

Abstract
Rho-kinase (ROCK) belongs to the AGC (protein kinase A/protein kinase G/protein kinase C, PKA/PKG/PKC) family of serine/threonine kinases and is a major downstream effector of small GTPase RhoA. Rho-kinase is involved in a wide range of fundamental cellular functions such as contraction, adhesion, migration, and proliferation. Two ROCK isoforms, ROCK1 and ROCK2, are assumed to be functionally redundant, based largely on the major common activators, the high degree of homology within the kinase domain, and studies from overexpression with kinase constructs and chemical inhibitors (e.g., Y27632 and fasudil), which inhibit both ROCK1 and ROCK2. Gene targeting and RNA interference approaches allow further dissection of distinct cellular, physiologic, and pathophysiologic functions of the two ROCK isoforms. This review focuses on the current understanding of ROCK isoform biology, with a particular emphasis on their functions in mouse development and the pathogenesis of heart failure.
Keywords: Genetic models, Heart failure, Rho-kinase, ROCK
Rho-kinase (ROCK) belongs to the AGC (protein kinase A/protein kinase G/protein kinase C, PKA/PKG/PKC) family of serine/threonine kinases [30, 38, 42, 46]. As a major downstream effector of the small GTPase RhoA, ROCK plays a major role in regulating rearrangement of the actomyosin cytoskeleton. The ROCK family contains two members, ROCK1 (also called ROKβ or p160 ROCK) and ROCK2 (also known as ROKα), that share 65% overall identity and 92% identity in the kinase domain. Both kinases contain a catalytic kinase domain at the N-terminus, followed by a central coiled-coil domain, including a Rho-binding domain (RBD) and a carboxyl-terminal pleckstrin-homology domain, with an internal cysteine-rich domain. In human and mouse, both ROCK1 and ROCK2 are ubiquitously expressed across tissues [46].
Since its discovery, the ROCK family has attracted much attention in various research fields. More than 7,000 articles have been published; many focused on ROCK function in the cardiovascular system, central nervous system, cancers, and embryonic development. Several excellent recent reviews have covered large aspects [4, 16, 25, 40, 44, 49].
The up-to-date progress in translational research has demonstrated that ROCK is an important therapeutic target for the treatment of various cardiovascular diseases and neurologic disorders, and cancers. In the studies, two relatively selective ROCK inhibitors, Y27632 [71] and fasudil [5], have been used extensively to dissect the roles of ROCK in cellular signaling and in animal disease models. These studies suggest that the inhibition of ROCK has great therapeutic potential. However, these inhibitors bind to the kinase domain and inhibit ROCK1 and ROCK2 with similar potency [7, 12, 31, 71]. The specific disruption of each ROCK isoform in mice offers a unique opportunity to analyze the physiologic and pathologic functions of ROCK1 and ROCK2 in vivo.
This review summarizes the current understanding of ROCK isoform biology, with a particular emphasis on their functions in mouse development and the pathogenesis of heart failure.
Common and Selective Interaction Partners of ROCK
Early ROCK studies support the paradigm that both ROCK isoforms are functionally redundant. Findings show that ROCK has autoinhibitory activity [2]. In the inactive form, the carboxyl terminal pleckstrin-homology domain and the RBD of ROCK interact with the kinase domain, forming an autoinhibitory loop. The RBD located in the coiled-coil domain interacts only with activated Rho GTPases including RhoA, RhoB, and RhoC [21]. The pleckstrin-homology domain is believed to interact with lipid mediators such as arachidonic acid [18] and sphingosylphosphorylcholine [18, 20, 66] and may also participate in protein localization [10, 32, 78]. RhoA and lipid mediators are common activators of both isoforms. Both isoforms phosphorylate the same major downstream substrates such as the myosin-binding subunit of myosin light chain phosphatase (MYPT1) [3, 33, 34], myosin light chain (MLC) [3, 36], LIM kinases [41, 50, 68], ezrin/radixin/moesin (ERM) [43], adducin [22], thereby modulating actin cytoskeleton organization, stress fiber formation, and cell contraction.
More than 20 ROCK substrates have been identified (see reviews in refs [4] and [61]). The majority of ROCK substrates have been identified from cell culture experiments. In most cases, only one ROCK isoform (more generally ROCK2) has been tested. Because ROCK1 and ROCK2 share 92% identity in the kinase domain, it is assumed that they share the same substrates.
A growing body of evidence indicates that both ROCK isoforms can have distinct interaction partners (regulators or substrates) in individual cell types and in turn can have distinct nonredundant functions. Caspase 3 cleaves ROCK1 at the cleavage site DETD1113 during apoptosis [11, 60]. This consensus sequence for caspase 3 cleavage is conserved in human, rat, and mouse but is not present in ROCK2.
On the other hand, during cytotoxic lymphocyte granule-induced cell death, human ROCK2 can be cleaved by the proapoptotic protease granzyme B at the IGLD1131 site, but this site is not present in ROCK1 [59]. The small GTP-binding protein RhoE interacts with the N-terminal region of ROCK1 (amino acids 1 to 420) but not ROCK2, and prevents Rho binding to RBD [55, 56]. By blocking RhoE association to ROCK1 in cancer cells, PDK1-kinase promotes ROCK1 (but not ROCK2), membrane localization, and activation [53]. In vascular smooth muscle cells, both ROCK isoforms modulate MYPT1 activity, but only ROCK2 binds directly to and phosphorylates MYPT1 [74].
Recent studies with an individual knockdown of ROCK1 and ROCK2 using short interfering RNA (siRNA)-based gene silencing or a genetic approach have shown that these two isoforms have nonredundant in vitro functions in fibroblasts [47, 78, 79], smooth muscle cells [74], endothelial cells [8, 45, 64], keratinocytes [39], and cancer cells [29]. Their functional differences could be explained by the fact that both isoforms are expressed at different levels or have different interaction partners in individual cell types. Their in vivo functional similarity and differences have been shown by mouse genetic studies during development and under pathologic conditions that this review highlights.
