

Abstract
Carcinogenesis is the result of mutations and subsequent clonal expansions of mutated, selectively advantageous cells. To investigate the relative contributions of mutation versus cell selection in tumorigenesis, we compared two mathematical models of carcinogenesis in two different cancer types: lung and colon. One approach is based on a population genetics model, the Wright-Fisher process, whereas the other approach is the two-stage clonal expansion model. We compared the dynamics of tumorigenesis predicted by the two models in terms of the time period until the first malignant cell appears, which will subsequently form a tumor. The mean waiting time to cancer has been calculated approximately for the evolutionary colon cancer model. Here, we derive new analytic approximations to the median waiting time for the two-stage lung cancer model and for a multistage approximation to the Wright-Fisher process. Both equations show that the waiting time to cancer is dominated by the selective advantage per mutation and the net clonal expansion rate, respectively, whereas the mutation rate has less effect. Our comparisons support the idea that the main driving force in lung and colon carcinogenesis is Darwinian cell selection.
Introduction
Studying the timing of carcinogenic events has provided important hints on the putative mechanisms of cancer onset, that is, of the processes that lead to the first malignant cell (M-cell). Research on mathematical models of carcinogenesis in the 1970s and 1980s was based on the assumptions that (a) carcinogenesis is the effect of initiation and promotion, and (b) carcinogens can be distinguished into those affecting early stages with an irreversible effect and those affecting late stages with a reversible effect. This distinction was largely based on experimental research conducted in the previous decades that had shown that initiation was likely to be due to mutations and that promotion was likely based on nongenotoxic mechanisms (1, 2).
Different models of carcinogenesis have been developed, and most involve mutation induction and cell expansion (3-10). Here, we compare two modeling approaches: an evolutionary model based on the Wright-Fisher (WF) process (11) and the MVK model of Moolgavkar, Venzon, and Knudson (7, 8), also known as the two-stage clonal expansion model (12). Earlier, the WF process has been used to describe the progression of a benign adenoma to an invasive carcinoma, and different values of the mutation and selection parameters have been explored based on tumor growth data (6). No analytic solution of the stochastic WF model is known, but an approximate closed-form expression of the mean waiting time until the first M-cell appears has been presented (6). In the present article, we derive a similar equation for the waiting time to the first M-cell based on the simplified MVK (S-MVK) model, a simplified deterministic variant of the stochastic MVK model (13).
We compare the dynamics predicted by the S-MVK model and the WF model in terms of the time it takes until the first M-cell occurs. The main result of our study is that for both models, we derive similar equations for the median waiting time to the first M-cell and that the expressions emphasize the dominating effect of cell selection (i.e., cell proliferation) on carcinogenesis. We also compare waiting times numerically using the same biological end point, that is, the same cancer site, for both models. Using parameter estimates obtained from fitting epidemiologic lung and colon cancer data, the MVK model predicts waiting times that are consistent across several previously reported data sets with those obtained from the WF model assuming basic genomic parameters such as mutation rate and selective advantage per mutation. Thus, while both mutation and selection are necessary for carcinogenesis (14-16), the model comparison suggests that the formation of the first M-cell is driven mainly by clonal expansion of selectively advantageous cells, whereas the effect of the mutation rate appears much smaller.
Materials and Methods
WF model
The WF process is a stochastic model of an evolving asexual population that has been applied to the cells of a tumor (11). In this model, cancer cells evolve in discrete, nonoverlapping generations, and each cell independently derives from a cell of the previous generation with a probability proportional to the fitness of the parent. Each cell is either identical to its parent, or hit by an additional mutation with probability u per gene location per cell division (Fig. 1). The expectation of the waiting time until the first M-cell with k mutations appears, denoted TWF, has been approximated (6) as
| (1) |
Here, s is the selective advantage per mutation. An exponential population growth is assumed from initially Ninit cells to Nfin cells when the first M-cell occurs. The number of putative genes involved in the process is denoted by d. The WF model has not been fitted to epidemiologic data. Therefore, we set the parameters to values obtained from published experimental data under additional assumptions as follows.
Figure 1.
