

Abstract
The spatial structure of host–parasite coevolution is shaped by population structure and genetic diversity of the interacting species. We analysed these population genetic parameters in three related ant species: the parasitic slavemaking ant Protomognathus americanus and its two host species Temnothorax longispinosus and T. curvispinosus. We sampled throughout their range, genotyped ants on six to eight microsatellite loci and an MtDNA sequence and found high levels of genetic variation and strong population structure in all three species. Interestingly, the most abundant species and primary host, T. longispinosus, is characterized by less structure, but lower local genetic diversity. Generally, differences between the species were small, and we conclude that they have similar evolutionary potentials. The coevolutionary interaction between this social parasite and its hosts may therefore be less influenced by divergent evolutionary potentials, but rather by varying selection pressures. We employed different methods to quantify and compare genetic diversity and structure between species and genetic markers. We found that Jost D is well suited for these comparisons, as long as mutation rates between markers and species are similar. If this is not the case, for example, when using MtDNA and microsatellites to study sex-specific dispersal, model-based inference should be used instead of descriptive statistics (such as D or GST). Using coalescent-based methods, we indeed found that males disperse much more than females, but this sex bias in dispersal differed between species. The findings of the different approaches with regard to genetic diversity and structure were in good accordance with each other.
Keywords: GST, host–parasite coevolution, Jost D, maximum likelihood methods, population genetics, social insects
Introduction
Host–parasite relationships are among the most widespread of all species interactions and important for the ecology and evolution of the interacting species. Host–parasite interactions can lead to coevolutionary arms races, where patterns of local adaptation can change over time and space. This spatial structure is often referred to as a geographic mosaic of coevolution (Thompson, 1994). We are interested in the coevolution of an obligate social parasite and its two host species and specifically in its spatial dimension. We use population genetic markers (microsatellites and MtDNA sequences) and explore different analytical methods to gain a better understanding of the coevolutionary dynamics of this host–parasite system. For many parasites, dispersal is tightly linked to host population structure either because parasites are transmitted directly from host to host or because the transmission stages can not survive long outside the host. This leads to the structure of host populations shaping parasite population structure, which has been indeed found in a number of parasite host systems (Nejsum et al., 2005; Prugnolle et al., 2005; Ren et al., 2008).
Theory predicts that relative levels of gene flow and population sizes of hosts and parasites determine their evolutionary potential and are therefore among the main determinants of the coevolutionary dynamics (Lively, 1999; Gandon & Michalakis, 2002). Given that migration rates are still moderate enough to permit population differentiation, the opponent with higher influx of new alleles through migration or mutation is expected to be locally adapted. Comparative studies of host and parasite population structure and genetic diversity have been conducted in various study systems, with very diverse outcomes. In different snail-trematode systems, population structure was consistently more pronounced in the snail host than in the parasite (Dybdahl & Lively, 1996; Davies et al., 1999; Keeney et al., 2009). The amount of genetic diversity was either higher for the host (Dybdahl & Lively, 1996), or for the parasite (Keeney et al., 2009) or it did not differ (Davies et al., 1999). Parasites in these systems appear to benefit from higher gene flow and albeit they were not always more genetically diverse, they were repeatedly reported to be locally adapted (Dybdahl & Lively, 1995; Manning et al., 1995; Mukaratirwa et al., 1996). A study on a parasitic plant also found stronger population structure for the host plant than for the parasite, similar levels of genetic diversity and local adaptation for the parasite (Mutikainen & Koskela, 2002). Unfortunately, it is not possible to compare structure and diversity between hosts and parasites directly, if different genetic markers are used for the different species, because the reported FST measures depend on levels of diversity (Jost, 2008).
Social parasites of social insects are usually closely related to their hosts and are likely to have similar mutation and migration rates and comparable population sizes. Indeed, two recent studies on comparative population genetics in social parasites and their hosts, found no significant differences in either diversity or structure between hosts and parasites (Hoffman et al., 2008; Foitzik et al., 2009b). On the other hand, two other studies detected stronger population structure for the parasites and higher genetic diversity in the hosts (Trontti et al., 2006, Vepsalainen et al., 2009). Social parasites and their hosts are interesting study systems for host–parasite interactions, because due to their relatedness, the main difference between host and parasite species is their life style, as host and parasite respectively. An additional advantage is that the same molecular markers can be used for the host and the parasite alike.
We study a host-social parasite system from the north-eastern United States and Canada where the two host species and their shared social parasite are closely related ant species (Emery, 1909; Beibl et al., 2005). The host species are Temnothorax longispinosus and T. curvispinosus, small ants which nest in acorns, hickory nuts or twigs on the forest floor. The parasite is the slavemaking ant Protomognathus americanus, which depends on enslaved host workers for all routine colony tasks such as brood care and foraging. This obligate social parasite exerts strong selection pressures on its hosts through frequent and destructive slave raids (Foitzik & Herbers, 2001). The host species display anti-parasite defence strategies such as enemy recognition, fighting and evacuation strategies and slave rebellion (Alloway, 1980; Foitzik et al., 2001, Brandt and Foitzik, 2005; Achenbach & Foitzik, 2009) and these defences may have triggered parasite counter-adaptations (Brandt et al., 2005).
In an earlier population genetic study, mainly based on MtDNA sequences, we found that the parasite P. americanus has higher levels of gene flow between sites, but overall less genetic diversity than its main host species T. longispinosus (Brandt et al., 2007). In this study, we conducted a more comprehensive population genetic study to take a fresh look at genetic diversity and the population structure of the parasite and its two host species. We used more genetic markers (eight microsatellite loci vs. four in the previous study), studied more individuals (781 independent individuals from separate colonies vs. 184 in the previous study) and conducted additional and more sophisticated genetic analyses. We expect to find the highest genetic diversity in T. longispinosus, because it has the highest nest densities, followed by the second host species T. curvispinosus and then the social parasite P. americanus, which has only a tenth of the density compared with its hosts. Additionally, based on earlier data (Brandt et al., 2007) and the biology of the species, we expect that the facultatively polygynous host species, where queens can return to the mother nest after mating, are more structured than the monogynous parasite species, P. americanus, where young queens found new colonies after dispersal and mating flights.
Our research objectives are, firstly, to compare genetic diversity and population structure between the three species and interpret these differences in the light of coevolution. Secondly, we aim to compare different methods which are available to study diversity and structure. For comparing genetic diversity between populations, species and loci, we focus on the effective number of alleles, which is a better measure of diversity than heterozygosity if the population-scaled mutation rate is high. To study population structure, we calculate D statistics (Jost, 2008), which are based on the effective number of alleles and are therefore better suited for highly variable markers. We apply a permutation test to ensure that D-values are significantly different from expectations under panmixia. To disentangle the effects of mutation and migration, we use two coalescent-based approaches to estimate population-wide mutation and migration rates.
Because the D statistic has not previously been applied to sequence data, we explore the effect of sequence length on D. For the MtDNA, we also use a test that takes into account genetic distances, because this may unveil different features of population structure. In addition, we analyse our data with the program Structure, which uses multi-locus genotypes to assign individuals to clusters (Pritchard et al., 2000; Falush et al., 2003).
Methods
Sampling sites and data collection
Colonies of the parasite P. americanus and its hosts T. longispinosus and T. curvispinosus were collected between 2002 and 2008 in 13 locations throughout the northwestern United States and at one site in Canada (Table 1, Fig. 1). Ants were found in acorns, nuts and twigs on the floor of deciduous forests.
Table 1.
Number of colonies sampled per species at each of the study sites. For geographic position of sampling locations see also Fig. 1
| Location | T. longispinosus | T. curvispinosus | P. americanus | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Abbreviation | Town or park | North | West | Microsatellites | Mt DNA | Microsatellites | Mt DNA | Microsatellites | Mt DNA |
| CT | Schenipsit, Connecticut | 41.95 | 72.40 | 4* | 5 | 10 | |||
| IL | Ottawa, Illinois | 41.37 | 88.83 | 50 | 6 | 48 | |||
| LI | Heckscher SP, Long Island | 40.70 | 73.16 | 2* | 2* | ||||
| MA | Belmont Rock Meadows Park, Boston, Massachusetts | 42.40 | 71.20 | 5 | 3* | 4* | 9 | ||
| MD | Maryland | 39.13 | 77.23 | 2* | 1* | 42 | 3* | 2* | |
| MI | Hell, Michigan | 42.42 | 83.97 | 3* | 2* | 48 | |||
| NY | Huyck Pres., Rensselaerville New York | 42.52 | 74.15 | 50 | 7 | 50 | 14 | ||
| OH | Harpersfield, Ohio | 41.75 | 80.95 | 50 | 4* | 50 | 5 | 50 | 20 |
| OH2 | Kraus Wild. Preserve, Ohio | 40.12 | 82.95 | 46 | 12 | ||||
| PA | Elliot State Park, Pennsyl. | 41.10 | 78.52 | 33 | 3* | 10 | 3* | ||
| QU | Gatineau, Quebec (Canada) | 45.48 | 75.85 | 2* | |||||
| VT | East Middlebury, Vermont | 38.88 | 78.20 | 32 | 9 | 2* | 4* | ||
| VA | Shenandoah NP, Virginia | 43.97 | 73.08 | 2* | 44 | 3* | 1* | 1* | |
| WV | Watoga SP, West Virginia | 38.10 | 80.12 | 50 | 7 | 50 | 15 | 49 | 16 |
Not used for most analysis, see Results section.