ROCK in Mouse Development
Both ROCK1 and ROCK2 are ubiquitously expressed in mouse embryos. However, they have distinct preferential expression patterns. In early mouse embryos (at stages E7.5 to E9.5), whole-mount in situ hybridization showed that ROCK1 is highly enriched in developing hearts, and ROCK2 is ubiquitously expressed [75]. In ROCK1-knockout (ROCK1−/−) embryos, which contain a knockin lacZ reporter gene, LacZ staining was detected in many locations throughout the embryo (E13.5 to E15.5) including the skin, heart, aorta, umbilical blood vessels, and dorsal root ganglia [65]. In ROCK2-knockout (ROCK2−/−) embryos with a knockin lacZ reporter gene, LacZ staining also was observed in many locations throughout the embryo (E13.5) including the heart, liver, umbilical blood vessels, and dorsal root ganglions. In addition, ROCK2 was highly expressed in the labyrinth layer of the placenta [69].
Data from our laboratory and others have shown that the genetic background affects the developmental phenotypes of ROCK1−/− mice [57, 65, 81] (Table 1). The ROCK1−/− mice with a C57BL/6 genetic background were born at expected Mendellian ratios but exhibited eyelids open at birth (EOB) and an omphalocele phenotype due to disorganization of actin filaments in the epithelial cells of the eyelids and of the umbilical ring [65].
Table 1.
Summary of developmental phenotype and survival rate of Rho-kinase (ROCK) knockout mice
| Genotype | Genetic background | Developmental phenotype | Survival ratea | Ref. |
|---|---|---|---|---|
| ROCK1−/− | C57BL/6 | Perinatal lethal with EOB and ompalocele | ~10% | [65] |
| Perinatal lethal with EOB and ompalocele | ~5% | [57] | ||
| Perinatal lethal with EOB and ompalocele | 3.5%b | d | ||
| FVB | Early embryonic lethal (before E9.5); survived mice have normal phenotype from E9.5 to adulthood |
40%b | [81] | |
| ROCK2−/− | C57BL/6 | Embryonic lethal at E13.5 with placental defects, also perinatal lethal with EOB and ompalocele |
<1%c | [70] |
| C57BL/6-129/SvJ | Embryonic lethal at E13.5 with placental defects | ~10%c | [69] | |
| CD1 | No developmental phenotype reported | ~100%c | [17] | |
| Embryonic lethal (stage and phenotype not determined); survived mice have normal phenotype |
~70% | [82] | ||
| ROCK1+/−/ROCK2+/− | C57BL/6 | Perinatal lethal with EOB and ompalocele | ~30% | [70] |
EOB, eyelids open at birth
Survival rate at weaning age
Same targeting vector for ROCK1 disruption
Same targeting vector for ROCK2 disruption
We analyzed 81 offspring obtained by intercrossing ROCK1+/− mice with a C57BL/6 background. Analysis of genotype distribution in offspring from heterozygous crosses showed that the homozygous ROCK1−/− mice were markedly underrepresented among littermates at the age of 3 weeks (29 ROCK1+/+, 56 ROCK1+/−, 1 ROCK1−/−, respectively)
The majority of ROCK1−/− mice (>90%) die soon after birth due to an omphalocele, with organs such as liver and gut protruding from the peritoneal cavity. However, EOB and omphalocele were not observed in ROCK1−/− mice with an FVB background, but the ratio of ROCK1−/− mice was sub-Mendellian because 60% died in utero before E9.5 [81]. The 40% survival rate was maintained for ROCK1−/− mice from E9.5 to adult stages, suggesting that ROCK1 acts on an early stage of embryonic development before organogenesis (before E9.5) in the FVB background.
To rule out the possibility that differences in targeting vector may contribute to the different phenotypes of ROCK1−/− mice generated by different laboratories [57, 65, 81], we backcrossed the ROCK1−/− mice from FVB into a C57BL/6 background for 10 generations. As expected, these ROCK1−/− mice with the C57BL/6 background exhibited EOB and omphalocele. Their survival rate at weaning age significantly dropped to less than 4% (Table 1). Thus, genetic background affects EOB and omphalocele in ROCK1−/− mice.
The developmental phenotypes of ROCK2−/− mice also depend on the genetic background. Findings show that ROCK2−/− mice with a mixed genetic background between 129/SvJ and C57BL/6 are embryonically lethal because of placental dysfunction from thrombus formation in the labyrinth layer of the placenta and have intrauterine growth retardation [69]. When these ROCK2−/− mice were backcrossed into a C57BL/6 genetic background, they exhibited not only the placental phenotype but also the EOB and omphalocele phenotype [70], indicating that genetic background affects the EOB and omphalocele phenotype in ROCK2−/− mice. In addition, when these ROCK2−/− mice were backcrossed into an outbred strain background, CD1 (C57BL/6xDba), they were born at close to Mendelian ratios, and most survived to adulthood [17]. High survival rates for ROCK2−/− mice with a CD1 background also were reported by another independent study (Table 1) [82].
The shared EOB and omphalocele phenotypes in ROCK1−/− and ROCK2−/− mice with a C57BL/6 genetic background indicate that they act together to regulate the assembly of actin bundles essential for closure of the eyelid and the ventricular body wall in mouse embryos. A common characteristic of ROCK1−/− and ROCK2−/− mice, regardless of their genetic background, is that they develop normally and are apparently healthy and fertile after surviving their intrauterine and perinatal period [57, 65, 69, 81], suggesting that each isoform is able to compensate functionally for the loss of the other during embryogenesis. In addition, no compensatory upregulation of the ROCK1 expression exists in ROCK2−/− mice or vice versa. Together, these genetic studies using ROCK1−/− and ROCK2−/− mice provide significant insights into the biologic functions of ROCK1 and ROCK2 isoforms, which appear to be largely redundant during development.