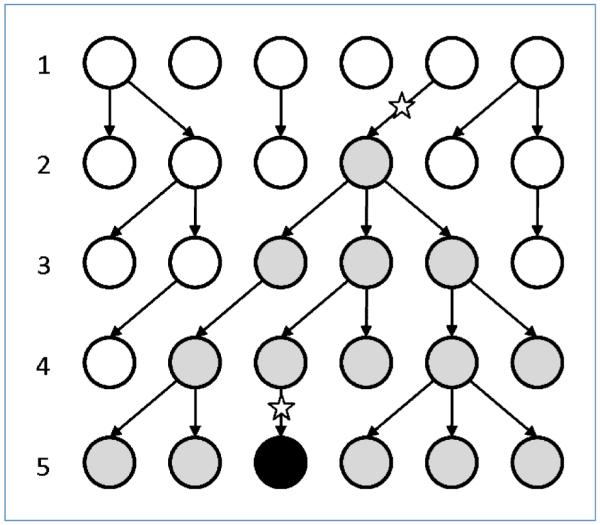
Schematic representation of the WF process. Illustrated are five generations of a single realization of the WF process with a constant population size of N = 6 cells. In each generation, cells are drawn randomly from the previous generation. Initially, in generation 1, all cells are wild-type (white). In generation 2, the first cell with one mutation appears (gray). Cells with additional mutations have a selective advantage: they are more likely to generate offspring and will, on average, outcompete cells with fewer mutations. In this realization, the first cell with two mutations occurs in generation 5 (black), and the waiting time for k = 2 mutations is TWF = 4 generations; see Eq. 1.
Based on genomic data (6, 17), we assume a total of d = 100 susceptible cancer-associated genes. We consider a cell initiated if k = 5 of those genes are mutated and malignant if k = 20. Both the number of driver genes (i.e., those that confer a selective advantage) available in the genome, d, and the number of required mutations per initiated or M-cell, k, are not known with certainty today and will eventually be determined in large cancer whole-genome sequencing projects. The values used here are motivated by the cancer genome study Sjöblom and colleagues (17), in which among 13,000 genes analyzed in 22 tumors, ~80 driver mutations have been identified, but only up to 18 and 23 per patient, for late-stage colon and breast cancer, respectively. These numbers were consistent with follow-up studies (18-20). Because only a few of those driver mutations occurred with high frequency (termed “mountains”) whereas most occurred at low frequencies (called “hills”), we assume that adenoma formation requires k = 5 mutated genes, which are likely to be more specific changes and therefore appear as mountains in the histogram provided by Sjöblom and colleagues (17). Assuming a normal mutation rate (no genetic instability), the average per-gene mutation rate has been estimated as u = 1 × 10−7 per generation (6). In general, no experimental data are available for the fitness advantage s conferred by a mutation. Plausible values of s have been determined as those that cause expected waiting times (Eq. 1) consistent with observed clinical progression times between 5 to 20 years. This approach suggests a selective advantage per mutation on the order of 1 × 10−3 to 1 × 10−4 (6).
Under the WF model, genetic progression proceeds in successive approximately equidistant mutational sweeps (6). Based on this regular arrival of new mutant waves and the near clonality of the population at any point in time, the stochastic WF process can be approximated by a linear multi-stage process with transition rate [2s lnNclone]/ln2[s/(ud)] in which stages correspond to clonal expansions (21, 22). Here, is the (geometric) mean number of adenoma cells. This deterministic approximation to the WF process can be regarded as an Armitage-Doll (AD) model with a series of transitions starting from a pool of N normal cells (N-cells; see Supplementary Material). The resulting approximated hazard function for this model is
| (2) |
The survival function is S(t) = exp[−H(t)], in which H(t)is the integrated hazard. The median waiting time to the first cancer cell, denoted τ, is the solution of the equation S(τ)= 1/2. It is the time when half of the population has acquired the first M-cell. The following expression for τ is derived in the Supplementary Material:
| (3) |
We denote this waiting time , if the starting point of the carcinogenic process is a set of N normal cells and Ninit = N. We also apply Eq. 3 to the process that starts with a clone of m adenoma cells and denote the resulting median waiting time .