Fig. 1.

Collection sites in the United States of America and Canada. The geographic position of the collection sites can be found in Table 1.
For microsatellite analysis, DNA was extracted from a total of 272 T. longispinosus, 338 T. curvispinosus and 171 P. americanus workers. Each worker came from a different nest (for sample size per location see Table 1). DNA was extracted using Puregene DNA extraction kit, Gentra System. Eight highly variable microsatellite loci were amplified with PCR using the primers L4 (Giraud et al., 1999), L5 and L18 (Foitzik et al., 1997), LXA GT1 (Bourke et al., 1997), LX GT218 and LX GT223 (Hamaguchi et al., 1993), MS86 (Azuma et al., 2005) and Myrt 3 (Evans, 1993). For P. americanus the loci GT223 and MS86 did not amplify reliably and were not used. The polymerase chain reactions were performed as described in Foitzik et al. (2009b).
For the MtDNA analysis, we amplified and sequenced two overlapping DNA fragments covering about 1400 bp of the mitochondrial Cytochrome Oxidase I and II genes, using the methods described in Brandt et al. (2007). We added new sequences to enlarge the dataset from Brandt et al. (2007) to a total of 50 (22) T. longispinosus, 36 (22) T. curvispinosus and 78 (60) P. americanus individuals (in brackets are the number of individuals that were already used in Brandt et al., 2007) (for sample sizes per location see Table 1).
Data analysis
For the microsatellite data, the observed allele lengths were rounded to integers (binned) using the software TANDEM (Matschiner & Salzburger, 2009). Summary statistics were calculated using R (R Development Core Team 2005), with the packages seqinr (Charif & Lobry, 2007), APE (Paradis et al., 2004) and fields (Furrer et al., 2009). The microsatellite data were tested for heterozygote deficiency (test for H–W equilibrium, using a U test (Rousset & Raymond, 1995) and for linkage disequilibrium between loci (Fisher exact test for each pair of loci across all locations) using genepop (webversion: http://genepop.curtin.edu.au).
Genetic diversity
To compare levels of genetic variation between species, locations and loci, the effective number of alleles, ne, was used, which is the reciprocal of homozygosity (J):
| (1) |
. HS was calculated following Nei & Chesser (1983). We prefer ne, because, unlike heterozygosity (H), it changes linearly with diversity. If a population goes from 4 to 2 to 1 alleles (always at equal frequencies), heterozygosity goes from 0.75 to 0.5 to 0 (first a decrease of 33% and then a decrease of 100%, although in both cases half of the alleles are lost). The effective number of alleles would go from 4 to 2 to 1, which better reflects genetic diversity (see Jost, 2008).
To test whether species, location or locus influenced ne, we fitted a linear model with ne as response variable and location, species and locus as explanatory variables using R. Separate analyses were performed for the microsatellite loci and MtDNA. We did not allow for interactions. For the linear model, we had to make the assumption that samples from different locations are independent, which is not strictly the case as they are related by genealogy. We repeated the same analysis with explanatory variables latitude and longitude.
Population structure
We use several methods to test for population structure in the three species. First of all, we use the D and GST statistics to quantify differentiation for both microsatellite and MtDNA sequence data and we apply a permutation test to test for significance. We then discuss the influence of sample sizes and the length of the DNA sequence. We quantify differentiation using genetic distances between MtDNA sequences and determine whether there is a pattern of isolation-by-distance. Finally, we apply the program Structure (Pritchard et al., 2000; Falush et al., 2003) to the microsatellite data to detect higher level geographic structure and compare the outcomes with the D analysis.
D and GST analysis
Standard F statistics and their relatives, such as GST, greatly underestimate differentiation when heterozygosity is high, which is usually the case for microsatellite markers (Hedrick, 2005; Jost, 2008). When heterozygosity is high, GST is automatically low, even when populations carry completely distinct sets of alleles. We therefore also calculated the D statistic, which was proposed by Jost (2008), and which is independent of within population diversity.
Jost D is usually written as follows:
| (2) |
(where N is the number of sampled subpopulations, HT is heterozygosity over all samples, HS is mean heterozygosity within the subpopulation samples). To get an intuitive understanding of D, it is useful to rewrite eqn 2 in terms of effective number of alleles (ne).
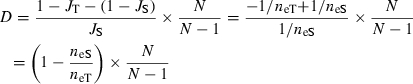 |
(3) |
From eqn 3, one can see that D depends on the ratio between neS and neT. In the finite-island model, D depends on the mutation rate and the migration rate between any two subpopulations (see eqn 22 in Jost, 2008). If mutation rates are expected to be the same between two species or loci, differences in D can be interpreted as differences in migration rates.
Permutation test for significance of D and GST
We tested the significance of the D and GST values with a simple permutation test (as described, for example, in Hudson et al., 1992), using 1000 randomized datasets. For each randomized dataset, individuals are redistributed over the subpopulations in such a way that all individuals are used (no replacement) and the sizes of the population samples do not change. We used individuals, and not alleles, as the unit for permutation, because the population samples were not in Hardy–Weinberg equilibrium, so using individuals is the conservative choice. From each randomized dataset, D and GST are calculated. The P-value of the test is the percentage of D (or GST) values from the randomized datasets that are higher than or equal to the real D (or GST) value. We used the same randomized datasets for D and GST and the resulting P-values are exactly the same for D and GST. Gerlach et al. (2010) also noted that statistical significance did not differ between D and GST. Note that this permutation test is not the same as the bootstrapping procedure as implemented in the web-based smogd software (Crawford, 2009). The bootstrapping procedure creates new datasets consisting of subsets of the original data, but without redistributing the individuals over the locations.
Statistical issues: sample sizes and length of MtDNA sequence
Simulations show that the power to detect population differentiation (with D or GST) is reduced when some of the samples are very small (Hudson et al., 1992). We therefore removed all samples with fewer than five individuals from the analysis (see Table 1).
For the MtDNA sequences, we calculated D and GST based on alleles. Longer sequences usually create more different alleles, and the values of D and GST will be influenced by the length of the DNA sequence which is used for a study. We studied this effect by analysing parts of the DNA sequence, as if we had only sequenced a shorter fragment. For each fragment length (100, 300, 500, 700, 900, 1100 bp), we took a fragment from our dataset, starting at a random location in the sequence and calculated D, GST and P-values. We repeated this 20 times for each fragment length and averaged over these 20 runs. For each randomly selected fragment, a P-value was estimated by 1000 permutations. We estimate the power to detect population structure as the percentage of these 20 fragments which showed a P-value of < 0.05.
Using genetic distances
The previous analyses of the microsatellites and MtDNA data are based on alleles, not genetic distance. When calculating D, there is no elegant way to include genetic distances, because it is based on the effective number of alleles.
In principle, one can calculate D for each nucleotide and then average over nucleotides, so that when more nucleotides have high D values, the average D value will be high. However, D per nucleotide is not very informative. For most nucleotides D will be 0, because any sequence which can be aligned will consist mostly of nucleotides which are monomorphic. Even for sites that are polymorphic, D will still be low because there are usually a maximum two states at each nucleotide. D is highest if each subpopulation carries unique alleles, but for single nucleotides this is not possible. In practice, this means that D per nucleotide must go down as more locations are sampled.
By contrast, genetic distances can be easily integrated in GST analyses by using heterozygosities (HT and HS) per nucleotide and averaging over all nucleotides (HTnuc and HSnuc). These heterozygosities increase with genetic distance between individuals. Using HTnuc and HSnuc, one can calculate  (Nei, 1982). Note that γST does not suffer from the same problems as GST does because there are usually only two and definitely not more than four allelic states per nucleotide, so heterozygosity cannot be high and is unlikely to reach a level where it does not increase (approximately) linearly with diversity. Significance can be estimated with the same permutation test as described before.
(Nei, 1982). Note that γST does not suffer from the same problems as GST does because there are usually only two and definitely not more than four allelic states per nucleotide, so heterozygosity cannot be high and is unlikely to reach a level where it does not increase (approximately) linearly with diversity. Significance can be estimated with the same permutation test as described before.