ROCK in Pathologic Cardiac Hypertrophy and Heart Failure
Both ROCK1 and ROCK2 are ubiquitously expressed across human, rat, and mouse tissues including vasculature and heart [30, 38, 46, 77]. Their expression, activation, or both can be subjected to distinct regulation under pathologic conditions in the heart. During pressure overload-induced cardiac hypertrophy [81] and failing hearts due to transgenic Gαq overexpression and pregnancy stress [63], ROCK1 mRNA and protein levels were increased and associated with increased ezrin/radixin/moesin phosphorylation, whereas ROCK2 expression levels remained unaltered. In addition, ROCK1 was found to be cleaved in failing human hearts, most likely via activated caspase 3, leading to ROCK1 activation by removal of the C-terminal autoinhibitory domain [9]. Studies using pharmacologic inhibitors Y27632 and fasudil suggest an in vivo role for ROCK in the pathogenesis of cardiac hypertrophy and remodeling [23, 26, 28, 35, 52, 58, 80]. However, as mentioned earlier, these inhibitors do not distinguish between ROCK1 and ROCK2 [71].
Studies have used ROCK1−/− mice with an FVB background to examine how this kinase contributes to pathologic cardiac hypertrophy and remodeling. Using several disease models that mimic chronic high blood pressure, partial or full ROCK1 deletion did not block the development of cardiomyocyte hypertrophy [57, 62, 63, 81]. Although ROCK1 is not required for the development of cardiac hypertrophy, ROCK1 deletion significantly reduces a number of structural and functional alterations attributable to pathologic hypertrophic remodeling including cardiac fibrosis [57, 81], cardiomyocyte apoptosis [9, 63], left ventricular dilation, and contractile dysfunction [62, 63].
The long-term impact of ROCK1 deficiency on the progression of heart failure was highlighted in a murine congestive heart failure model [63]. Transgenic mice with cardiac-restricted overexpression of Gαq experienced lethal cardiomyopathy after pregnancy or at old age [1]. Deletion of ROCK1 completely abolished animal death and preserved cardiac function in peripartum or 1-year-old Gαq mice [63]. On the other hand, transgenic cardiac-restricted overexpression of ROCK1 accelerated progression to heart failure in Gαq hearts in the absence of stress accompanied by increased cardiac fibrosis and cardiomyocyte apoptosis [63]. Together, these studies provide in vivo evidence for an important role of ROCK1 in hypertrophic decompensation.
The cellular and molecular mechanisms underlying the fibrotic role of ROCK1 in hypertrophic decompensation remain to be defined. The reduced fibrosis in ROCK1-deficient mice may be due to reduced induction of fibrogenic cytokines such as transforming growth factor-beta 2 (TGF-β2) and connective tissue growth factor (CTGF) released from cardiomyocytes in response to pathologic stimuli [81]. Alternatively, ROCK1 may mediate cardiac fibrosis by regulating cardiac fibroblast differentiation and activation. Clearly, ROCK1 is involved in monocytic fibroblast precursor cell differentiation because ROCK1 deletion resulted in markedly lower numbers of myofibroblasts and monocytic fibroblast precursors in a murine ischemic/reperfusion cardiomyopathy model [27].
According to a recent study, ROCK1 may regulate myocardin-related transcription factor A (MRTF-A)-mediated myofibroblast activation because the findings showed that TGF-β1 induced nuclear accumulation of MRTF-A in a ROCK-dependent manner in cardiac fibroblasts, leading to the activation of serum response factor and collagen synthesis [67]. Further studies with cell-type-specific deletion of ROCK1 will help to examine the contribution of different cell types to ROCK1-mediated fibrosis.
How ROCK1 contributes to cardiomyocyte apoptosis is another important question raised by in vivo studies mentioned earlier. The anti-apoptotic effects of ROCK1 deletion in hypertrophic hearts were found to be associated with enhanced extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK) and/or Akt activation [9, 63], suggesting a role for ROCK1 in modulating the activity of these survival pathways under pathologic conditions. Both the pro-apoptotic and anti-apoptotic roles of ROCK have been extensively reported in a variety of in vitro and in vivo studies. But how the basic components of the apoptotic machinery are regulated by ROCK is not completely understood in many instances and likely differs depending on the cell type and the apoptotic stimulus [61]. In cultured cardiomyocytes, acute activation of RhoA/ROCK (<24 h) inhibited apoptosis through the focal adhesion kinase (FAK)/PI3K/Akt survival pathway, whereas more sustained activation of Rho/ROCK (48–72 h) induced apoptosis through activation of the p53/Bax-mediated mitochondrial death pathway [14, 15]. Clearly, more studies are needed for an understanding of the molecular mechanisms through which chronic ROCK1 deletion inhibits apoptosis in hypertrophic hearts.
The preventive effects of ROCK1 deletion on contractile dysfunction in hypertrophic hearts can be attributed to reduced remodeling events including fibrosis, apoptosis, and chamber dilation. In addition, rescuing the expression of types 5 and 6 adenylyl cyclases (AC5/6) by ROCK1 deletion also may contribute to the improved β-adrenergic receptor signaling and preserved contractile function in the Gαq transgenic model of dilated cardiomyopathy [62, 63].
Most of the studies using ROCK inhibitors address the involvement of ROCK in cardiac hypertrophy and remodeling, but the role of ROCK in cardiac contraction still is not clear. Opposite mechanisms have been proposed. On the one hand, ROCK is reported to mediate α1-adregergic receptor agonist-stimulated contraction in hearts through the MYPT/MLC pathway (similar to smooth muscle cells) [13, 24, 54]. On the other hand, phosphorylation of cardiac troponin I/T by ROCK resulted in impaired contraction [72].
The studies with ROCK1-deficient mice have shown critical contributions of ROCK1 in the pathogenesis of heart failure (Fig. 1), representing an important advance in our understanding in ROCK1 isoform biology. These findings also highlight gaps in our understanding of ROCK2 isoform biology in pathologic cardiac hypertrophy and remodeling. The aforementioned studies did not use ROCK2−/− mice. The finding that ROCK1 is not required for the development of cardiac hypertrophy is in contrast to the observed antihypertrophic effects of ROCK inhibitors Y27632 and fasudil [26, 28, 35, 58], suggesting that ROCK2 may play a dominant role in regulating hypertrophic response or that the antihypertrophic effects of ROCK inhibitors are not exclusively the result of ROCK inhibition.