Two-stage model with clonal expansion
In the MVK model (7, 8), normal stem cells can be transformed into cells of an intermediate form at an event rate μ1, the first mutation rate. These intermediate cells can divide symmetrically into two intermediate cells at rate α, die, or differentiate at rate β, and divide asymmetrically into one initiated cell (I-cell) and one M-cell at rate μ2 (Fig. 2). Clonal expansion of I-cells is formulated as a stochastic process (7, 8, 12). M-cells are assumed to develop into a detectable tumor after a constant lag time, tlag. The hazard function of the MVK model has been derived (23, 24) and can be used to describe cancer incidence and cancer mortality rates (25). It is
| (4) |
with , δ2 = α(B – A), h(0) = 0, and . If the background estimates for μ1, μ2, α, and β are used (spontaneous rates), then the hazard function describes the baseline cancer incidence. Recently, the MVK model has been fitted to lung cancer incidence data from the large European Prospective Investigation into Cancer and Nutrition cohort study (26).
Figure 2.
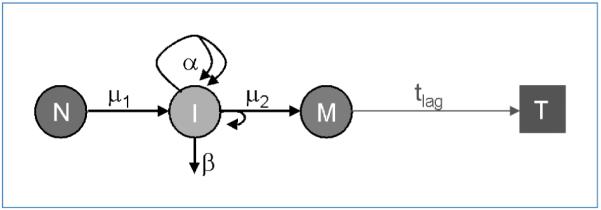
Conceptual view of the two-stage model with clonal expansion. Normal cells (N) can develop into initiated (or intermediate) cells (I). I-cells have sustained the first rate-limiting event in the pathway to malignancy at rate μ1, the parameter defining the rate of critical genomic events involved in initiation. An I-cell can divide into two I-cells with rate α; it dies or differentiates with rate β; it divides asymmetrically into one I-cell and one cell that has sustained the second event [malignant cells (M)] with rate μ2. The M-cells are assumed to develop into a tumor (T) after a deterministic lag time, tlag.
The S-MVK model (13) is defined by the hazard
| (5) |
in which N is the number of normal cells. The S-MVK model is unbound at high ages, that is, , whereas the hazard of the MVK model reaches a plateau at high ages because of the stochasticity of the birth-death process of I-cells. The S-MVK model can be derived from the MVK model (Eq. 4) by assuming that μ2 is negligible (13).
In the MVK model, the expected waiting time to cancer can be calculated as the expected value of the time T to the first M-cell, . A closed-form expression for the survival function, P(T > t), has been reported (7, 24), but the integral E[T] cannot be solved analytically. By contrast, we obtained the following closed-form expression for the median waiting time from a pool of N-cells to the first M-cell for the S-MVK model (see Supplementary Material),
| (6) |
We are also interested in the time to the first M-cell starting with a clone of m I-cells, that is, in the transition from adenoma to carcinoma, in the MVK model. Such an expression would be analogous to Eq. 1, which gives an approximation to the expected waiting time to the first M-cell for the WF process starting with an adenoma (a clone of mutated cells). However, for the MVK model, an analogous expression for the mean time to the first M-cell is not defined because intermediate cells have a positive probability of becoming extinct (Supplementary Material). Under the condition that the clone of initiated cells does not become extinct, the mean time from the birth of a premalignant clone to malignancy has been calculated (27; see Discussion for more details). Here, we have derived an expression for the median time to the first M-cell starting from a clone of m I-cells (Supplementary Material),
| (7) |
with δ2 = α(B – A).
Comparison of the two models
The WF and the MVK model are different stochastic models of carcinogenesis (Figs. 1 and 2). In the MVK model, carcinogenesis is viewed as the result of two critical, irreversible, rate-limiting, and hereditary (at the level of somatic cells) events (23). By contrast, the WF model describes carcinogenesis explicitly as a series of genetic changes in a reproducing cell population. The genetic progression stages that eventually lead to the first M-cell are defined by the number of mutated cancer-associated genes. These mutant types sweep through the population in several subsequent clonal waves (6). Tumor growth is also modeled differently. In the S-MVK model, the net clonal expansion rate (α – β) describes the growth of initiated cells, whereas in the WF model, a deterministic exponential growth from adenoma to carcinoma is assumed. The background rate μ0 in the MVK model corresponds to the composite parameter u × d in the WF model because in the WF process, no carcinogenic environmental factors such as smoking or radiation are included. Likewise, the growth rate, (α – β), plays a similar role in the MVK model as the selective advantage, s, in the WF model. The term “initiated cell” used in the MVK model corresponds to the notion of “adenoma cell” in the context of the WF model.