Isolation by distance
To test for isolation by distance, a Mantel test was performed over the whole dataset using the web based software ibdws (Jensen et al., 2005) for both microsatellites and MtDNA. For the microsatellites, we used pairwise D values, for the MtDNA we used genetic distances (mean number of pairwise differences) directly.
Structure analysis
We analysed the microsatellite data with the program Structure 2.3.1 (Pritchard et al., 2000; Falush et al., 2003). We did the standard analysis with admixture, a burn in period of 50 000 iterations, then 100 000 iterations of the Markov chain. K (number of clusters) ranged from 1 to 6 for all species and we did four repeats per K value and used delta K (Evanno et al., 2005) to decide on the ‘true’ value of K. To determine delta K we used Structure Harvester (http://taylor0.biology.ucla.edu/struct_harvest/). We compared the outcome of Structure with pairwise D values averaged over loci.
Estimating population-wide mutation and migration rates
The previous analyses describe the observable pattern of population structure, which is created by processes such as mutation and migration. Ideally, we would like to infer the parameters of these underlying processes using a model based approach (Beaumont et al., 2010). This is possible, in principle, using a likelihood or Bayesian framework, as implemented in programs such as migrate-n (Beerli & Felsenstein, 2001) or Migraine (Rousset & Leblois, 2007). Realistically, parameter estimation is only possible in a simplified model with fewer parameters than the natural system, for example by ignoring unsampled demes, which means that the estimates obtained may be biased by model misspecification. Some of these model misspecifications have been studied. Specifically Beerli (2004), Slatkin (2005) and Rousset & Leblois (2007) found that even if unsampled demes are ignored, it is often still possible to estimate well the product of θ (population-wide mutation rate) per subpopulation and NT (the number of subpopulations).
For the microsatellite data, we use Migraine (Rousset & Leblois, 2007), which is based on the importance sampling algorithm by De Iorio & Griffiths (2004a,b);, to estimate θ*NT and the migration rate M for each locus and each species. We assume that our data are from a finite-island model (with equal population sizes in each subpopulation and equal migration rates between all subpopulations). Two aspects of our data make us confident that the finite-island model is not too far from reality for our species. (i) Our samples from different locations do not differ significantly in genetic diversity, which indicates that local population sizes are similar. (ii) Values of pairwise differentiation are similar between all locations (see Fig. 6), which suggests that migration rates are relatively symmetric. We ignore unsampled subpopulations and assume that mutations follow a K-allele model and that population size is constant. We run Migraine for each locus separately with the following settings: the shape parameter, g, is fixed at 1, so that migration is independent of distance and the model is a finite-island model. We first estimated likelihoods by 10 runs at each of 2000 points in the parameter range 2Nμ [0,40] and 2Nm
[0,40] and 2Nm [0,100], and in a second round, 30 runs at 512 points in a range of high likelihoods as determined by the program in the first round. In one case (locus Myrt3 for T. longispinosus), the original range did not include the whole confidence interval, so we reran with 2Nm
[0,100], and in a second round, 30 runs at 512 points in a range of high likelihoods as determined by the program in the first round. In one case (locus Myrt3 for T. longispinosus), the original range did not include the whole confidence interval, so we reran with 2Nm [0,100]. The output of Migraine for the mutation rate is divided by the number of samples, N, so we have to multiply the output by N to get θ*NT. For the migration rate, the output is also divided by N, which we use as is.
[0,100]. The output of Migraine for the mutation rate is divided by the number of samples, N, so we have to multiply the output by N to get θ*NT. For the migration rate, the output is also divided by N, which we use as is.
Fig. 6.
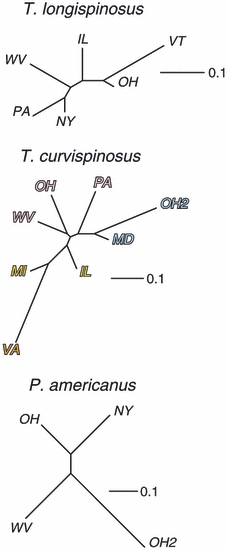
Neighbour-joining trees using pairwise D values. Pairwise D values were averaged over the eight microsatellite loci (six loci for Protomognathus americanus). Names at the tips of the trees correspond to the locations of the samples (see Table 1). The colours of the labels in Fig. 6b reflect the four clusters in b.
For the MtDNA data, we run migrate-n (Beerli & Felsenstein, 2001), which uses an MCMC algorithm to find maximum likelihood estimates. We assume a finite-island model. We ran the program with the following settings: we ran 10 short-chains, with 50 000 sampled trees, and one long chain with 1 000 000 sampled trees, a 100 step increment in both cases and a burn in of 100 000 steps. migrate-n also outputs an estimate of θ, whereas we are interested in θ*NT. We therefore multiply the estimated θ value with N (the number of subpopulations). Unlike Migraine, migrate-n estimates migration rates between all sampled populations (e.g. the migration rate from subpopulation 1 to subpopulation 2). We multiply the estimated migration rate with the number of neighbouring populations to get the total migration rate M.
To test whether θ*NT or M are different between the species of interest, we fit a linear model with the estimated values for θ*NT and M as response variables and species and locus as explanatory variables using R.
Results
Binning of the microsatellite data
The authors of TANDEM suggest that good loci have an average rounding error which is below 10% of the repeat size. Only three of the eight loci were below this threshold for T. longispinosus (Myrt3, GT223 and MS86), two of the six for P. americanus and also two of eight for T. curvispinosus were below this threshold (L5 and L18 for both species). Nevertheless, we decided to keep all loci in the analysis. For the demography and the population structure analysis, we found no differences between the high and the low quality loci. For the genetic diversity analysis, the data of only the high quality loci are not enough to make a comparison between the species, but also for this analysis, the results seem consistent over loci.
Linkage disequilibrium and Hardy–Weinberg
Hardy–Weinberg equilibrium over all subpopulations and loci was rejected because of heterozygote deficiency for all three species (P< 0.0001). The underlying reason could be the presence of null-alleles and/or inbreeding. We proceeded to estimate the proportion of null-alleles for each species, locus and population, using the method by Chakraborty et al. (1992) under the assumption that heterozygote deficiency was entirely because of null-alleles. Averaging the proportion over populations gave us upper limits of the proportion of null-alleles for each locus and species which ranged from 0 to 0.39 with an average of 0.10. When performing the permutation test for D and GST we will therefore permute individuals and not alleles.
Significant linkage disequilibrium was found in all three species: T. longispinosus had six significant locus pairs (P< 0.01) out of 28 (L5 & GT1, L5 & L18, GT1 & L18, L5 & Myrt3, GT1 & Myrt3, L18 & Myrt3), T. curvispinosus seven of 28 (L5 & GT1, L5 & L18, L18 & Myrt3, L5 & GT223, Myrt3 & GT223) and P. americanus nine of 15 (L5 & GT1, L5 & L18, GT1 & L18, L5 & Myrt3, GT1 & Myrt3, L18 & Myrt3, L18 & GT218, Myrt3 & GT218, L18 & L4). This could mean that the different loci are not independent and not too much weight should be placed on evidence which comes from different loci.
Genetic diversity
The mean effective number of alleles (ne) averaged over loci and locations varied between 7 and 11 for the three species for the microsatellites (Table 2, Fig. 2a) and between 6 and 10 for the MtDNA data (Table 2, Fig. 2b). For the microsatellites, we found that T. longispinosus has significantly lower ne than the other two species (P< 0.005). The different loci had different values of ne (all P< 0.05), but the study sites were not significantly different (P= 0.98). We found no effect of longitude and latitude (P= 0.60). For the MtDNA dataset, we found no effect of species or location (all P> 0.05).
Table 2.
Comparison of ne (effective number of alleles) and H (heterozygosity) values for all loci (eight microsatellite loci and MtDNA) and species. For the MtDNA, we also report π (mean pairwise differences per nucleotide). In all cases, values are calculated within each population and averaged over all populations
| Species | L5 | GT1 | L18 | Myrt3 | GT218 | L4 | GT223 | MS86 | Mean microsatellites | MtDNA |
|---|---|---|---|---|---|---|---|---|---|---|
| T. longispinosus | ||||||||||
| ne | 11.0 | 12.9 | 8.3 | 6.1 | 3.2 | 12.5 | 3.3 | 1.6 | 7.4 | 9.7 |
| H | 0.88 | 0.92 | 0.88 | 0.83 | 0.65 | 0.92 | 0.57 | 0.31 | 0.74 | 0.86 |
| π | 0.020 | |||||||||
| T. curvispinosus | ||||||||||
| ne | 4.8 | 22.2 | 7.6 | 11.9 | 4.8 | 15.4 | 9.4 | 2.9 | 9.8 | 7.3 |
| H | 0.75 | 0.95 | 0.86 | 0.91 | 0.74 | 0.90 | 0.86 | 0.50 | 0.81 | 0.76 |
| π | 0.005 | |||||||||
| P. americanus | ||||||||||
| ne | 10.7 | 16.4 | 11.3 | 13.2 | 4.3 | 10.9 | – | – | 11.1 | 6.4 |
| H | 0.91 | 0.91 | 0.90 | 0.92 | 0.66 | 0.89 | – | – | 0.86 | 0.83 |
| π | 0.010 | |||||||||
Fig. 2.