Fig. 1.
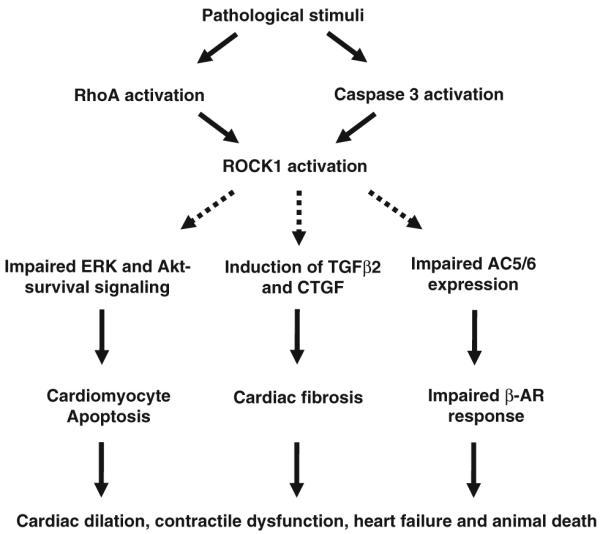
Schematic diagram showing pathologic roles of Rho-kinase 1 (ROCK1) in cardiac remodeling of pathologic hypertrophy. The studies with ROCK1-deficient mice have shown critical contributions of ROCK1 in the pathogenesis of heart failure. Pathologic stimuli activate ROCK1 through RhoA or caspase 3-dependent cleavage. Although ROCK1 is not required for the development of cardiomyocyte hypertrophy, it contributes to the pathologic remodeling events including cardiac fibrosis, cardiomyocyte apoptosis, and contractile dysfunction, leading to cardiac dilation, heart failure, and animal death. Moreover, ROCK1 appears to mediate impaired ERK- and Akt-dependent survival signaling, increased fibrogenic cytokines (TGFβ2 and CTGF), and altered AC5/6 expression induced by hypertrophic stimuli. Further studies are needed to define the signaling pathways downstream of ROCK1 that are involved in regulating heart failure progression (broken lines)
The findings that deletion of ROCK1 reduced cardiac fibrosis and heart failure progression are consistent with the results derived from ROCK inhibitor studies and validate ROCK1 as a potential therapeutic target in limiting heart failure progression. These observations suggest that ROCK1 and ROCK2 may have nonredundant functions in pathologic hypertrophy, with ROCK1 perhaps involved in cardiac fibrosis and apoptosis and ROCK2 possibly involved in hypertrophy. However, whether these two ROCK isoforms are qualitatively different or whether their function differences are due to their different expression levels in individual cell or tissue types remains to be determined.
It is worth noting that in contrast to the observed effects in the heart, ROCK1 deletion did not prevent renal fibrosis induced by unilateral ureteral obstruction, suggesting that ROCK2 may regulate fibrosis in this renal fibrosis model [19]. Further studies with systemic and conditional ROCK2 knockout mice are needed to determine the contribution of ROCK2 to cardiac hypertrophy, fibrosis, apoptosis, and contraction.
ROCK in Other Pathologic Conditions
Homozygous and heterozygous ROCK1 and ROCK2 knockout mice have been used for examination of their contributions to several pathologic conditions in addition to cardiac hypertrophy and decompensation. For some diseases, such as glaucoma, both ROCK isoforms contribute to the regulation of intraocular pressure [76]. On the other hand, ROCK1 appears to play a predominant role in vascular inflammation diseases [48].
In many studies, only one isoform has been investigated. For example, ROCK1-deficient mice exhibited systemic insulin resistance with impaired insulin signaling in skeletal muscle, suggesting that ROCK1 regulates glucose homeostasis and insulin sensitivity [37]. Moreover, ROCK1 deficiency resulted in increased recruitment and migration of macrophages and neutrophils in vitro and in vivo during acute inflammation through regulation of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) phosphorylation and stability [73]. In the study of Ongusaha et al. [51], ROCK1 haplodeficient mice exhibited decreased ultraviolet (UV)-mediated activation of c-Jun N-terminal kinase (JNK) and apoptosis in keratinocytes through regulation of phosphorylation of JNK-interacting protein 3. Zhou et al. [82] showed that ROCK2 knockout mice have impaired spine morphology and synaptic function, supporting the conclusion that ROCK2/cofilin signaling is critical in the regulation of neuronal actin, spine properties, and synapse density. However, ROCK2-deficient mice have shown improved functional recovery after spinal cord injury, suggesting a role for ROCK2 in limiting axonal growth after trauma within the adult mouse spinal cord [17]. Although these studies suggest a major contributory role of one isoform to these pathophysiologic conditions, future studies are required to determine the contribution of another isoform.
Conclusions and Future Directions
Not only do ROCK1 and ROCK2 function in a redundant manner, they also have their own distinct roles in some tissues under certain pathophysiologic conditions. A number of factors could contribute to their distinct functions including the differences in their expression level, tissue distribution, subcellular localization, activation by upstream signals, and interaction with downstream molecules. Evidence from knockout mice and the experimental and clinical use of ROCK inhibitors support the conclusion that ROCK is a potential therapeutic target for many human diseases.
The use of more refined mouse models, including conditional and inducible deletion of ROCK1 and ROCK2, will lead to a deeper understanding of each isoform’s relative importance in a disease context. As mentioned earlier, most published ROCK inhibitors are equally potent for both isoforms. Only one ROCK2-selective compound, SLx-2119, has been described [6] and currently is being tested in clinical trials. A further understanding of the physiologic and pathophysiologic roles of each ROCK isoform is much desired for a better evaluation of the beneficial and side effects of ROCK pan-inhibitors in animal and clinical studies and will be informative in efforts to generate new isoform-selective ROCK inhibitors.
Acknowledgments
This work was supported by the Riley Children’s Foundation, the Indiana University Department of Pediatrics (Cardiology), and the National Institutes of Health (NIH P01 HL085098 to LW).