Results
Despite their differences, the WF model and the MVK model are conceptually similar. Both explain the appearance of the first malignant cancer cell by accumulating mutations with a selective advantage in a cell population, and both distinguish the generation of a new cell type (mutation) from its growth (clonal expansion). The dynamics of the carcinogenic process can be directly compared between the two models in terms of the time to appearance of the first M-cell. Because the expectation of this waiting time cannot be calculated analytically for the MVK model, we have calculated the median waiting time to the first M-cell, and , for the WF model and the S-MVK model, respectively (see Materials and Methods and Supplementary Material),
| (8) |
| (9) |
The two equations are strikingly similar. In both expressions the parameter governing the growth of clones harboring advantageous mutations, namely the selective advantage, s, and the net clonal expansion rate, (α – β), appears in the denominator, whereas all other parameters enter only logarithmically. Thus, in both models, the selection parameters have the strongest impact on the waiting time distributions, suggesting that cell selection is the major driving force of carcinogenesis.
The observed prominent role of cell selection is confirmed by numerical evaluation of the waiting time equations using best estimates from various model fits to individuals with full grown tumors. Sets of best estimated values, which were obtained from several studies by fitting the MVK or two-stage clonal expansion model to lung (26, 28-31) and colon cancer data (32, 33), were used to calculate (Supplementary Material, section 4; Table 1). In the present study, no model fits were performed. We have analyzed whether the predicted time from a clone of I-cells to the first M-cell is consistent between the two models. We performed calculations using m = 1, that is, a clone consisting of one I-cell and β = 0, assuming non-extinction (see Section 5 of the Supplementary Material for the effect of setting β = 0 on the parameter estimates). For lung cancer, we obtained values for between 99 and 192 years, and for colon cancer between 62 and 69 years (Table 1). For the WF process, we evaluated the same median waiting time with N = Ninit = 1, Nfin = 1 × 109, and k = 15, because initiated cells are assumed to harbor 5 of the 20 required mutations already (see Methods). With s = 1 × 10−3 and 1 × 10−4, the resulting waiting times are 17.6 and 43.9 years, respectively. We also evaluated Eq. 1 for the same parameter values and obtained E[TWF] = 42.0 and 105.1 years for s = 1 × 10−3 and 1 × 10−4, respectively. Given that the WF model has not been fitted to any cancer incidence or mortality data, the values for and E[TWF] based on the WF model compare well to those for based on the MVK model. Our values for also compare very well with those for the sojourn time, Ts, from the study by Meza and colleagues (27). For colon cancer, those authors found values between 55 and 64 years. For m = 1, we obtain = 62 and 69 years for the two colon cancer data sets (Table 1).
Table 1.
Median times to failure
| References and exact information from which sets of best estimates values were used for the calculations of τ |
Cancer | ||
|---|---|---|---|
| Meza and colleagues (28); “NHS” from Table 2 in cited article | 109.8 | 119.5 | Lung |
| Schöllnberger and colleagues (26); fit-RIVM1 from Table 4 in cited article | 192.3 | 134.8 | Lung |
| Hazelton and colleagues (29); “CPS-I males” from Table 2 in cited article | 99.7 | 132.0 | Lung |
| Moolgavkar and colleagues (30); “Model B” from Table 2 in cited article | 99.3 | 128.6 | Lung |
| Moolgavkar and colleagues (31); “Full Model” from Table 2 in cited article | 126.1 | 107.6 | Lung |
| Luebeck and Moolgavkar (32); “White males, 2 stage (k = 2)” from Table 3 in appendix to cited article |
68.7 | 87.5 | Colon |
| Moolgavkar and Luebeck (33); “Males, Two-mutation model” from Table 1 in cited article | 61.7 | 106.4 | Colon |
NOTE: Values for median time to the first M-cell (Eqs. 7 and 9) calculated from different sets of best estimated values to lung and colon cancer. We calculated for m = 1 and β = 0, i.e., starting with one initiated cell and assuming that there is no extinction of clones. For the lung cancer calculations of N = 1 × 107; for colon cancer, N = 1 × 108. These values are taken from the referenced studies. Further details of the calculations are provided in section 4 of the Supplementary Material.