(a) Effective number of alleles (ne) for microsatellites. Values are averaged over populations and loci. Bars indicate standard errors. Temnothorax longispinosus has a lower effective number of alleles than the other two species. (b) Effective number of alleles (ne) for MtDNA sequences for all three species. Values are averaged over populations. Bars indicate standard errors.
Population structure
GST and D
GST values for the microsatellite loci were low for all three species, on average 0.03 for T. longispinosus, 0.07 for T. curvispinosus and 0.03 for P. americanus, as expected for highly variable loci. The values, however, were always higher than expected under panmixia (P-values between 0.001 and 0.034, Table 3 and Fig. 3a). Jost D values were much higher than the GST values: T. longispinosus: 0.25, T. curvispinosus: 0.35, P. americanus: 0.58. D values were significantly lower for T. longispinosus than for the other two species (P= 0.022), indicating a higher migration rate for this species. For the MtDNA data, both GST and D values were consistently higher than for the microsatellite loci (Table 3 and Fig. 3a). The P-values were below 0.05 for all species (Table 3 and Fig. 3b).
Table 3.
Comparison of the D and GST values for all loci and species
| Species | L5 | GT1 | L18 | Myrt3 | GT218 | L4 | GT223 | MS86 | Mean | mtDNA |
|---|---|---|---|---|---|---|---|---|---|---|
| T. longispinosus | ||||||||||
| D | 0.62 | 0.23 | 0.16 | 0.07 | 0.32 | 0.24 | 0.31 | 0.02 | 0.25 | 0.96 |
| GST | 0.05 | 0.01 | 0.01 | 0.01 | 0.05 | 0.01 | 0.07 | 0.01 | 0.03 | 0.11 |
| P-values | 0.000 | 0.000 | 0.000 | 0.007 | 0.000 | 0.000 | 0.003 | 0.034 | 0.0001 | |
| T. curvispinosus | ||||||||||
| D | 0.28 | 0.29 | 0.23 | 0.25 | 0.12 | 0.76 | 0.57 | 0.34 | 0.35 | 1.00 |
| GST | 0.07 | 0.01 | 0.03 | 0.02 | 0.03 | 0.07 | 0.07 | 0.24 | 0.07 | 0.19 |
| P-values | 0.000 | 0.001 | 0.010 | 0.003 | 0.029 | 0.000 | 0.000 | 0.001 | 0.010 | |
| P. americanus | ||||||||||
| D | 0.60 | 0.67 | 0.33 | 0.59 | 0.55 | 0.75 | – | – | 0.58 | 0.81 |
| GST | 0.03 | 0.03 | 0.02 | 0.03 | 0.08 | 0.04 | – | – | 0.04 | 0.12 |
| P-values | 0.001 | 0.000 | 0.05 | 0.001 | 0.001 | 0.01 | – | – | 0.000 | |
Fig. 3.
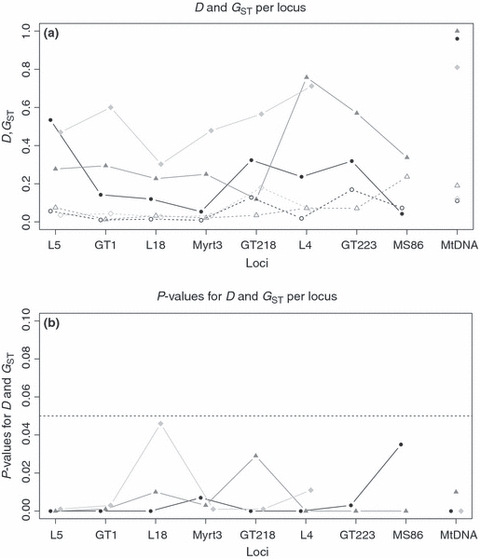
(a) D and GST values for the different microsatellite loci and all three species. Black circles: T. longispinosus, dark grey triangles: T. curvispinosus, light grey diamonds: P. americanus. Closed symbols: D values, open symbols GST values. (b) Significance of the D and GST values given in (a). Black circles: T. longispinosus, dark grey triangles: T. curvispinosus, light grey diamonds: P. americanus. Dashed line: 0.05 significance level. P-values are based on 1000 permutations and are the same for D and GST (see text for details).
Effect of length of fragment for MtDNA
For short fragment lengths, D values were low and the permutation test often gave nonsignificant results. As an example, we show the result for P. americanus (Fig. 4). The results for the other species are similar (not shown). We show the average D and GST value for a given fragment length and the proportion of tests that gave a significant (P< 0.05) result. The number of base pairs that were needed for consistently significant results was 500 for P. americanus, 300 for T. longispinosus and 900 for T. curvispinosus.
Fig. 4.
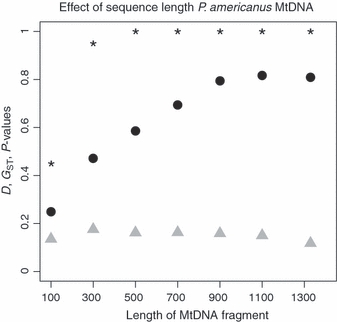
Effect of the length of the MtDNA sequence on D, GST and the power to detect population structure using the data from Protomognathus americanus. Circles: D values, triangles: GST values, asterisks: power to detect population structure. Each datapoint for D and GST is based on 20 randomly chosen fragments of the total data. For each randomly selected fragment, significance was estimated by 1000 permutation. The power is the percentage of these 20 fragments which showed a P-value of < 0.05.
From 300 to 1305 base pairs, GST slowly goes down because heterozygosity goes up when longer sequences are considered. D goes up with longer fragments, as it is more likely that mutations which make subpopulations differentiated are found. However, if the mutation rate increases even more (had we sampled an even longer fragment), the P-values would go up and power goes down (unpublished data). The reason is that if (almost) every individual carries a different allele, there is no power to detect population structure. This is not the case in our dataset.
Using genetic distances
We find that T. longispinosus shows the highest γST (Table 4), which is significantly different from expectations under panmixia (P= 0.01), T. curvispinosus has a lower value of γST which is borderline significant (P= 0.05). P. americanus has even lower γST, which is not significant (P= 0.25).
Table 4.
Distance based measure of population differentiation
| γST | P-value | |
|---|---|---|
| T. longispinosus | 0.23 | 0.01 |
| T. curvispinosus | 0.18 | 0.05 |
| P. americanus | 0.03 | 0.25 |
Isolation-by-distance
We found no significant correlations between genetic and geographic distances in T. longispinosus or P. americanus for the microsatellite or the MtDNA data (P-values > 0.05). For T. curvispinosus, we found a negative correlation for the microsatellites: subpopulations that are further away from each other tend to have lower D values (r = −0.39, P= 0.04), but a positive one for the MtDNA (r = 0.99, P< 0.001). The latter, however, is based on only three subpopulations (OH, WV and CT).
Structure analysis
For all three species, we find the highest delta K for K = 4 (Table 5, Fig. 5). For T. longispinosus, we did not detect meaningful structure and most individuals could not be convincingly assigned to either of the clusters (Fig. 5a). By contrast, T. curvispinosus is clearly structured, each of the subpopulations is relatively homogeneous, and some subpopulations cluster together (Fig. 5b). For P. americanus, we also find the highest delta K for K = 4, which is equal to the number of locations we have samples from. The clusters are not very clear, but subpopulations do seem to be differentiated (Fig. 5c).
Table 5.
Results of the structure analysis. The Log likelihood of the data given K (the number of subpopulations), averaged over four independent runs is given. For the settings of the program, see text
| K | T. longispinosus | T. curvispinosus | P. americanus |
|---|---|---|---|
| 1 | −8324 | −12050 | −4666 |
| 2 | −8256 | −11797 | −4530 |
| 3 | −8204 | −11529 | −4357 |
| 4 | −8098 | −11304 | −4242 |
| 5 | −8074 | −11189 | −4285 |
| 6 | −7944 | −11077 | −4206 |
Fig. 5.

Structure analysis results for three species. Shown are the estimates of Q (estimated membership coefficient for each individual) for each cluster. The most probable number of genetic populations present in the data is K = 4 for Temnothorax longispinosus (a), K = 4 for T. curvispinosus (b) and K = 4 for Protomognathus americanus (c). The vertical lines are broken into coloured segments showing the proportion of each individual assigned to each of the inferred clusters. Names above the graph correspond to the state of the various sample sites (see Table 1).