Contributor Information
Jianjian Shi, Riley Heart Research Center, Herman B Wells Center for Pediatric Research, Department of Pediatrics, Indiana University, School of Medicine, 1044 West Walnut Street, Indianapolis, IN 46202-5225, USA.
Lumin Zhang, Riley Heart Research Center, Herman B Wells Center for Pediatric Research, Department of Pediatrics, Indiana University, School of Medicine, 1044 West Walnut Street, Indianapolis, IN 46202-5225, USA.
Lei Wei, Riley Heart Research Center, Herman B Wells Center for Pediatric Research, Department of Pediatrics, Indiana University, School of Medicine, 1044 West Walnut Street, Indianapolis, IN 46202-5225, USA.
References
- 1.Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, Chien KR, Brown JH, Dorn GW., II Enhanced Galphaq signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci USA. 1998;95:10140–10145. doi: 10.1073/pnas.95.17.10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amano M, Chihara K, Nakamura N, Kaneko T, Matsuura Y, Kaibuchi K. The COOH terminus of rho-kinase negatively regulates rho-kinase activity. J Biol Chem. 1999;274:32418–32424. doi: 10.1074/jbc.274.45.32418. [DOI] [PubMed] [Google Scholar]
- 3.Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and activation of myosin by rho-associated kinase (rho-kinase) J Biol Chem. 1996;271:20246–20249. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- 4.Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton Hoboken. 2010;67:545–554. doi: 10.1002/cm.20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asano T, Ikegaki I, Satoh S, Suzuki Y, Shibuya M, Takayasu M, Hidaka H. Mechanism of action of a novel antivasospasm drug, HA1077. J Pharmacol Exp Ther. 1987;241:1033–1040. [PubMed] [Google Scholar]
- 6.Boerma M, Fu Q, Wang J, Loose DS, Bartolozzi A, Ellis JL, McGonigle S, Paradise E, Sweetnam P, Fink LM, Vozenin-Brotons MC, Hauer-Jensen M. Comparative gene expression profiling in three primary human cell lines after treatment with a novel inhibitor of rho-kinase or atorvastatin. Blood Coagul Fibrinolysis. 2008;19:709–718. doi: 10.1097/MBC.0b013e32830b2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breitenlechner C, Gassel M, Hidaka H, Kinzel V, Huber R, Engh RA, Bossemeyer D. Protein kinase A in complex with rho-kinase inhibitors Y-27632, Fasudil, and H-1152P: structural basis of selectivity. Structure. 2003;11:1595–1607. doi: 10.1016/j.str.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Bryan BA, Dennstedt E, Mitchell DC, Walshe TE, Noma K, Loureiro R, Saint-Geniez M, Campaigniac JP, Liao JK, D’Amore PA. RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. FASEB J. 2010;24:3186–3195. doi: 10.1096/fj.09-145102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang J, Xie M, Shah VR, Schneider MD, Entman ML, Wei L, Schwartz RJ. Activation of rho-associated coiled-coil protein kinase 1 (ROCK-1) by caspase-3 cleavage plays an essential role in cardiac myocyte apoptosis. Proc Natl Acad Sci USA. 2006;103:14495–14500. doi: 10.1073/pnas.0601911103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen XQ, Tan I, Ng CH, Hall C, Lim L, Leung T. Characterization of rhoA-binding kinase ROKalpha implication of the pleckstrin homology domain in ROKalpha function using region-specific antibodies. J Biol Chem. 2002;277:12680–12688. doi: 10.1074/jbc.M109839200. [DOI] [PubMed] [Google Scholar]
- 11.Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- 12.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis JS, Hassanzadeh S, Winitsky S, Lin H, Satorius C, Vemuri R, Aletras AH, Wen H, Epstein ND. The overall pattern of cardiac contraction depends on a spatial gradient of myosin regulatory light chain phosphorylation. Cell. 2001;107:631–641. doi: 10.1016/s0092-8674(01)00586-4. [DOI] [PubMed] [Google Scholar]
- 14.Del Re DP, Miyamoto S, Brown JH. RhoA/rho-kinase upregulates Bax to activate a mitochondrial death pathway and induce cardiomyocyte apoptosis. J Biol Chem. 2007;282:8069–8078. doi: 10.1074/jbc.M604298200. [DOI] [PubMed] [Google Scholar]
- 15.Del Re DP, Miyamoto S, Brown JH. Focal adhesion kinase as a rhoA-activable signaling scaffold mediating Akt activation and cardiomyocyte protection. J Biol Chem. 2008;283:35622–35629. doi: 10.1074/jbc.M804036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong M, Yan BP, Liao JK, Lam YY, Yip GW, Yu CM. Rho-kinase inhibition: a novel therapeutic target for the treatment of cardiovascular diseases. Drug Discov Today. 2010;15:622–629. doi: 10.1016/j.drudis.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duffy P, Schmandke A, Schmandke A, Sigworth J, Narumiya S, Cafferty WB, Strittmatter SM. Rho-associated kinase II (ROCKII) limits axonal growth after trauma within the adult mouse spinal cord. J Neurosci. 2009;29:15266–15276. doi: 10.1523/JNEUROSCI.4650-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng J, Ito M, Kureishi Y, Ichikawa K, Amano M, Isaka N, Okawa K, Iwamatsu A, Kaibuchi K, Hartshorne DJ, Nakano T. Rho-associated kinase of chicken gizzard smooth muscle. J Biol Chem. 1999;274:3744–3752. doi: 10.1074/jbc.274.6.3744. [DOI] [PubMed] [Google Scholar]
- 19.Fu P, Liu F, Su S, Wang W, Huang XR, Entman ML, Schwartz RJ, Wei L, Lan HY. Signaling mechanism of renal fibrosis in unilateral ureteral obstructive kidney disease in ROCK1 knockout mice. J Am Soc Nephrol. 2006;17:3105–3114. doi: 10.1681/ASN.2005121366. [DOI] [PubMed] [Google Scholar]