In addition to the waiting times that start with a clone of I-cells, we also evaluated the median waiting time, , which starts with a pool of N-cells. For lung cancer, ranges from 108 to 135 years. For colon cancer, the calculated waiting times are also consistent across data sets (88–106 y; Table 1).
Discussion
We have applied two different mathematical models to study the relative contribution of mutation induction and cell selection to carcinogenesis. The WF model is based on an evolutionary theory of carcinogenesis (6, 34), and the MVK model is a stochastic two-stage model with clonal expansion (7, 8). Both models share essential biologically motivated characteristics, namely the generation of new cell types with increased replication capacities (Figs. 1 and 2). However, some specific model features are conceptually very different. For example, the MVK model applies two stages to malignancy with typical values for the background mutation rates of 1 × 10−8 to 1 × 10−6 per year and background net clonal expansion rates of 0.01 to 0.1 per year (26, 33). In general, the two mutation rates μ1 and μ2 in the MVK model do not refer to genomic mutation rates, but rather to transition rates between biophysical states of the carcinogenic process that might be the result of several genetic changes. This interpretation is in line with the original definition of μ1 and μ2 as event rates (7, 8) and consistent with Moolgavkar’s interpretation of Vogelstein’s multistage genetic model for colorectal tumorigenesis within the two-stage model (35). By contrast, the WF process assumes ~20 genetically defined stages, and the mutation rate between these stages directly reflects the DNA mutation rate in humans. Typical values for the selective advantage s are 0.01 and 0.001 per mutation, that is, 1% and 0.1% selective advantage, respectively. Because s can also be regarded as a growth rate (refer to Eq. 9 in the Supplementary Material of ref. 6), these values correspond to 3.65 and 0.365 per year, respectively. The normal mutation rate of u = 1 × 10−7 per gene per cell division corresponds to a total of u × d = 3.65 × 10−3 mutations per year. The mutation and selection parameters of the MVK and the WF model are not directly comparable because they refer to differently defined stages of carcinogenesis. However, obviously, the 20 stages of the WF model need to be taken much faster than the two stages of the MVK model to arrive at similar waiting times, implying higher values for the mutation rate or the selective advantage (or both) in the WF model, consistent with the values for s and u × d mentioned above.
We find that the MVK and the WF model have similar dynamics in the development of the first M-cell mainly driven by cell selection, that is, clonal expansions of cells harboring selectively advantageous mutations. This conclusion is based on comparing Eqs. 8 and 9, the median times to the first M-cell in the WF model and in the S-MVK model, respectively. Both waiting times are inversely proportional to the selective advantage, s, and the net clonal expansion rate, (α – β), respectively, whereas all other parameters enter the equations only logarithmically. Thus, in both models, the waiting time is very sensitive to parameters associated with promotion, but less sensitive to all other parameters, including mutation rates. Formulas have also been developed for the waiting time to the first M-cell starting with a clone of I-cells (Eqs. 1 and 7). Again, these expressions both show the strongest dependence on cell selection confirming the main finding of the study. Other approximations to the average time between initiation and promotion and more generally to the speed of evolution in large asexual populations have been reported elsewhere (34, 36).
Our findings are in line with the results of several earlier approaches (14-16). The major role of selection, in the form of promotion, was indicated as the mechanism of caloric reduction in preventing experimental cancer 65 years ago (37). Rodin and Rodin (14) analyzed mutation spectra and concluded that the main driver of lung carcinogenesis is cell selection, not mutagenesis. Cell selection was also shown to be the driving force in spontaneous transformation of cells in culture (38, 39). Tomlinson and colleagues (16) mathematically analyzed the role of the mutation rate in the growth of sporadic colorectal cancer and concluded that selection without increased mutation rates is sufficient to explain the evolution of tumors (15), challenging the concept of a mutator phenotype proposed by Loeb and colleagues (40, 41).
The dominance of cell selection over mutation is also visible in the hazard functions (Eqs. 2 and 5; Supplementary Material). For the WF process (Eq. 2), the mutation rate enters only logarithmically, whereas the selective advantage enters in a much stronger way as sk. For the S-MVK model (Eq. 5), the mutation rates enter linearly, whereas the net clonal expansion rate, (α – β), enters exponentially. These qualitative differences reflect our finding based on analyzing the median times to the first M-cell. For the S-MVK model, the much stronger influence of clonal expansion of initiated cells over mutations is a consequence of the exponential growth rate implied by the model structure. The WF process, which is based on the basic evolutionary mechanisms of mutation and selection, comes to the same conclusion. Therefore, both the S-MVK and the WF model provide independent support for this conclusion.
