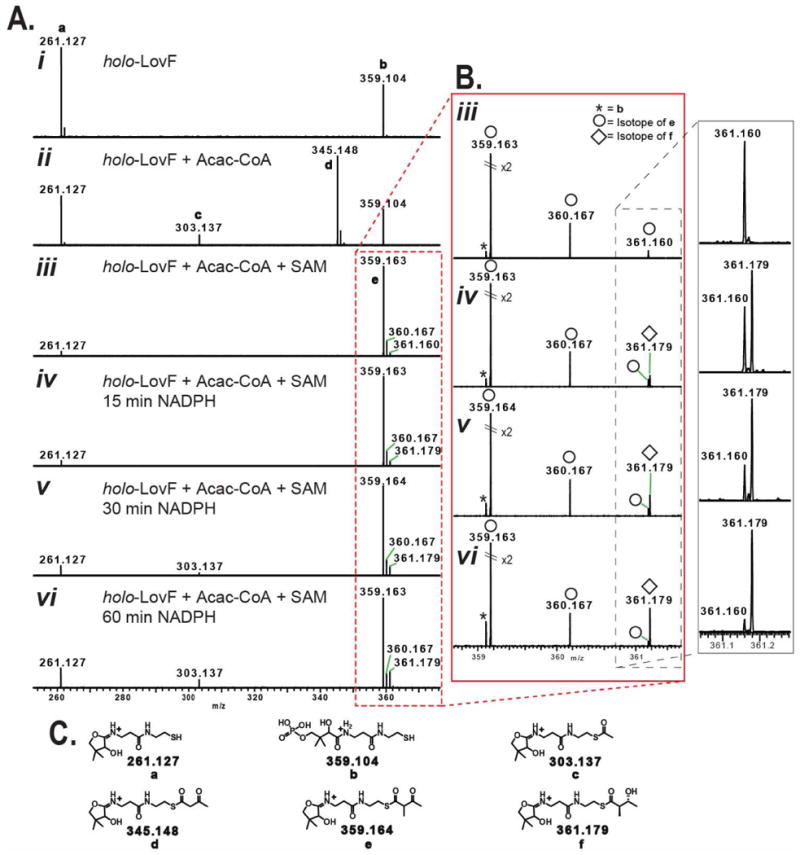
Comparison by FT-ICR-MS2 of various intermediate-loaded PPant ejection ions from holo-LovF following various in vitro incubation conditions. (A) PPant ejection ions resulting from specified incubation conditions. (B) Detail of ejection ions observed between 358-362 m/z by FT-ICR-MS. High resolution capabilities permit the differentiation of the α-methyl-acetoacetyl-loaded pantetheine ejection ion (e) and the phosphopantetheine ejection ion (b). Most critical is ability to detect the time-dependant formation and increase of low-abundance the β-hydroxy-α-methylbutyryl intermediate (361.179 m/z) and to distinguish this ion from an isotope 34S-containing isotope (361.160 m/z) of the α-methyl-acetoacetyl-loaded pantetheine ejection ion (e). An isotope of e containing a 13C was detectable at 360.167 m/z. (C) Detailed structures of various PPant ejection ions observed in the above spectra.