- 20.Fu X, Gong MC, Jia T, Somlyo AV, Somlyo AP. The effects of the rho-kinase inhibitor Y-27632 on arachidonic acid-, GTPgammaS-, and phorbol ester-induced Ca2+-sensitization of smooth muscle. FEBS Lett. 1998;440:183–187. doi: 10.1016/s0014-5793(98)01455-0. [DOI] [PubMed] [Google Scholar]
- 21.Fujisawa K, Fujita A, Ishizaki T, Saito Y, Narumiya S. Identification of the rho-binding domain of p160ROCK, a rho-associated coiled-coil containing protein kinase. J Biol Chem. 1996;271:23022–23028. doi: 10.1074/jbc.271.38.23022. [DOI] [PubMed] [Google Scholar]
- 22.Fukata Y, Oshiro N, Kinoshita N, Kawano Y, Matsuoka Y, Bennett V, Matsuura Y, Kaibuchi K. Phosphorylation of adducin by rho-kinase plays a crucial role in cell motility. J Cell Biol. 1999;145:347–361. doi: 10.1083/jcb.145.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukui S, Fukumoto Y, Suzuki J, Saji K, Nawata J, Tawara S, Shinozaki T, Kagaya Y, Shimokawa H. Long-term inhibition of rho-kinase ameliorates diastolic heart failure in hypertensive rats. J Cardiovasc Pharmacol. 2008;51:317–326. doi: 10.1097/FJC.0b013e31816533b7. [DOI] [PubMed] [Google Scholar]
- 24.Grimm M, Haas P, Willipinski-Stapelfeldt B, Zimmermann WH, Rau T, Pantel K, Weyand M, Eschenhagen T. Key role of myosin light chain (MLC) kinase-mediated MLC2a phosphorylation in the alpha 1-adrenergic positive inotropic effect in human atrium. Cardiovasc Res. 2005;65:211–220. doi: 10.1016/j.cardiores.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 25.Hahmann C, Schroeter T. Rho-kinase inhibitors as therapeutics: from pan inhibition to isoform selectivity. Cell Mol Life Sci. 2010;67:171–177. doi: 10.1007/s00018-009-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hattori T, Shimokawa H, Higashi M, Hiroki J, Mukai Y, Tsutsui H, Kaibuchi K, Takeshita A. Long-term inhibition of rho-kinase suppresses left ventricular remodeling after myocardial infarction in mice. Circulation. 2004;109:2234–2239. doi: 10.1161/01.CIR.0000127939.16111.58. [DOI] [PubMed] [Google Scholar]
- 27.Haudek SB, Gupta D, Dewald O, Schwarz RJ, Wei L, Trial J, Entman ML. Rho-kinase-1 mediates cardiac fibrosis by regulating fibroblast precursor cell differentiation. Cardiovasc Res. 2009;83:511–518. doi: 10.1093/cvr/cvp135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Higashi M, Shimokawa H, Hattori T, Hiroki J, Mukai Y, Morikawa K, Ichiki T, Takahashi S, Takeshita A. Long-term inhibition of rho-kinase suppresses angiotensin II-induced cardiovascular hypertrophy in rats in vivo: effect on endothelial NAD(P)H oxidase system. Circ Res. 2003;93:767–775. doi: 10.1161/01.RES.0000096650.91688.28. [DOI] [PubMed] [Google Scholar]
- 29.Inaba N, Ishizawa S, Kimura M, Fujioka K, Watanabe M, Shibasaki T, Manome Y. Effect of inhibition of the ROCK isoform on RT2 malignant glioma cells. Anticancer Res. 2010;30:3509–3514. [PubMed] [Google Scholar]
- 30.Ishizaki T, Maekawa M, Fujisawa K, Okawa K, Iwamatsu A, Fujita A, Watanabe N, Saito Y, Kakizuka A, Morii N, Narumiya S. The small GTP-binding protein rho binds to and activates a 160-kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. Embo J. 1996;15:1885–1893. [PMC free article] [PubMed] [Google Scholar]
- 31.Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, Narumiya S. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol Pharmacol. 2000;57:976–983. [PubMed] [Google Scholar]
- 32.Kawabata S, Usukura J, Morone N, Ito M, Iwamatsu A, Kaibuchi K, Amano M. Interaction of rho-kinase with myosin II at stress fibres. Genes Cells. 2004;9:653–660. doi: 10.1111/j.1356-9597.2004.00749.x. [DOI] [PubMed] [Google Scholar]
- 33.Kawano Y, Fukata Y, Oshiro N, Amano M, Nakamura T, Ito M, Matsumura F, Inagaki M, Kaibuchi K. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by rho-kinase in vivo. J Cell Biol. 1999;147:1023–1038. doi: 10.1083/jcb.147.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by rho and rho-associated kinase (rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi N, Horinaka S, Mita S, Nakano S, Honda T, Yoshida K, Kobayashi T, Matsuoka H. Critical role of rho-kinase pathway for cardiac performance and remodeling in failing rat hearts. Cardiovasc Res. 2002;55:757–767. doi: 10.1016/s0008-6363(02)00457-1. [DOI] [PubMed] [Google Scholar]
- 36.Kureishi Y, Kobayashi S, Amano M, Kimura K, Kanaide H, Nakano T, Kaibuchi K, Ito M. rho-associated kinase directly induces smooth muscle contraction through myosin light chain phosphorylation. J Biol Chem. 1997;272:12257–12260. doi: 10.1074/jbc.272.19.12257. [DOI] [PubMed] [Google Scholar]
- 37.Lee DH, Shi J, Jeoung NH, Kim MS, Zabolotny JM, Lee SW, White MF, Wei L, Kim YB. Targeted disruption of ROCK1 causes insulin resistance in vivo. J Biol Chem. 2009;284:11776–11780. doi: 10.1074/jbc.C900014200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leung T, Chen XQ, Manser E, Lim L. The p160 rhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol Cell Biol. 1996;16:5313–5327. doi: 10.1128/mcb.16.10.5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lock FE, Hotchin NA. Distinct roles for ROCK1 and ROCK2 in the regulation of keratinocyte differentiation. PLoS One. 2009;4:e8190. doi: 10.1371/journal.pone.0008190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loirand G, Pacaud P. The role of rho protein signaling in hypertension. Nat Rev Cardiol. 2010;7:637–647. doi: 10.1038/nrcardio.2010.136. [DOI] [PubMed] [Google Scholar]