The approximations made in the derivation of τWF (Eq. 3), namely the approximation of the stochastic WF process by a linear multistep process using approximate transition rates, together with the fact that it had to be evaluated with parameter values that were not determined by fitting incidence or mortality data, make the comparison with the MVK model difficult. It is therefore not surprising that somewhat different numerical values are obtained for compared with . However, all results are clearly in the same order of magnitude, and given the strong approximations in the derivation of Eq. 3, they appear in reasonable agreement.
The two-stage clonal expansion model has recently been advanced to include preinitiation stages (27, 32). It has been shown that this new multistage clonal expansion model is consistent with the linear phase of cancer incidence in the Surveillance Epidemiology and End Results (SEER) data (27). The authors also presented a formula for the mean duration, Ts, from the birth of a premalignant clone (i.e., m = 1) to its eventual development into a malignant tumor, conditional on nonextinction (equivalent to assuming that β = 0): Ts ≈ ln(α/μ2)/α (here, we applied β = 0 in Eq. 3 of ref. 27). It can be shown that for β = 0 and m = 1, Eq. 7 yields the same expression: (refer to Supplementary Material, Section 6 for the mathematical details). It is plausible that because of the approximations necessary to obtain this closed-form expression, the median time to the first M-cell is identical to the mean time Ts. Values for Ts between 55 and 64 years obtained with the multistage clonal expansion model for colon cancer have been reported (27). These values are based on fitting three- and four-stage models to the SEER data. Their values for Ts need to be compared with evaluations of Eq. 7 for m = 1 and β = 0. For colon cancer, we obtained values between = 62 and 69 years (Table 1). We suggest that the differences from the Ts values in ref. (27) stem from the fact that these authors used different models and data compared with (32, 33). Given these differences, it seems that their values are in excellent agreement with ours.
We remark that a hazard function of the AD-type has been shown to provide a poorer fit to age-specific incidence data of colorectal cancer and pancreatic cancer in the SEER registry than more complex models (27). We nevertheless used the AD-model here because it has been shown (21) that the stochastic WF process can be approximated by a linear multistage process (i.e., an AD-type model) in which stages correspond to clonal expansions. In addition, the formula for τWF (Eq. 3), which was derived using an AD-model, was evaluated using values from ref. (6) and not with values obtained with the multistage clonal expansion model of Meza and colleagues (27).
The values for the median time to the first M-cell for the S-MVK model, , are consistent among various sets of best estimates for lung cancer (Table 1). Values between 110 and 140 years [i.e., + tlag with tlag = 5, respectively, 3.5 y (refer to Supplementary Material, Section 4)] for half of a population of nonsmokers to get lung cancer are compatible with epidemiologic studies (26, 42). We also calculated with parameter estimates relating to a male smoker who smokes 20 cigarettes per day starting at the age of 20 years for life (α – β = 0.133 y−1, β = 0, μ1 = μ0 = 2.03 × 10−7 y−1, μ2 = 5.06 × 10−7 y−1; these parameter estimates were taken from ref. 26). At an age of + tlag = 75.6 years, half of these life-long smokers would die from lung cancer. This model prediction is similar to the 88 years estimated in ref. (26) for the same event to happen. Two suitable sets of best estimates from MVK model fits to colon cancer were also identified. The calculated values for are not much different from each other (Table 1) and are consistent with extrapolations from epidemiologic studies. For example, Fig. 3 in ref. (33) provides the lifetime probability of colon cancer in the male population of Birmingham, Alabama represented in the SEER data. When this curve is extrapolated to higher ages (after fitting a suitable function to selected data points on that curve), it is found that after ~120 years half of the population has died from cancer. This is consistent with = 106 years from Table 1.
Summarizing, it can be said that the analytic and numerical analyses of the waiting times derived from two different mathematical models of carcinogenesis support the claim that cancer onset is dominated by the clonal expansion of mutated cells, that is, by cell selection, and that mutation induction has a smaller influence. We expect this conclusion to hold also for other solid tumors.