There were two instances in which FT-ICR-MS capabilities were crucial for accurately identifying pantetheine ejection ions bearing covalent intermediates. First, we were able to determine the identity of the α-methylbutyryl-loaded ejection ion, which co-purified following heterologous expression of holo-LovF, by FT-ICR-MS from its exact mass (345.184 m/z). Following acylation by acetoacetyl-CoA, the pantetheine ejection assay yielded an ion with a nominal mass of 345 m/z which, by FT-ICR, was found to have an exact mass of 345.148 m/z (Figure 10A ii). When attempting to observe the low intensity of the MS3 fragmentation patterns of the α-methylbutyryl-loaded and acetoacetyl-loaded ejection ions, it was necessary to use of the high sensitivity linear ion trap detector. However, the MS3 fragmentation patterns of these two ejection ions were identical. Following incubation of holo-LovF with acetoacetyl-CoA and SAM, the formation of the α-methyl-acetoacetyl intermediate was evident on the intact ACP active site tryptic peptide. The resulting pantetheine ejection ion bearing this intermediate (359.164 m/z) could be distinguished by FT-ICR-MS from the unloaded phospho-pantheine ejection ion (359.104 m/z). When optimizing the direct acylation reaction conditions, the ability to detect that a small amounta of the α-methylated-acetoacetyl intermediate was present and to distinguish it from the phosphopantetheine ejection ion (both of which were present only low quantities), was critical for determining the efficacy of the various reaction conditions attempted. This feat would not have been possible on a low resolution instrument alone. Following our discovery that direct acylation of holo-LovF by acetoacetyl-CoA in the presence of SAM resulted in a highly-abundant, highly-stable intermediate, the addition of NADPH did not immediately seem to reveal the presence of β-hydroxy-α-methylbutyryl present of the ACP active site. However, closer inspection of the pantetheine ejection ions following these reactions revealed that this intermediate was in low-abundance and the monoisotopic peak of the β-hydroxy-α-methylbutyryl-loaded pantetheine ejection ion nearly overlapped with an isotope of the highly-abundance α-methylacetoacetyl-loaded pantetheine ejection ion (Figure 10B iii-vi). The two ions differed in mass by a mere 0.018 amu. Subsequent MS3 fragmentation pattern unambiguously confirmed the ion at 361.1 m/z as ions being a pantetheine ejection ion loaded with the β-hydroxy-α-methylbutyryl intermediate (Fig SI-1).
Discussion
Highly-reducing and partially-reducing fungal polyketide synthases hold a number of very intriguing mysteries (34). The lovastatin biosynthetic pathway possesses very unique functional components, such as the trans-acting enoyl reductase LovC (14, 35), which works in unison with LovB, and the broadly acting acyltrasferase LovD. The inner workings of the polyketide synthases LovB and LovF themselves possess intriguing y not fully understood features, such as control of chain length, regioselectivity of methylation, and β-ketoreduction at select positions (36, 37). Here we detailed the utilization of high-resolution, high-mass accuracy FT-ICR-MS as a tool to investigate the biosynthesis by LovF of α-methylbutyrate, which decorates the cholesterol lowering agent lovastatin, using the ACP active site as the window through which to view the process. In these experiments, we sought to detect the biosynthetic steps performed by an intact megasynthase. The fact that the biosynthesis of α-methylbutyrate, a diketide, involves only a handful of catalytic steps made this fungal type I polyketide synthase an attractive system to study by FT-ICR-MS. Many of the experimental hurdles that were overcome during the analysis of LovF are being used to guide ongoing mass spectrometric investigations of LovB and other iterative type I PKSs.
Deconstructed/reconstituted type I PKS system are widely utilized to study the catalytic processes responsible for the creation of diverse array of compounds, and for good reason: the expression of separate domains as individual proteins allows for the fine-tuning of molar ratios of different pieces of a PKS pathway in order to facilitate examination of the functional role or substrate tolerance of a given component, much the same way type II PKSs are studied (30, 38, 39). In addition, it is possible in some cases to determine the kinetics of individual catalytic steps (40). However, keys to understanding the basis for controlling many of the catalytic steps, during successive iterations, may only be gained by studying fully-intact megasythases.
After we determined the trypsin digestion conditions that consistently generated abundant quantities of the ACP active site peptide, we anticipated that incubation of holo-LovF with malonyl-CoA would yield readily detectable quantities of several acyl-intermediates. Incubation with 1mM malonyl-CoA alone resulted primarily in a high quantity of malonyl-S-ACP, and to a lesser extent acetyl-S-ACP. Much to our surprise (and annoyance), there was no detectable acetoacetyl-S-ACP under these reaction conditions. Inclusion of SAM and NADPH did not change this observed trend, nor did shortening or increasing malonyl-CoA incubation times. Our initial hypothesis was that 1mM concentration of malonyl-CoA was simply too high relative to the quantity of holo-LovF in the in vitro reaction, resulting in congestion of malonyl at the ACP, wherein the ACP was immediately re-occupied by malonyl after decarboxylative condensation between the starter and extender unit and transfer of the resulting diketide to the KS active site. However, lowering the concentration of malonyl-CoA during incubation with LovF failed produce detectable levels of neither acetoacetyl-S-LovF, nor α-methylbutyryl-S-LovF. Therefore, the diketide intermediate may remain at the KS active site following condensation of the starter and extender units, and upon exiting the KS active site the intermediate undergoes α-methylation, β-ketoreducation, dehydration, and enoyl reduction very rapidly to form α-methylbutyrate. This hypothesis is supported by our previous observation that after in vitro reaction of LovF, LovD, monacolin J acid, and the complement of various substrates & cofactors, only α-methylbutyrate (not acetoacetate, nor α-methyl-acetoacetate) was transferred to monacolin J acid by LovD.