Using pairwise D values, we created neighbour-joining trees (Fig. 6), which can be compared with Structure outcomes. For the one species for which Structure detected higher level structure, T. curvispinosus, we find that that Structure and D agree quite well (see Figs 5b and 7b). Both Structure and D cluster OH2 and MD together, just like OH and WV. D clusters MI, IL and VA together, whereas Structure only clusters MI and IL together. However, when Structure is run for K = 3, it also consistently places VA with IL and MI (data not shown). The colours in the population labels in Fig. 6b are chosen to reflect the clustering in Structure (Fig. 5b).
Estimating population size and migration rate
For the microsatellites, the maximum likelihood estimate for θ*NT, averaged over loci as estimated with Migraine is 2.7 for T. longispinosus, 4.0 for T. curvispinosus and 6.3 for P. americanus (Fig. 7a). Population-wide mutation rates (θ*NT) did not differ between species (P= 0.2), but locus had an almost significant effect (P= 0.05). The maximum likelihood estimates for M, averaged over loci, is 32.0 for T. longispinosus, 17.6 for T. curvispinosus and 6.5 for P. americanus (Fig. 7b). We found differences between species in migration rates (P = 0.02), although none of the one-by-one comparisons between the three species were significant. As expected, locus did not affect migration rate (P= 0.08).
Fig. 7.

Estimated values of mutation and migration rates from the program Migraine and migrate-n for all three species. Black circles: T. longispinosus, dark grey triangles: T. curvispinosus, light grey diamonds: P. americanus. Arrows indicate 95% confidence intervals as estimated by the programs. (a) Population-wide mutation rates (θ*NT), (b) migration rates (M).
For MtDNA, the maximum likelihood estimate for θ*NT (per nucleotide), as estimated by migrate-n is 0.004 for T. longispinosus, 0.005 for T. curvispinosus and 0.003 for P. americanus (Fig. 7a). The maximum likelihood estimates for M are 1.1 for T. longispinosus, 1.1 for T. curvispinosus and 0.84 for P. americanus (Fig. 7b). The confidence intervals for the estimates of the three species strongly overlap, so that species differences were not apparent.
Discussion
Our population genetic analyses reveal that all three interacting ant species have high genetic diversity and strongly structured populations. From these results and earlier evidence for strong reciprocal selection pressures (Foitzik et al., 2001), we conclude that spatially structured coevolutionary arms races are expected, which could lead to local adaptation. Indeed, chemical analyses and field and laboratory manipulations demonstrated that the social parasite P. americanus is locally adapted to its host species (Achenbach & Foitzik, 2009; Foitzik et al., 2009a). Behavioural experiments also revealed clear population differences, but no local adaptation in either host or parasite (Foitzik et al., 2001; Brandt & Foitzik, 2004).
Overall, the three species resemble each other with respect to migration and mutation patterns and we conclude that they have similar evolutionary potentials. Adaptation not only depends on the availability of genetic variation, but also on the strength of selection. Therefore our results suggest that to gain a better understanding of who leads the coevolutionary arms race in this host–parasite system, we should focus on reciprocal selection pressures rather than on population genetics. In contrast to other comparative studies on host and parasite population genetics (Dybdahl & Lively, 1996; Keeney et al., 2009; Trontti et al., 2006; Vepsalainen et al., 2009), the differences we find between our study species are very small. Similar levels of genetic diversity and structure between host and parasite was also found in two other social parasite systems of a slavemaking ant and a socially parasitic wasp (Hoffman et al., 2008; Foitzik et al., 2009b). In the social wasp system, neither host nor parasite was clearly structured, but sampling was performed on a much smaller scale than in our studies. In other host–parasite systems, large differences, often 10-fold, in either diversity or structure were found and this was also the case for two other host-social parasite systems (Trontti et al., 2006; Vepsalainen et al., 2009), in which the parasite was much less variable and more structured. Host-social parasite systems with similar levels of diversity and structure are therefore interesting exceptions, which are worth studying because the opponents are on equal footing in evolutionary terms (current study, Foitzik et al., 2009b; Hoffman et al., 2008).
Albeit the overall pattern was one of similarity, the species did differ from each other in some surprising aspects. For example, although T. longispinosus has the highest population densities, we find that it has lower genetic diversity at the microsatellite loci in the subpopulations. Moreover, we expected the parasite P. americanus to be less structured than the other two species, because it is strictly monogynous, but it turned out to be as structured as T. curvispinosus and more structured than T. longispinosus (at the microsatellite loci) and its estimated migration rates were lowest. Only the MtDNA analyses based on genetic distances revealed the expected pattern of structure in the host species but not in the parasite.
There are several limitations to the inferences we can make based on our data. Naturally, we have data from a limited number of subpopulations and a limited number of microsatellite loci. The mitochondrial genome is nonrecombining and therefore only ‘shows’ one genealogical history, which can be greatly influenced by chance events. In addition, we detected linkage disequilibrium and an excess of homozygotes. The presence of linkage disequilibrium means that different loci are not entirely independent, and evidence based on different loci should therefore be treated with caution. The excess of homozygotes could be attributed to null-alleles or inbreeding, which could make subpopulations look more differentiated than they really are. Keeping in mind these limitations, we will now focus on each of the species separately.
T. longispinosus
At the microsatellite loci, we found a significantly lower effective number of alleles (ne) for the host species T. longispinosus compared with the other two species. The same trend (though not significant) was found for the estimates of θ*NT by the software Migraine. This is surprising, given that T. longispinosus occurs at 10-fold higher densities than the parasite P. americanus (Herbers & Foitzik, 2002; Brandt & Foitzik, 2004) and is also more abundant in its native range than T. curvispinosus (Mackay, 2000). Possibly, for reasons unknown to us, only relatively few nests contribute to the next generation, leading to higher drift in T. longispinosus compared with the other species. The effective population size of Hymenopteran species is generally lower because of haplodiploidy and even more so if complementary sex determination leads to the production of diploid males (Zayed, 2004). Indeed, diploid males have been detected in T. longispinosus, but not in the two other study species (Foitzik et al., 2004).
Another possible explanation for the lower genetic diversity in T. longispinosus is that the population has only recently grown to its current size. Such a population expansion leaves a specific genetic signature, which can be detected with the k-test of Reich & Goldstein (1998). We found indeed that the species T. longispinosus had eight negative k values (exact binomial test P= 0.008), which can be interpreted as evidence for a population size expansion in this species. In the other two species, we found no significant deviation from expectations (T. curvispinosus: six negative values out of eight, P= 0.29, P. americanus: four negative values out of six, P= 0.69). It is therefore likely that the T. longispinosus population has recently increased in size in which case the observed genetic diversity will reflect ancient population sizes, rather than current. The evolutionary potential, however, is determined largely by its current population size (Charlesworth, 2009, Karasov et al., 2010). An empirical example for an increase in the evolutionary potential because of a recent population expansion stems from the comparative population genetic analyses of a host–parasite system between a fungal parasite and an introduced host plant (Linde et al., 2010). Genetic diversity should therefore be used with caution to predict the evolutionary potential of a species.
Temnothorax longispinosus shows clear evidence for population structure, but at the microsatellite loci it has the lowest D values of the three species (Fig. 3a), and the highest migration rates (Fig. 7b). The Structure analysis confirms the low level of population differentiation in this species. The MtDNA sequence data for T. longispinosus show clear population structure and high diversity (Figs 2 and 3), similar to what was found in the previous study (Brandt et al., 2007). The much clearer population structure for the MtDNA when compared with the microsatellite loci suggests that in this species most dispersal is done by males.
T. curvispinosus
The host T. curvispinosus has lower population density than T. longispinosus, but higher diversity in the subpopulations. It also shows stronger population structure than T. longispinosus in every analysis: it has higher D values (Fig. 3a), lower maximum likelihood estimates of migration rates (Fig. 7b) and the Structure analysis shows clearer structuring (see Fig. 5). However, the subpopulations, which are clustered by Structure, are surprising: Northern Ohio and West-Virginia cluster together and Southern Ohio and Maryland. Geographically these clusters are not close to each other at all. When we test for isolation by distance, using pairwise D values (for the microsatellites) we find a negative correlation between geographic distance and D values; the populations that are further away tend to be closer genetically. This is clearly a surprising finding. However, other phylogeographic studies found that postglacial resettlement routes can lead to a divergent evolutionary history of close populations, for example, because they stem from different pleistocene refuges. For example, a population genetic analysis of the shrew Sorex antinorii detected two resettlement routes in the European Alps. These two genetic lineages came into secondary contact in the Rhone valley and neighbouring shrew populations are genetically clearly distinct (Yannic et al., 2008). For our study species, more detailed population genetic analyses (including more populations) would be necessary to reconstruct similar phylogeographic patterns. In a previous study (Brandt et al., 2007), we detected no significant population structure for T. curvispinosus at the microsatellite loci, but this is attributed to the fact that we only had samples from two locations which, in hindsight, turned out to have a very low pairwise D value (Northern Ohio and West Virginia).