Supplementary Material
Acknowledgments
We thank Dr. Annette Kopp-Schneider, German Cancer Research Center, for the fruitful discussions and Dr. Petra Wark, Imperial College London, for reading an earlier version of the manuscript.
Grant Support
Grant from the European Union to the ECNIS Network of Excellence ((FOOD-CT-2005-513943), P. Vineis) and grants from the Austrian Science Fund FWF (P18055-N02 and P21630-N22) and financial support from Stiftungs- und Förderungsgesellschaft of Salzburg University (H. Schöllnberger).
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Day NE. Epidemiological data and multistage carcinogenesis. IARC Sci Publ. 1984;56:339–57. [PubMed] [Google Scholar]
- 2.Day NE, Brown CC. Multistage models and primary prevention of cancer. J Natl Cancer Inst. 1980;64:977–89. [PubMed] [Google Scholar]
- 3.Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 1954;8:1–12. doi: 10.1038/bjc.1954.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armitage P, Doll R. A two-stage theory of carcinogenesis in relation to the age distribution of human cancer. Br J Cancer. 1957;11:161–9. doi: 10.1038/bjc.1957.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gatenby RA, Vincent TL. An evolutionary model of carcinogenesis. Cancer Res. 2003;63:6212–20. [PubMed] [Google Scholar]
- 6.Beerenwinkel N, Antal T, Dingli D, et al. Genetic progression and the waiting time to cancer. PLoS Comput Biol. 2007;3:2239–46. doi: 10.1371/journal.pcbi.0030225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moolgavkar SH, Venzon D. Two-event models for carcinogenesis: incidence curves for childhood and adult tumors. Math Biosci. 1979;47:55–77. [Google Scholar]
- 8.Moolgavkar SH, Knudson AG., Jr. Mutation and cancer: a model for human carcinogenesis. J Natl Cancer Inst. 1981;66:1037–52. doi: 10.1093/jnci/66.6.1037. [DOI] [PubMed] [Google Scholar]
- 9.Michor F, Iwasa Y, Nowak MA. Dynamics of cancer progression. Nat Rev Cancer. 2004;4:197–205. doi: 10.1038/nrc1295. [DOI] [PubMed] [Google Scholar]
- 10.Vineis P, Schatzkin A, Potter JD. Models of carcinogenesis: an overview. Carcinogenesis. 2010 May 11; doi: 10.1093/carcin/bgq087. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ewens WJ. Mathematical population genetics. Springer; New York: 2004. [Google Scholar]
- 12.Heidenreich WF. On the parameters of the clonal expansion model. Radiat Environ Biophys. 1996;35:127–29. doi: 10.1007/BF02434036. [DOI] [PubMed] [Google Scholar]
- 13.Chen CW. Armitage-Doll two-stage model: implications and extension. Risk Anal. 1993;13:273–9. doi: 10.1111/j.1539-6924.1993.tb01079.x. [DOI] [PubMed] [Google Scholar]
- 14.Rodin SN, Rodin AS. Origins and selection of p53 mutations in lung carcinogenesis. Semin Cancer Biol. 2005;15:103–12. doi: 10.1016/j.semcancer.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 15.Tomlinson I, Bodmer W. Selection, the mutation rate and cancer: ensuring that the tail does not wag the dog. Nat Med. 1999;5:11–2. doi: 10.1038/4687. [DOI] [PubMed] [Google Scholar]
- 16.Tomlinson IPM, Novelli MR, Bodmer WF. The mutation rate and cancer. Proc Natl Acad Sci U S A. 1996;93:14800–3. doi: 10.1073/pnas.93.25.14800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sjöblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 18.Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 19.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerstung M, Beerenwinkel N. Waiting time models of cancer progression. Math Popul Stud. 2010;17:115–35. [Google Scholar]
- 22.Maley CC. Multistage carcinogenesis in Barrett’s esophagus. Cancer Lett. 2007;245:22–32. doi: 10.1016/j.canlet.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 23.Moolgavkar SH, Dewanji A, Venzon DJ. A stochastic two-stage model for cancer risk assessment. I. The hazard function and the probability of tumor. Risk Anal. 1988;8:383–92. doi: 10.1111/j.1539-6924.1988.tb00502.x. [DOI] [PubMed] [Google Scholar]