Further clues regarding the location of the acetoacetyl intermediate could be gleaned from the direct acylation experiments. We found that Sfp was unable to efficiently transfer phosphopantetheine to apo-LovF, while it readily converted a LovF apo-ACP construct to holo-ACP. The failure of promiscuous phosphopantetheinyl transferase (PPTase) Sfp, from Bacillus subtillus, to effectively transfer acetoacetyl-S-phosphopantetheine to apo-LovF led us to seek an alternative. As others have recently noted (33), direct acylation serves as a reliable alternative for generating acyl-S-ACPs. Direct acylation of the acetoacetate from acetoacetyl-CoA proved to be a very efficient means for generating the acetoacetyl-S-LovF, although the ACP active site was at most only 60% occupied in the presence of an ∼80× molar excess of acetoacetyl-CoA relative to holo-LovF.
While it was possible to generate acetoacetyl-S-LovF by direct acylation, this intermediate did not stably remain on the ACP active site. With time, the ACP active site quickly returned to its unoccupied holo-form even in the presence of a substantial molar excess of acetoacetyl-CoA. Whether return of the ACP active site to the unoccupied holo-state is due to transfer of the acetoacetyl intermediate to the KS active site or due to hydrolysis of the intermediate off of the ACP active site remains unclear at this point. Following direct acylation to form acetoacetyl-S-LovF, we observed the reverse reaction of the acetoacetyl intermediate to acetyl-S-LovF. This provides further insight into why we did not detect acetoacetyl-S-LovF following incubation of LovF with malonyl-CoA, as it appears that the diketide may be susceptible to a retro-aldol cleavage. Ultimately, it was clear from these experiments is that under in vitro conditions, the diketide acectoacetate does not stably reside at the holo-LovF ACP active site under steady state conditions.
Our ability to monitor the occupancy of the active site of LovF provided us with an unprecedented snapshot of several of the catalytic steps carried out by this megasythase. First, a small amount of α-methylbutyrate that purified with the overproduced protein in yeast was detected by FT-ICR-MS. However, 13C-labeling verified that α-methylbutyrate synthesized by LovF originated from the malonyl-CoA alone under in vitro reaction conditions. Also under these same conditions, the majority of the holo-LovF ACP active site peptides were occupied by the malonyl-S-thioester starter unit, and to a lesser degree, the acetyl-intermediate. The small amount of α-methylbutyryl observed relative to these two ACP-bound species, in addition to lack of all other LovF intermediates, suggested that the acetoacetyl-intermediate quickly progressed through all subsequent enzymatic tailoring reactions (α-methylation, β-ketoreducation, dehydration, and enoyl reduction) when it was present on the ACP active site. We observed that incubation of holo-LovF with acetoacetyl-CoA and SAM resulted in the abundant formation of stable α-methyl-acetoacetyl-S-LovF. The large quantity of α-methyl-acetoacetyl-S-LovF observed, after incubation of LovF with acetoacetyl-CoA and SAM, relative to all other intermediates, strongly suggested that α-methylation of the diketide, catalyzed by the MT domain, prevented the retrograde movement of the intermediate to the KS active site, or from hydrolysis. In the case of LovF, methylation of the diketide may act as one measure of quality-control in which the enzyme prevents the additional chain-elongation by limiting the accessibility of this intermediate to the KS active site. We had previously shown that under in vitro conditions LovD was capable of transferring both non-methylated intermediates from LovF to the C8 position of monacolin J (via LovD), in addition to the α-methyl-acetoacetyl intermediate (27). Of the various lovastatin (monacolin K) analogues formed, the C8-α-methyl-acetoacetyl-monacolin J (monacolin X) was the predominant shunt product. Monacolin X is also produced by a mutant strain of the lovastatin producer Monascus ruber (41). Our observation presented in this manuscript, that α-methylation of the diketide essentially locked-in the methylated intermediate at the ACP active site (>95% as α-methyl-acetoacetyl-S-Lov), provides additional insight into our why the monacolin X tends to be the predominant shunt product of the lovastatin biosynthetic pathway.
Supplementary Material
Acknowledgments
This work was supported by the NIH (5R01GM85128).