P. americanus
The social parasite P. americanus has a slightly smaller range than T. longispinosus and only 10% of its host density. Still, we find that it has as high diversity as T. curvispinosus and higher genetic diversity in the subpopulations than T. longispinosus for the microsatellites (see Fig. 2). It also has the highest maximum likelihood estimate of θ*NT (Fig. 7a).
All three species conduct mating flights, but their reproductive biology differs. P. americanus colonies invariably contain a single queen, whereas both host species are facultatively polygynous. This means that in the two host species, some host queens can return to the mother nest and are re-adopted (and thus have zero dispersal). Strongly structured populations have been found for example in polygynous ant species of the genus Myrmica and Formica (Seppa & Pamilo, 1995; Sundstrom et al., 2005). Moreover, an association between social organization and genetic structure has also been found for the ant Petalomyrmex phylax, which shows inter-populational differences in the degree of polygyny. (Dalecky et al., 2007). In P. americanus queens always disperse and found new colonies by invading host nests. We therefore expected P. americanus to have higher migration rates, at least for the females, which would be reflected in the MtDNA. And indeed we found that this species has a lower D value for the MtDNA compared with the other two species. Besides, the genetic distance based test is not significant for P. americanus, whereas it is for the two facultatively polygynous host species, which fits with the expectation. However, the maximum likelihood estimates of the migration rate (from migrate-n, using the MtDNA data) were not different between the three species (see Fig. 7b).
Surprisingly, we found that P. americanus had the strongest population structure of the three species for the microsatellite loci (though D values were not significantly higher than those for T. curvispinosus) (Fig. 3a), and the lowest estimated migration rates (Fig. 7b). We hypothesize that this may be because dispersal could be more risky for P. americanus, because it needs a suitable abiotic habitat and a host colony to settle successfully in a new patch. The density of the species is also lower, so males may be less likely to find a mate if they disperse. If dispersal related death is higher, optimal dispersal rates are lower and effective dispersal rates may be even lower, leading to higher differentiation between subpopulations (Gros et al., 2006). We recently found that in a European host-social parasite system, structure was also strongest for the parasite (Foitzik et al., 2009b) and another recent study on a slavemaker species found even much stronger population structuring (Sanllorente et al., 2010). This could mean that it is a general pattern that obligate social parasites exhibit low dispersal because this is a more risky endeavour for them.
MtDNA vs. microsatellites
We find consistently higher GST and D values for MtDNA than microsatellites, as did many other population genetic studies on ants (Doums et al., 2002; Clemencet et al., 2005; Brandt et al., 2007; Goropashnaya et al., 2007). A common conclusion from this finding was that ant queens disperse less than males. However, we think that this conclusion based only on different levels of differentiation is incorrect, because such a difference could also be caused by differences in mutation rates between the marker systems. Indeed, our results show that the mutation rates are very different for the two marker systems. If we multiply the estimated per nucleotide population-wide mutation rates for the MtDNA (from migrate-n) with the number of nucleotides we have sequenced, we find an average population wide mutation rate of 22 for the MtDNA locus, whereas the microsatellite loci have an average population wide mutation rate of 4. The per-individuum mutation rates are even more different than this comparison suggests, because population size for MtDNA is 4-fold lower than for nuclear markers. This means that the differences in D values between the marker systems could be entirely attributed to differences in mutation rates.
If one is interested in sex-specific dispersal, one could compare the maximum likelihood estimates of migration rates between marker systems. For this comparison, we have to multiply the estimate of M for MtDNA with a factor 4 to adjust for the four-fold lower population size. If we do that we find that T. longispinosus and T. curvispinosus have clearly higher migration rates for the nuclear markers (T.l. 32 vs. 4.6, T.c. 17.6 vs. 4.5), whereas for P. americanus the difference is not so large (6.5 vs. 3.4). Note that M for microsatellites depends on the average migration rates of males and females. Under the assumption of similar effective population sizes for males and females, the comparisons suggest that in the two host species males have migration rates that are between 6 and 15-fold higher than that of queens. In the monogynous parasite, this difference appears to be only three-fold.
Methods to infer population structure
In our population genetic analysis we used several approaches: the newly introduced D statistic (Jost, 2008), the Bayesian clustering algorithm Structure (Pritchard et al., 2000; Falush et al., 2003) and the maximum likelihood estimates of mutation and migration rates based on coalescent theory (migrate-n by Beerli & Felsenstein (2001) and Migraine by Rousset & Leblois (2007).
The statistic D is by far the easiest of the methods we used, as it can simply be calculated from the data and it does not require elaborate computations. However, there are two main drawbacks of using D. First, it depends on both migration and mutation rates (Jost, 2008; Ryman & Leimar, 2009), which make observed differences between species or loci hard to interpret. Second, it only uses part of the data (namely the effective number of alleles, ne). The first drawback is not too severe for our dataset because we used the same loci for different species and found similar mutation rates between the loci we used. However, it makes comparisons between different marker systems (MtDNA vs. nuclear markers) difficult. We stress that whenever D values are compared between different loci, species or studies, it is necessary to consider the effect of mutation rates. The variability of the loci determines part of the outcome: very variable loci or long sequences lead to high D values, suggesting strong differentiation, whereas less variable loci or short sequences tend to underestimate structuring. Moreover, permutation tests are needed to determine whether D values are significantly higher than expected under panmixia.
We see no intuitive way to include genetic distances in the calculation of D, but this may not be necessary because there are elegant ways to include genetic distances in GST-like statistics. We used γST (see Fig. 5), which does not suffer from the problems that other heterozygosity-based GST statistics experience, because they use heterozygosity per nucleotide, which is naturally low.
We analysed the microsatellite data with Structure and found evidence for four clusters in each of the three species (see Fig. 6). Only in the host species T. curvispinosus, the Structure analysis reveals clear structuring. When comparing these Structure results with pairwise D values, we find good agreement. The subpopulations that are clustered together by Structure are mostly the ones that would also be clustered based on pairwise D values (averaged over loci). However, the neighbour-joining tree for T. curvispinosus shows that the clustering of subpopulations is not very strong: the tree is quite starlike and no two populations are really close to each other, but this information is not clear from the Structure analysis. For the data and the questions we have, the Structure analysis does not provide novel insights.
To estimate population wide mutation and migration rates, we used two programs, Migraine and migrate-n, which are both based on coalescent theory and which use a maximum likelihood framework to estimate population demographic parameters from genetic data from different locations. In principle, the two programs should find the same results. However, we found that for the microsatellite data Migraine performed better. For migrate-n, even when running the program for 1 000 000 steps and with adding heated chains, outcomes still depended on starting values for the MCMC chain and the program did not converge. We did not experience this problem for the MtDNA data. Results from Migraine and migrate-n will always be biased because of model misspecification. For example, we do not know how many subpopulations there really are in the species we study, and we chose to ignore all unsampled populations in the statistical model. Model misspecification is a difficult problem, but several studies suggest that inference can still be made (Beerli, 2004; Slatkin, 2005; Rousset & Leblois, 2007). Additionally, we can hope that the species we study are similar enough, so that we misspecified the model in the same way for each of the three species and results are therefore comparable.
Theoretical studies suggest that population size and migration rates are key parameters in coevolutionary arms races, because these parameters together determine how many new beneficial mutations enter a population in a given generation (Gandon & Michalakis, 2002). In this study, we compared genetic variation and population structure between a social parasite and its two main host species. We find similarities and differences and conclude that the three species probably do not differ much in terms of the evolutionary potential. However, we also find that the methods used in this and similar studies are necessarily based on simplified assumptions. For example, historical population size changes may have a larger impact on the observed genetic diversity than current effective population size does. If this is the case, then the observed genetic diversity may not be related at all to a species’ evolutionary potential. This is known for well-studied species such as Drosophila melanogaster and humans (Charlesworth, 2009, Karasov et al., 2010) and it is likely to be the case for many other species. However, to take into account the demographic history of the species of interest, much more data are needed and more complex models would need to be used to estimate the relevant parameters.
Acknowledgments
We would like to thank Andreas Gros, Joachim Hermisson, Tobias Pamminger, Tony Wilson and Justyna Wolinska for discussion and comments on the manuscript, and François Rousset for help with the software Migraine. Three anonymous reviewers made useful comments. This study was funded by the DFG grants Fo 298/7 and 9. We thank the E.N. Huyck Preserve for supporting our research. Part of the computations in this paper was run on the Odyssey cluster supported by the FAS Sciences Division Research Computing Group at Harvard.