- 24.Hoogenveen RT, Clewell HJ, Andersen ME, Slob W. An alternative exact solution to the two-stage clonal growth model of cancer. Risk Anal. 1999;19:9–14. [Google Scholar]
- 25.Luebeck EG, Heidenreich WF, Hazelton WD, et al. Biologically based analysis of the data for the Colorado uranium miners cohort: age, dose and dose-rate effects. Radiat Res. 1999;152:339–51. [PubMed] [Google Scholar]
- 26.Schöllnberger H, Manuguerra M, Bijwaard H, et al. Analysis of epidemiological cohort data on smoking effects and lung cancer with multistage cancer models. Carcinogenesis. 2006;27:1432–44. doi: 10.1093/carcin/bgi345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meza R, Jeon J, Moolgavkar SH, Luebeck EG. Age-specific incidence of cancer: phases, transitions, and biological implications. Proc Natl Acad Sci U S A. 2008;105:16284–9. doi: 10.1073/pnas.0801151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meza R, Hazelton WD, Colditz GA, Moolgavkar SH. Cancer Causes Control. 2008;19:317–28. doi: 10.1007/s10552-007-9094-5. [DOI] [PubMed] [Google Scholar]
- 29.Hazelton WD, Clements MS, Moolgavkar SH. Multistage carcinogenesis and lung cancer mortality in three cohorts. Cancer Epidemiol Biomarkers Prev. 2005;14:1171–81. doi: 10.1158/1055-9965.EPI-04-0756. [DOI] [PubMed] [Google Scholar]
- 30.Moolgavkar SH, Luebeck EG, Krewski D, et al. Radon, cigarette smoke, and lung cancer: a re-analysis of the Colorado Plateau uranium miners’ data. Epidemiology. 1993;4:204–17. [PubMed] [Google Scholar]
- 31.Moolgavkar SH, Dewanji A, Luebeck G. Cigarette smoking and lung cancer: reanalysis of the British doctors’ data. J Natl Cancer Inst. 1989;81:415–20. doi: 10.1093/jnci/81.6.415. [DOI] [PubMed] [Google Scholar]
- 32.Luebeck EG, Moolgavkar SH. Multistage carcinogenesis and the incidence of colorectal cancer. Proc Natl Acad Sci U S A. 2002;99:15095–100. doi: 10.1073/pnas.222118199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moolgavkar SH, Luebeck EG. Multistage carcinogenesis: population-based model for colon cancer. J Natl Cancer Inst. 1992;84:610–18. doi: 10.1093/jnci/84.8.610. [DOI] [PubMed] [Google Scholar]
- 34.Park S-C, Simon D, Krug J. The speed of evolution in large asexual populations. J Stat Phys. 2010;138:381–410. [Google Scholar]
- 35.Moolgavkar SH. Carcinogenesis models: An overview. In: Glass WA, Varma MN, editors. Physical and chemical mechanisms in molecular radiation biology. Plenum Press; New York: 1991. pp. 387–99. [Google Scholar]
- 36.Herrero-Jimenez P, Tomita-Mitchell A, Furth EE, et al. Population risk and physiological rate parameters for colon cancer. The union of an explicit model for carcinogenesis with the public health records of the United States. Mutat Res. 2000;447:73–116. doi: 10.1016/s0027-5107(99)00201-8. [DOI] [PubMed] [Google Scholar]
- 37.Tannenbaum A. The dependence of the genesis of induced skin tumors on the caloric intake during different stages of carcinogenesis. Cancer Res. 1944;4:673–7. [Google Scholar]
- 38.Rubin H. Selected cell and selective microenvironment in neoplastic development. Cancer Res. 2001;61:799–807. [PubMed] [Google Scholar]
- 39.Rubin H. Cell-cell contact interactions conditionally determine suppression and selection of the neoplastic phenotype. Proc Natl Acad Sci U S A. 2008;105:6215–21. doi: 10.1073/pnas.0800747105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc Natl Acad Sci U S A. 2003;100:776–81. doi: 10.1073/pnas.0334858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991;51:3075–9. [PubMed] [Google Scholar]
- 42.Peto R, Darby S, Deo H, et al. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ. 2000;321:323–9. doi: 10.1136/bmj.321.7257.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.