Abbreviations
- ACP
Acyl Carrier Protein
- Acac-CoA
Acetoacetyl Coenzyme A
- CID
Collision-Induced Dissociation
- CoA
Coenzyme A
- DH
Dehydratase
- DKS
Diketide Synthase
- ER
Enoyl Reductase
- FT-ICR-MS
Fourier Transform Ion Cyclotron Resonance Mass Spectrometry
- HMG-CoA
3-Hydroxy-3-Methyl-Glutaryl Coenzyme A
- IRMPD
Infrared Multi-Photon Dissociation
- KR
Ketoreductase
- KS
Ketosynthase
- MAT
Malonyl-CoA:ACP Acyl-Transferase
- MS
Mass Spectrometry
- MSn
Tandem MS (n = number of stages of fragmentation)
- MT
Methyltransferase
- NADPH
Nicotinamide Adenine Dinucleotide Phosphate
- NKS
Nonaketide Synthase
- NRPS
Non-Ribosomal Peptide Synthetase
- Pant
Pantetheine
- PKS
Polyketide Synthase
- PPant
Phosphopantetheine
- SAM
S-adenosylmethionine
Footnotes
Supporting Information Available
Figure SI-1: Confirmation of pantetheine loaded intermediates by linear ion trap MS3 of loaded phosphopantetheine ejection ions.
Figure SI-2: Detection of minor quantity of α-methylbutyryl-loaded PPant ejection ion active site of LovF.
This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Nat Prod Rep. 2001;18(4):380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 2.Cortes J, Haydock SF, Roberts GA, Bevitt DJ, Leadlay PF. An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora erythraea. Nature. 1990;348(6297):176–178. doi: 10.1038/348176a0. [DOI] [PubMed] [Google Scholar]
- 3.Schwecke T, et al. The biosynthetic gene cluster for the polyketide immunosuppressant rapamycin. Proc Natl Acad Sci. 1995;92(17):7839–7843. doi: 10.1073/pnas.92.17.7839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grimm A, Madduri K, Ali A, Hutchinson CR. Characterization of the Streptomyces peucetius ATCC 29050 genes encoding doxorubicin polyketide synthase. Gene. 1994;151(1-2):1–10. doi: 10.1016/0378-1119(94)90625-4. [DOI] [PubMed] [Google Scholar]
- 5.Moore RN, Bigam G, Chan JK, Hogg AM, Nakashima TT, Vederas JC. Biosynthesis of the hypocholesterolemic agent mevinolin by Aspergillus terreus. Determination of the origin of carbon, hydrogen, and oxygen atoms by carbon-13 NMR and mass spectrometry. J Am Chem Soc. 1985;107(12):3694–3701. [Google Scholar]
- 6.Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106(8):3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 7.Walsh CT, Gehring AM, Weinreb PH, Quadri LE, Flugel RS. Post-translational modification of polyketide and nonribosomal peptide synthases. Curr Opin Chem Biol. 1997;1(3):309–315. doi: 10.1016/s1367-5931(97)80067-1. [DOI] [PubMed] [Google Scholar]
- 8.Mootz HD, Finking R, Marahiel MA. 4′-phosphopantetheine transfer in primary and secondary metabolism of Bacillus subtilis. J Biol Chem. 2001;276(40):37289–37298. doi: 10.1074/jbc.M103556200. [DOI] [PubMed] [Google Scholar]
- 9.Cox RJ. Polyketides, proteins and genes in fungi: programmed nano-machines begin to reveal their secrets. Org Biomol Chem. 2007;5:2010–2026. doi: 10.1039/b704420h. [DOI] [PubMed] [Google Scholar]
- 10.Endo A. Monacolin K, a new hypocholesterolemic agent that specifically inhibits 3-hydroxy-3-methylglutaryl coenzyme A reductase. J Antibiot (Tokyo) 1980;33:334–336. doi: 10.7164/antibiotics.33.334. [DOI] [PubMed] [Google Scholar]
- 11.Yoshizawa Y, Witter DJ, Liu Y, Vederas JC. Revision of the biosynthetic origin of oxygens in mevinolin (lovastatin), a hypocholesterolemic drug from Aspergillus terreus MF 4845. J Am Chem Soc. 1994;116:2693–2694. [Google Scholar]
- 12.Alberts AW, et al. Mevinolin: a highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc Natl Acad Sci USA. 1980;77:3957–3961. doi: 10.1073/pnas.77.7.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manzoni M, Rollini M. Biosynthesis and biotechnological production of statins by filamentous fungi and application of these cholesterol-lowering drugs. Appl Microbiol Biotechnol. 2002;58:555–564. doi: 10.1007/s00253-002-0932-9. [DOI] [PubMed] [Google Scholar]