- 41.Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- 42.Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein rho. Embo J. 1996;15:2208–2216. [PMC free article] [PubMed] [Google Scholar]
- 43.Matsui T, Maeda M, Doi Y, Yonemura S, Amano M, Kaibuchi K, Tsukita S, Tsukita S. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J Cell Biol. 1998;140:647–657. doi: 10.1083/jcb.140.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miyamoto S, Del Re DP, Xiang SY, Zhao X, Florholmen G, Brown JH. Revisited and revised: is rhoA always a villain in cardiac pathophysiology? J Cardiovasc Transl Res. 2010;3:330–343. doi: 10.1007/s12265-010-9192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mong PY, Wang Q. Activation of rho-kinase isoforms in lung endothelial cells during inflammation. J Immunol. 2009;182:2385–2394. doi: 10.4049/jimmunol.0802811. [DOI] [PubMed] [Google Scholar]
- 46.Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, two isoforms of rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996;392:189–193. doi: 10.1016/0014-5793(96)00811-3. [DOI] [PubMed] [Google Scholar]
- 47.Noguchi M, Hosoda K, Fujikura J, Fujimoto M, Iwakura H, Tomita T, Ishii T, Arai N, Hirata M, Ebihara K, Masuzaki H, Itoh H, Narumiya S, Nakao K. Genetic and pharmacological inhibition of rho-associated kinase II enhances adipogenesis. J Biol Chem. 2007;282:29574–29583. doi: 10.1074/jbc.M705972200. [DOI] [PubMed] [Google Scholar]
- 48.Noma K, Rikitake Y, Oyama N, Yan G, Alcaide P, Liu PY, Wang H, Ahl D, Sawada N, Okamoto R, Hiroi Y, Shimizu K, Luscinskas FW, Sun J, Liao JK. ROCK1 mediates leukocyte recruitment and neointima formation following vascular injury. J Clin Invest. 2008;118:1632–1644. doi: 10.1172/JCI29226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nunes KP, Rigsby CS, Webb RC. RhoA/rho-kinase and vascular diseases: what is the link? Cell Mol Life Sci. 2010;67:3823–3836. doi: 10.1007/s00018-010-0460-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K. Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem. 2000;275:3577–3582. doi: 10.1074/jbc.275.5.3577. [DOI] [PubMed] [Google Scholar]
- 51.Ongusaha PP, Qi HH, Raj L, Kim YB, Aaronson SA, Davis RJ, Shi Y, Liao JK, Lee SW. Identification of ROCK1 as an upstream activator of the JIP-3 to JNK signaling axis in response to UVB damage. Sci Signal. 2008;1:ra14. doi: 10.1126/scisignal.1161938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Phrommintikul A, Tran L, Kompa A, Wang B, Adrahtas A, Cantwell D, Kelly DJ, Krum H. Effects of a rho-kinase inhibitor on pressure overload induced cardiac hypertrophy and associated diastolic dysfunction. Am J Physiol Heart Circ Physiol. 2008;294:H1804–H1814. doi: 10.1152/ajpheart.01078.2007. [DOI] [PubMed] [Google Scholar]
- 53.Pinner S, Sahai E. PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by rhoE. Nat Cell Biol. 2008;10:127–137. doi: 10.1038/ncb1675. [DOI] [PubMed] [Google Scholar]
- 54.Rajashree R, Blunt BC, Hofmann PA. Modulation of myosin phosphatase targeting subunit and protein phosphatase 1 in the heart. Am J Physiol Heart Circ Physiol. 2005;289:H1736–H1743. doi: 10.1152/ajpheart.00318.2004. [DOI] [PubMed] [Google Scholar]
- 55.Riento K, Guasch RM, Garg R, Jin B, Ridley AJ. RhoE binds to ROCK I and inhibits downstream signaling. Mol Cell Biol. 2003;23:4219–4229. doi: 10.1128/MCB.23.12.4219-4229.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Riento K, Totty N, Villalonga P, Garg R, Guasch R, Ridley AJ. RhoE function is regulated by ROCK I-mediated phosphorylation. Embo J. 2005;24:1170–1180. doi: 10.1038/sj.emboj.7600612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rikitake Y, Oyama N, Wang CY, Noma K, Satoh M, Kim HH, Liao JK. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/− haploinsufficient mice. Circulation. 2005;112:2959–2965. doi: 10.1161/CIRCULATIONAHA.105.584623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Satoh S, Ueda Y, Koyanagi M, Kadokami T, Sugano M, Yoshikawa Y, Makino N. Chronic inhibition of rho-kinase blunts the process of left ventricular hypertrophy leading to cardiac contractile dysfunction in hypertension-induced heart failure. J Mol Cell Cardiol. 2003;35:59–70. doi: 10.1016/s0022-2828(02)00278-x. [DOI] [PubMed] [Google Scholar]
- 59.Sebbagh M, Hamelin J, Bertoglio J, Solary E, Breard J. Direct cleavage of ROCK II by granzyme B induces target cell membrane blebbing in a caspase-independent manner. J Exp Med. 2005;201:465–471. doi: 10.1084/jem.20031877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol. 2001;3:346–352. doi: 10.1038/35070019. [DOI] [PubMed] [Google Scholar]