Footnotes
Data deposited at Dryad: doi: 10.5061/dryad.8134
References
- Achenbach A, Foitzik S. First evidence for slave rebellion: enslaved ant workers systematically kill the brood of their social parasite Protomagnathus americanus. Evolution. 2009;63:1068–1075. doi: 10.1111/j.1558-5646.2009.00591.x. [DOI] [PubMed] [Google Scholar]
- Alloway TM. Origins of slavery in Leptothoracine ants (Hymenoptera, Formicidae) Am. Nat. 1980;115:247–261. [Google Scholar]
- Azuma N, Takahashi J, Kikuchi T, Yoshimura M, Onoyama K, Higashi S. Microsatellite loci for Myrmica kotokui and their application in some congeneric ant species distributed in northern Japan. Mol. Ecol. Notes. 2005;5:118–120. [Google Scholar]
- Beaumont MA, Nielsen R, Robert C, Hey J, Gaggiotti O, Knowles L, et al. In defence of model-based inference in phylogeography. Mol. Ecol. 2010;19:436–446. doi: 10.1111/j.1365-294X.2009.04515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli P. Effect of unsampled populations on the estimation of population sizes and migration rates between sampled populations. Mol. Ecol. 2004;13:827–836. doi: 10.1111/j.1365-294x.2004.02101.x. [DOI] [PubMed] [Google Scholar]
- Beerli P, Felsenstein J. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc. Natl Acad. Sci. USA. 2001;98:4563–4568. doi: 10.1073/pnas.081068098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beibl J, Stuart RJ, Heinze J, Foitzik S. Six origins of slavery in formicoxenine ants. Insect. Soc. 2005;52:291–297. [Google Scholar]
- Bourke AFG, Green HAA, Bruford MW. Parentage, reproductive skew and queen turnover in a multiple-queen ant analysed with microsatellites. Proc. R Soc. Lond. B Biol. Sci. 1997;264:277–283. doi: 10.1098/rspb.1997.0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt M, Foitzik S. Community context and specialization influence coevolution between a slavemaking ant and its hosts. Ecology. 2004;85:2997–3009. [Google Scholar]
- Brandt M, Foitzik S, Fischer-Blass B, Heinze J. The coevolutionary dynamics of obligate ant social parasite system-between prudence and antagonism. Biol. Rev. 2005;80:251–267. doi: 10.1017/s1464793104006669. [DOI] [PubMed] [Google Scholar]
- Brandt M, Fischer-Blass B, Heinze J, Foitzik S. Population structure and the co-evolution between social parasites and their hosts. Mol. Ecol. 2007;16:2063–2078. doi: 10.1111/j.1365-294X.2007.03300.x. [DOI] [PubMed] [Google Scholar]
- Chakraborty R, Deandrade M, Daiger SP, Budowle B. Apparent heterozygote deficiencies observed in DNA typing data and their implications in forensic applications. Ann. Hum. Genet. 1992;56:45–57. doi: 10.1111/j.1469-1809.1992.tb01128.x. [DOI] [PubMed] [Google Scholar]
- Charif D, Lobry JR. SeqinR 1.0-2: a contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis. 2007.
- Charlesworth B. Effective population size and patterns of molecular evolution and variation. Nat. Rev. Gen. 2009;10:195–205. doi: 10.1038/nrg2526. [DOI] [PubMed] [Google Scholar]
- Clemencet J, Viginier B, Doums C. Hierarchical analysis of population genetic structure in the monogynous ant Cataglyphis cursor using microsatellite and mitochondrial DNA markers. Mol. Ecol. 2005;14:3735–3744. doi: 10.1111/j.1365-294X.2005.02706.x. [DOI] [PubMed] [Google Scholar]
- Crawford NG. smogd: software for the measurement of genetic diversity. Mol. Ecol. Resour. 2010;10:556–557. doi: 10.1111/j.1755-0998.2009.02801.x. [DOI] [PubMed] [Google Scholar]
- Dalecky A, Debout G, Estoup A, McKey DB, Kjellberg F. Changes in mating system and social structure of the ant Petalomyrmex phylax are associated with range expansion in Cameroon. Evolution. 2007;61:579–595. doi: 10.1111/j.1558-5646.2007.00044.x. [DOI] [PubMed] [Google Scholar]
- Davies CM, Webster JP, Kruger O, Munatsi A, Ndamba J, Woolhouse MEJ. Host–parasite population genetics: a cross-sectional comparison of Bulinus globosus and Schistosoma haematobium. Parasitology. 1999;119:295–302. doi: 10.1017/s0031182099004722. [DOI] [PubMed] [Google Scholar]
- De Iorio M, Griffiths RC. Importance sampling on coalescent histories. I. Adv. Appl. Probab. 2004a;36:417–433. [Google Scholar]
- De Iorio M, Griffiths RC. Importance sampling on coalescent histories. II: subdivided population models. Adv. Appl. Probab. 2004b;36:434–454. [Google Scholar]
- Doums C, Cabrera H, Peeters C. Population genetic structure and male-biased dispersal in the queenless ant Diacamma cyaneiventre. Mol. Ecol. 2002;11:2251–2264. doi: 10.1046/j.1365-294x.2002.01619.x. [DOI] [PubMed] [Google Scholar]
- Dybdahl MF, Lively CM. Host–parasite interactions – infection of common clones in natural populations of a fresh-water snail (Potamopyrgus antipodarum) Proc. R Soc. Lond. B Biol. Sci. 1995;260:99–103. [Google Scholar]
- Dybdahl MF, Lively CM. The geography of coevolution: comparative population structures for a snail and its trematode parasite. Evolution. 1996;50:2264–2275. doi: 10.1111/j.1558-5646.1996.tb03615.x. [DOI] [PubMed] [Google Scholar]
- Emery C. Ueber den Ursprung der dulotischen, parasitischen und myrmekophilen Ameisen. Biol. Centralbl. 1909;29:352–362. [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Evans JD. Parentage analyses in ant colonies using simple sequence repeat loci. Mol. Ecol. 1993;2:393–397. doi: 10.1111/j.1365-294x.1993.tb00032.x. [DOI] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foitzik S, Herbers JM. Colony structure of a slavemaking ant. I. Intracolony relatedness, worker reproduction, and polydomy. Evolution. 2001;55:307–315. doi: 10.1111/j.0014-3820.2001.tb01295.x. [DOI] [PubMed] [Google Scholar]
- Foitzik S, Haberl M, Gadau J, Heinze J. Mating frequency of Leptothorax nylanderi ant queens determined by microsatellite analysis. Insectes Sociaux. 1997;44:219–227. [Google Scholar]
- Foitzik S, DeHeer CJ, Hunjan DN, Herbers JM. Coevolution in host–parasite systems: behavioural strategies of slave-making ants and their hosts. Proc. R Soc. Lond. B Biol. Sci. 2001;268:1139–1146. doi: 10.1098/rspb.2001.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foitzik S, Backus VL, Trindl A, Herbers JM. Ecology of Leptothorax ants: impact of food, nest sites, and social parasites. Behav. Ecol. Sociobiol. 2004;55:484–493. [Google Scholar]
- Foitzik S, Achenbach A, Brandt M. Locally adapted social parasite affects density, social structure, and life history of its ant hosts. Ecology. 2009a;90:1195–1206. doi: 10.1890/08-0520.1. [DOI] [PubMed] [Google Scholar]
- Foitzik S, Bauer S, Laurent S, Pennings PS. Genetic diversity, population structure and sex-biased dispersal in three co-evolving species. J. Evol. Biol. 2009b;22:2470–2480. doi: 10.1111/j.1420-9101.2009.01863.x. [DOI] [PubMed] [Google Scholar]
- Furrer R, Nychka D, Sain S. fields: tools for spatial data. R package version 6.01. 2009.