- 14.Kennedy J, Auclair K, Kendrew SG, Park C, Vederas JC, Hutchinson CR. Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science. 1999;284(5418):1368–1372. doi: 10.1126/science.284.5418.1368. [DOI] [PubMed] [Google Scholar]
- 15.Hendrickson L, Davis CR, Roach C, Nguyen DK, Aldrich T, McAda PC, Reeves CD. Lovastatin biosynthesis in Aspergillus terreus: characterization of blocked mutants, enzyme activities and a multifunctional polyketide synthase gene. Chem Biol. 1999;6:429–439. doi: 10.1016/s1074-5521(99)80061-1. [DOI] [PubMed] [Google Scholar]
- 16.Auclair K, Sutherland A, Kennedy J, Witter DJ, Van den Heever JP, Hutchinson CR, Vederas JC. Lovastatin nonaketide synthase catalyzes an intramolecular Diels–Alder reaction of a substrate analogue. J Am Chem Soc. 2000;122(46):11519–11520. [Google Scholar]
- 17.Ma SM, Tang Y. Biochemical characterization of the minimal polyketide synthase domains in the lovastatin nonaketide synthase LovB. FEBS. 2007;274(11):2854–2864. doi: 10.1111/j.1742-4658.2007.05818.x. [DOI] [PubMed] [Google Scholar]
- 18.Ma SM, Li JWH, Choi JW, Zhou H, Lee KK, Moorthie VA, Xie X, Kealey JT, Da Silva NA, Vederas JC, Tang Y. Complete reconstitution of a highly reducing iterative polyketide synthase. Science. 2009;326(5952):589–592. doi: 10.1126/science.1175602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie X, Watanabe K, Wojcicki WA, Wang CC, Tang Y. Biosynthesis of lovastatin analogs with a broadly specific acyltransferase. Chem & Bio. 2006;13(11):1161–1169. doi: 10.1016/j.chembiol.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 20.Hicks LM, Mazur MT, Miller LM, Dorrestein PC, Schnarr NA, Khosla C, Kelleher NL. Investigating nonribosomal peptide and polyketide biosynthesis by direct detection of intermediates on >70 kDa polypeptides by using Fourier-transform mass spectrometry. Chem Bio Chem. 2006;7(6):904–907. doi: 10.1002/cbic.200500416. [DOI] [PubMed] [Google Scholar]
- 21.Gu L, Geders TW, Wang B, Gerwick WH, Håkansson K, Smith JL, Sherman DH. GNAT-like strategy for polyketide chain initiation. Science. 2007;318(5852):970–974. doi: 10.1126/science.1148790. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Van Lanen SG, Ju J, Liu W, Dorrestein PC, Li W, Kelleher NL, Shen B. A phosphopantetheinylating polyketide synthase producing a linear polyene to initiate enediyne antitumor antibiotic biosynthesis. Proc Natl Acad Sci USA. 2008;105(5):1460–1465. doi: 10.1073/pnas.0711625105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hicks LM, Balibar CJ, Walsh CT, Kelleher NL, Hillson NJ. Probing intra-versus interchain kinetic preferences of L-Thr acylation on dimeric VibF with mass spectrometry. Biophys J. 2006;91:2609–2619. doi: 10.1529/biophysj.106.084848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, Kelleher NL. Facile detection of acyl and peptidyl intermediates on thiotemplate carrier domains via phosphopantetheinyl elimination reactions during tandem mass spectrometry. Biochemistry. 2006;45(42):12756–12766. doi: 10.1021/bi061169d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dorrestein PC, Kelleher NL. Dissecting non-ribosomal and polyketide biosynthetic machineries using electrospray ionization Fourier-Transform mass spectrometry. Nat Prod Rep. 2006;23:893–918. doi: 10.1039/b511400b. [DOI] [PubMed] [Google Scholar]