- 61.Shi J, Wei L. Rho-kinase in the regulation of cell death and survival. Arch Immunol Ther Exp Warsz. 2007;55:61–75. doi: 10.1007/s00005-007-0009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi J, Zhang YW, Summers LJ, Dorn GW, II, Wei L. Disruption of ROCK1 gene attenuates cardiac dilation and improves contractile function in pathological cardiac hypertrophy. J Mol Cell Cardiol. 2008;44:551–560. doi: 10.1016/j.yjmcc.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shi J, Zhang YW, Yang Y, Zhang L, Wei L. ROCK1 plays an essential role in the transition from cardiac hypertrophy to failure in mice. J Mol Cell Cardiol. 2010;49:819–828. doi: 10.1016/j.yjmcc.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shimada H, Rajagopalan LE. Rho-kinase-2 activation in human endothelial cells drives lysophosphatidic acid-mediated expression of cell adhesion molecules via NF-kappaB p65. J Biol Chem. 2010;285:12536–12542. doi: 10.1074/jbc.M109.099630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shimizu Y, Thumkeo D, Keel J, Ishizaki T, Oshima H, Oshima M, Noda Y, Matsumura F, Taketo MM, Narumiya S. ROCK-I regulates closure of the eyelids and ventral body wall by inducing assembly of actomyosin bundles. J Cell Biol. 2005;168:941–953. doi: 10.1083/jcb.200411179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shirao S, Kashiwagi S, Sato M, Miwa S, Nakao F, Kurokawa T, Todoroki-Ikeda N, Mogami K, Mizukami Y, Kuriyama S, Haze K, Suzuki M, Kobayashi S. Sphingosylphosphorylcholine is a novel messenger for rho-kinase-mediated Ca2+ sensitization in the bovine cerebral artery: unimportant role for protein kinase C. Circ Res. 2002;91:112–119. doi: 10.1161/01.res.0000026057.13161.42. [DOI] [PubMed] [Google Scholar]
- 67.Small EM, Thatcher JE, Sutherland LB, Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara K, Olson EN. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res. 2010;107:294–304. doi: 10.1161/CIRCRESAHA.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sumi T, Matsumoto K, Nakamura T. Specific activation of LIM kinase 2 via phosphorylation of threonine 505 by ROCK, a rho-dependent protein kinase. J Biol Chem. 2001;276:670–676. doi: 10.1074/jbc.M007074200. [DOI] [PubMed] [Google Scholar]
- 69.Thumkeo D, Keel J, Ishizaki T, Hirose M, Nonomura K, Oshima H, Oshima M, Taketo MM, Narumiya S. Targeted disruption of the mouse rho-associated kinase 2 gene results in intrauterine growth retardation and fetal death. Mol Cell Biol. 2003;23:5043–5055. doi: 10.1128/MCB.23.14.5043-5055.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thumkeo D, Shimizu Y, Sakamoto S, Yamada S, Narumiya S. ROCK-I and ROCK-II cooperatively regulate closure of eyelid and ventral body wall in mouse embryo. Genes Cells. 2005;10:825–834. doi: 10.1111/j.1365-2443.2005.00882.x. [DOI] [PubMed] [Google Scholar]
- 71.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 72.Vahebi S, Kobayashi T, Warren CM, de Tombe PP, Solaro RJ. Functional effects of rho-kinase-dependent phosphorylation of specific sites on cardiac troponin. Circ Res. 2005;96:740–747. doi: 10.1161/01.RES.0000162457.56568.7d. [DOI] [PubMed] [Google Scholar]
- 73.Vemula S, Shi J, Hanneman P, Wei L, Kapur R. ROCK1 functions as a suppressor of inflammatory cell migration by regulating PTEN phosphorylation and stability. Blood. 2009;115:1785–1796. doi: 10.1182/blood-2009-08-237222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Y, Zheng XR, Riddick N, Bryden M, Baur W, Zhang X, Surks HK. ROCK isoform regulation of myosin phosphatase and contractility in vascular smooth muscle cells. Circ Res. 2009;104:531–540. doi: 10.1161/CIRCRESAHA.108.188524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wei L, Roberts W, Wang L, Yamada M, Zhang S, Zhao Z, Rivkees SA, Schwartz RJ, Imanaka-Yoshida K. Rho-kinases play an obligatory role in vertebrate embryonic organogenesis. Development. 2001;128:2953–2962. doi: 10.1242/dev.128.15.2953. [DOI] [PubMed] [Google Scholar]
- 76.Whitlock NA, Harrison B, Mixon T, Yu XQ, Wilson A, Gerhardt B, Eberhart DE, Abuin A, Rice DS. Decreased intraocular pressure in mice following either pharmacological or genetic inhibition of ROCK. J Ocul Pharmacol Ther. 2009;25:187–194. doi: 10.1089/jop.2008.0142. [DOI] [PubMed] [Google Scholar]
- 77.Wibberley A, Chen Z, Hu E, Hieble JP, Westfall TD. Expression and functional role of rho-kinase in rat urinary bladder smooth muscle. Br J Pharmacol. 2003;138:757–766. doi: 10.1038/sj.bjp.0705109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yoneda A, Multhaupt HA, Couchman JR. The rho-kinases I and II regulate different aspects of myosin II activity. J Cell Biol. 2005;170:443–453. doi: 10.1083/jcb.200412043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yoneda A, Ushakov D, Multhaupt HA, Couchman JR. Fibronectin matrix assembly requires distinct contributions from rho-kinases I and -II. Mol Biol Cell. 2007;18:66–75. doi: 10.1091/mbc.E06-08-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang J, Bian HJ, Li XX, Liu XB, Sun JP, Li N, Zhang Y, Ji XP. ERK-MAPK signaling opposes rho-kinase to reduce cardiomyocyte apoptosis in heart ischemic preconditioning. Mol Med. 2010;16:307–315. doi: 10.2119/molmed.2009.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang YM, Bo J, Taffet GE, Chang J, Shi J, Reddy AK, Michael LH, Schneider MD, Entman ML, Schwartz RJ, Wei L. Targeted deletion of ROCK1 protects the heart against pressure overload by inhibiting reactive fibrosis. Faseb J. 2006;20:916–925. doi: 10.1096/fj.05-5129com. [DOI] [PubMed] [Google Scholar]
- 82.Zhou Z, Meng Y, Asrar S, Todorovski Z, Jia Z. A critical role of rho-kinase ROCK2 in the regulation of spine and synaptic function. Neuropharmacology. 2009;56:81–89. doi: 10.1016/j.neuropharm.2008.07.031. [DOI] [PubMed] [Google Scholar]