- Gandon S, Michalakis Y. Local adaptation, evolutionary potential and host–parasite coevolution: interactions between migration, mutation, population size and generation time. J. Evol. Biol. 2002;15:451–462. [Google Scholar]
- Gerlach G, Jueterbock A, Kraemer P, Deppermann J, Harmand P. Calculations of population differentiation based on G(ST) and D: forget G(ST) but not all of statistics! Mol. Ecol. 2010;19:3845–3852. doi: 10.1111/j.1365-294X.2010.04784.x. [DOI] [PubMed] [Google Scholar]
- Giraud T, Blatrix R, Solignac M, Jaisson P. Polymorphic microsatellite DNA markers in the ant Gnamptogenys striatula. Mol. Ecol. 1999;8:2143–2145. doi: 10.1046/j.1365-294x.1999.00802-2.x. [DOI] [PubMed] [Google Scholar]
- Goropashnaya AV, Fedorov VB, Seifert B, Pamilo P. Phylogeography and population structure in the ant Formica exsecta (Hymenoptera, Formicidae) across Eurasia as reflected by mitochondrial DNA variation and microsatellites. Ann. Zool. Fenn. 2007;44:462–474. [Google Scholar]
- Gros A, Poethke HJ, Hovestadt T. Evolution of local adaptations in dispersal strategies. Oikos. 2006;114:544–552. [Google Scholar]
- Hamaguchi K, Ito Y, Takenaka O. GT dinucleotide repeat polymorphisms in a polygynous ant, Leptothorax spinosior and their use for measurement of relatedness. Naturwissenschaften. 1993;80:179–181. doi: 10.1007/BF01226379. [DOI] [PubMed] [Google Scholar]
- Hedrick PW. A standardized genetic differentiation measure. Evolution. 2005;59:1633–1638. [PubMed] [Google Scholar]
- Herbers JM, Foitzik S. The ecology of slavemaking ants and their hosts in north temperate forests. Ecology. 2002;83:148–163. [Google Scholar]
- Hoffman EA, Kovacs JL, Goodisman MAD. Genetic structure and breeding system in a social wasp and its social parasite. BMC Evol. Biol. 2008;8 doi: 10.1186/1471-2148-8-239. art. 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Boos DD, Kaplan NL. A statistical test for detecting geographic subdivision. Mol. Biol. Evol. 1992;9:138–151. doi: 10.1093/oxfordjournals.molbev.a040703. [DOI] [PubMed] [Google Scholar]
- Jensen JL, Bohonak AJ, Kelley ST. Isolation by distance, web service. BMC Genet. 2005;6 doi: 10.1186/1471-2156-6-13. art. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost L. G(ST) and its relatives do not measure differentiation. Mol. Ecol. 2008;17:4015–4026. doi: 10.1111/j.1365-294x.2008.03887.x. [DOI] [PubMed] [Google Scholar]
- Karasov T, Messer PW, Petrov DA. Evidence that adaptation in Drosophila is not limited by mutation at single sites. PLoS Genetics. 2010;6:6. doi: 10.1371/journal.pgen.1000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney DB, King TM, Rowe DL, Poulin R. Contrasting mtDNA diversity and population structure in a direct-developing marine gastropod and its trematode parasites. Mol. Ecol. 2009;18:4591–4603. doi: 10.1111/j.1365-294X.2009.04388.x. [DOI] [PubMed] [Google Scholar]
- Linde CC, Liles JA, Thrall PH. Expansion of genetic diversity in randomly mating founder populations of Alternaria brassicicola infecting Cakile maritima in Australia. Appl. Environ. Microbiol. 2010;76:1946–1954. doi: 10.1128/AEM.01594-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lively CM. Migration, virulence, and the geographic mosaic of adaptation by parasites. Am. Nat. 1999;153:S34–S47. doi: 10.1086/303210. [DOI] [PubMed] [Google Scholar]
- Mackay WP. A review of the New World ants of the subgenus Myrafant, (Genus Leptothorax) (Hymenoptera: Formicidae) Sociobiology. 2000;36:265–434. [Google Scholar]
- Manning SD, Woolhouse MEJ, Ndamba J. Geographic compatibility of the fresh-water snail Bulinus globosus and schistosomes from the Zimbabwe Highveld. Int. J. Parasitol. 1995;25:37–42. doi: 10.1016/0020-7519(94)00097-8. [DOI] [PubMed] [Google Scholar]
- Matschiner M, Salzburger W. TANDEM: integrating automated allele binning into genetics and genomics workflows. Bioinformatics. 2009;25:1982–1983. doi: 10.1093/bioinformatics/btp303. [DOI] [PubMed] [Google Scholar]
- Mukaratirwa S, Siegismund HR, Kristensen TK, Chandiwana SK. Genetic structure and parasite compatibility of Bulinus globosus (Gastropoda: Planorbidae) from two areas of different endemicity of Schistosoma haematobium in Zimbabwe. Int. J. Parasitol. 1996;26:269–280. doi: 10.1016/0020-7519(95)00130-1. [DOI] [PubMed] [Google Scholar]
- Mutikainen P, Koskela T. Population structure of a parasitic plant and its perennial host. Heredity. 2002;89:318–324. doi: 10.1038/sj.hdy.6800142. [DOI] [PubMed] [Google Scholar]
- Nei M. Evolution of human races at the gene level. In: Bonne-Tamir B, editor. Progress in Clinical and Biological Research, Vol. 103a. Human Genetics Part a: the Unfolding Genome; Proceedings of the 6th International Congress, Jerusalem, Israel, Sept. 13–18, 1981. New York, N.Y., USA: Alan R. Liss, Inc.; 1982. Lii+531p. Illus: P 167–182. [Google Scholar]
- Nei M, Chesser RK. Estimation of fixation indexes and gene diversities. Ann. Hum. Genet. 1983;47:253–259. doi: 10.1111/j.1469-1809.1983.tb00993.x. [DOI] [PubMed] [Google Scholar]
- Nejsum P, Frydenberg J, Roepstorff A, Parker ED. Population structure in Ascaris suum (Nematoda) among domestic swine in Denmark as measured by whole genome DNA fingerprinting. Hereditas. 2005;142:7–14. doi: 10.1111/j.1601-5223.2005.01864.x. [DOI] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prugnolle F, Theron A, Pointier JP, Jabbour-Zahab R, Jarne P, Durand P, et al. Dispersal in a parasitic worm and its two hosts: consequence for local adaptation. Evolution. 2005;59:296–303. [PubMed] [Google Scholar]
- RDC Team . R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2005. [Google Scholar]
- Reich DE, Goldstein DB. Genetic evidence for a Paleolithic human population expansion in Africa. Proc. Natl Acad. Sci. USA. 1998;95:8119–8123. doi: 10.1073/pnas.95.14.8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren ZM, Zhu B, Wang DJ, Ma E, Su DM, Zhong Y. Comparative population structure of Chinese sumac aphid Schlechtendalia chinensis and its primary host-plant Rhus chinensis. Genetica. 2008;132:103–112. doi: 10.1007/s10709-007-9153-6. [DOI] [PubMed] [Google Scholar]
- Rousset F, Leblois R. Likelihood and approximate likelihood analyses of genetic structure in a linear habitat: performance and robustness to model mis-specification. Mol. Biol. Evol. 2007;24:2730–2745. doi: 10.1093/molbev/msm206. [DOI] [PubMed] [Google Scholar]
- Rousset F, Raymond M. Testing heterozygote excess and deficiency. Genetics. 1995;140:1413–1419. doi: 10.1093/genetics/140.4.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman N, Leimar O. G(ST) is still a useful measure of genetic differentiation – a comment on Jost's D. Mol. Ecol. 2009;18:2084–2087. doi: 10.1111/j.1365-294X.2009.04187.x. [DOI] [PubMed] [Google Scholar]
- Sanllorente O, Hammond R, Ruano F, Keller L, Tinaut A. Extreme population differentiation in a vulnerable slavemaking ant with a fragmented distribution. Conserv. Genet. 2010;11:1701–1710. [Google Scholar]
- Seppa P, Pamilo P. Gene flow and population viscosity in Myrmica ants. Heredit. 1995;74:200–209. [Google Scholar]
- Slatkin M. Seeing ghosts: the effect of unsampled populations on migration rates estimated for sampled populations. Mol. Ecol. 2005;14:67–73. doi: 10.1111/j.1365-294X.2004.02393.x. [DOI] [PubMed] [Google Scholar]
- Sundstrom L, Seppa P, Pamilo P. Genetic population structure and dispersal patterns in Formica ants – a review. Ann. Zool. Fenn. 2005;42:163–177. [Google Scholar]
- Thompson JN. The coevolutionary process. Chicago: University of Chicago Press; 1994. xi+376p. [Google Scholar]
- Trontti K, Aron S, Sundstrom L. The genetic population structure of the ant Plagiolepis xene – implications for genetic vulnerability of obligate social parasites. Conserv. Genet. 2006;7:241–250. [Google Scholar]
- Vepsalainen K, Ebsen JR, Savolainen R, Boomsma JJ. Genetic differentiation between the ant Myrmica rubra and its microgynous social parasite. Insect. Soc. 2009;56:425–437. [Google Scholar]
- Yannic G, Basset P, Hausser J. Phylogeography and recolonization of the Swiss Alps by the Valais shrew (Sorex antinorii), inferred with autosomal and sex-specific markers. Mol. Ecol. 2008;17:4118–4133. doi: 10.1111/j.1365-294x.2008.03888.x. [DOI] [PubMed] [Google Scholar]
- Zayed A. Effective population size in Hymenoptera with complementary sex determination. Heredity. 2004;93:627–630. doi: 10.1038/sj.hdy.6800588. [DOI] [PubMed] [Google Scholar]