- 26.Bumpus SB, Kelleher NL. Accessing natural product biosynthetic processes by mass spectrometry. Curr Op in Chem Bio. 2008;12(5):475–482. doi: 10.1016/j.cbpa.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie X, Meehan MJ, Xu W, Dorrestein PC, Tang Y. Acyltransferase mediated polyketide release from a fungal megasynthase. J Am Chem Soc. 2009;131(24):8388–8389. doi: 10.1021/ja903203g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crawford JM, Thomas PM, Scheerer JR, Vagstad AL, Kelleher NL, Townsend CA. Deconstruction of iterative multidomain polyketide synthase function. Science. 2008;320(5873):243–246. doi: 10.1126/science.1154711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meluzzi D, Zheng WH, Hensler M, Nizet V, Dorrestein PC. Top-down mass spectrometry on low-resolution instruments: Characterization of phosphopantetheinylated carrier domains in polyketide and non-ribosomal biosynthetic pathways. Bioorg & Med Chem Ltrs. 2008;18(10):3107–3111. doi: 10.1016/j.bmcl.2007.10.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong H, Leadlay PF, Staunton J. The changing patterns of covalent active site occupancy during catalysis on a modular polyketide synthase multienzyme revealed by ion-trap mass spectrometry. FEBS Journal. 2009;276:7057–7069. doi: 10.1111/j.1742-4658.2009.07418.x. [DOI] [PubMed] [Google Scholar]
- 31.Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, Walsh CT. Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry. 1998;37:1585–1595. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- 32.Mootz HD, Schörgendorfer K, Marahiel MA. Functional characterization of 4′-phosphopantetheinyl transferase genes of bacterial and fungal origin by complementation of Saccharomyces cerevisiae lys5. FEMS Microbiol Lett. 2002;213(1):51–57. doi: 10.1111/j.1574-6968.2002.tb11285.x. [DOI] [PubMed] [Google Scholar]
- 33.Amoroso JW, Borketey LS, Prasad G, Schnarr NA. Direct Acylation of Carrier Proteins with Functionalized β-Lactones. Org Ltrs. 2010;12(10):2330–2333. doi: 10.1021/ol100684s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weissman KJ, Leadlay PF. Combinatorial biosynthesis of reduced polyketides. Nat Rev Microbiol. 2005;3(12):925–936. doi: 10.1038/nrmicro1287. [DOI] [PubMed] [Google Scholar]
- 35.Auclair K, Kennedy J, Hutchinson CR, Vederas JC. Conversion of cyclic nonaketides to lovastatin and compactin by a lovC deficient mutant of Aspergillus terreus. Bioorg & Med Chem Ltrs. 2001;11(12):1527–1531. doi: 10.1016/s0960-894x(01)00290-6. [DOI] [PubMed] [Google Scholar]
- 36.Hoffmeister D, Keller NP. Natural products of filamentous fungi: enzymes, genes, and their regulation. Nat Prod Rep. 2007;24(2):393–416. doi: 10.1039/b603084j. [DOI] [PubMed] [Google Scholar]
- 37.Weissman KJ. Peering into the black box of fungal polyketide biosynthesis. Chem Bio Chem. 2010;11(4):485–488. doi: 10.1002/cbic.201000023. [DOI] [PubMed] [Google Scholar]
- 38.Calderone CT, Bumpus SB, Kelleher NL, Walsh CT, Magarvey NA. A ketoreductase domain in the PksJ protein of the bacillaene assembly line carries out both α- and β- ketone reduction during chain growth. Proc Nat Acac Sci. 2008;105:12809–12814. doi: 10.1073/pnas.0806305105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalaitzis JA, Cheng Q, Thomas PM, Kelleher NL, Moore BS. In vitro biosynthesis of unnatural enterocin and wailupemycin polyketides. J Nat Prod. 2009;72:469–472. doi: 10.1021/np800598t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu L, Wang B, Kulkamid A, Gehret JJ, Lloyd KR, Gerwick L, Gerwick WH, Wipf P, Hakansson K, Smith JL, Sherman DH. Polyketide decarboxylative chain termination preceded by O-sulfonation in curacin A biosynthesis. J Am Chem Soc. 2009;131(44):16033–16035. doi: 10.1021/ja9071578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Endo A, Hasumi K, Nakamura T, Kunishima M, Masuda M. Dihydromonacolin L and monacolin X, new metabolites which inhibit cholesterol biosynthesis. J Antibiot(Tokyo) 1985;38(3):321–327. doi: 10.7164/antibiotics.38.321. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.