

Abstract
Urtica dioica agglutinin (UDA) is a small plant monomeric lectin, 8.7 kDa in size, with an N-acetylglucosamine specificity that inhibits viruses from Nidovirales in vitro. In the current study, we first examined the efficacy of UDA on the replication of different SARS-CoV strains in Vero 76 cells. UDA inhibited virus replication in a dose-dependent manner and reduced virus yields of the Urbani strain by 90% at 1.1 ± 0.4 μg/ml in Vero 76 cells. Then, UDA was tested for efficacy in a lethal SARS-CoV-infected BALB/c mouse model. BALB/c mice were infected with two LD50 (575 PFU) of virus for 4 h before the mice were treated intraperitoneally with UDA at 20, 10, 5 or 0 mg/kg/day for 4 days. Treatment with UDA at 5 mg/kg significantly protected the mice against a lethal infection with mouse-adapted SARS-CoV (p < 0.001), but did not significantly reduce virus lung titers. All virus-infected mice receiving UDA treatments were also significantly protected against weight loss (p < 0.001). UDA also effectively reduced lung pathology scores. At day 6 after virus exposure, all groups of mice receiving UDA had much lower lung weights than did the placebo-treated mice. Thus, our data suggest that UDA treatment of SARS infection in mice leads to a substantial therapeutic effect that protects mice against death and weight loss. Furthermore, the mode of action of UDA in vitro was further investigated using live SARS-CoV Urbani strain virus and retroviral particles pseudotyped with SARS-CoV spike (S). UDA specifically inhibited the replication of live SARS-CoV or SARS-CoV pseudotyped virus when added just before, but not after, adsorption. These data suggested that UDA likely inhibits SARS-CoV infection by targeting early stages of the replication cycle, namely, adsorption or penetration. In addition, we demonstrated that UDA neutralizes the virus infectivity, presumably by binding to the SARS-CoV spike (S) glycoprotein. Finally, the target molecule for the inhibition of virus replication was partially characterized. When UDA was exposed to N-acetylglucosamine and then UDA was added to cells just prior to adsorption, UDA did not inhibit the virus infection. These data support the conclusion that UDA might bind to N-acetylglucosamine-like residues present on the glycosylated envelope glycoproteins, thereby preventing virus attachment to cells.
Keywords: BALB/c mouse, SARS-CoV, Urtica dioica agglutinin (UDA)
1. Introduction
Severe acute respiratory syndrome coronavirus (SARS-CoV) is the causative agent of an emerging human infectious disease, severe acute respiratory syndrome (SARS) (Drosten et al., 2003, Ksiazek et al., 2003, Peiris et al., 2003b, Rota et al., 2003). SARS originated in Southern China at the end of 2002, and was characterized by high mortality and morbidity in infected individuals. By July 31, 2003, more than 8000 SARS cases and nearly 800 SARS-related deaths were reported around the world. Studies on the molecular evolution of SARS-CoV suggest that the virus emerged from non-human sources (Guan et al., 2003). The civet cat coronavirus was shown to have a sequence identity of more than 99% to the SARS-CoV with only a limited number of deletions and mutations between both viruses. SARS-CoV was shown to have a deletion of 29 nucleotides relative to the civet cat virus, possibly suggesting that there was direct transmission from civet to humans (Guan et al., 2003). Virus transmission to humans may have occurred when civet cats, probably infected by bats, were traded on Chinese wet markets (Lau et al., 2005, Li et al., 2005b).
Infections caused by SARS-CoV pose a serious threat to the human population and represent a challenge for antiviral drug development and administration (Groneberg et al., 2003, Groneberg et al., 2004) because there are no proven or approved efficacious agents to treat SARS-CoV infection. Ribavirin in combination with corticosteroids was the most frequently administered antiviral agent during the SARS outbreak (Booth et al., 2003, Ho et al., 2003, Peiris et al., 2003a, Tsang et al., 2003, Tsui et al., 2003). However, ribavirin alone at nontoxic concentrations was found to have little in vitro activity against SARS-CoV (Cinatl et al., 2003), although an improved clinical outcome was reported among SARS patients receiving early administration of Kaletra plus ribavirin and corticosteroids (Tsang and Seto, 2004). At present, no clear evidence has been provided to support these clinical observations yet (Barnard and Kumaki, 2009). In several mouse models and in vitro, ribavirin was even found to enhance the SARS-CoV infection (Barnard et al., 2006a, Barnard et al., 2006b, Day et al., 2009). Therefore, development of new anti-SARS-CoV agents is urgently needed should SARS-CoV, or a virus closely related to it, emerge or re-emerge to cause disease.
Numerous types of agents have been tested against SARS-CoV in vitro. For example, antibodies to the SARS-CoV spike protein have been shown to block entry (Sui et al., 2004) and small peptides derived from the heptad repeat (HR) regions of SARS-CoV S protein have been shown to inhibit SARS-CoV infection by the interference of SARS-CoV fusion with target cells (Bosch et al., 2004, Ho et al., 2006). Additionally, the main protease of SARS, which is essential for the replication cycle of SARS-CoV, has been a key target for developing anti-SARS drugs (Anand et al., 2003, Barnard and Kumaki, 2009, Yang et al., 2003). Antisense RNA and RNA interference (RNAi) technologies have shown potential in treating some severe diseases including SARS-CoV infection (Leonard and Schaffer, 2006). Interferons have also been considered as the first line of defense against viral infections. In earlier studies, we evaluated a few compounds approved for therapeutic use in humans and some in vitro inhibitors of SARS-CoV for inhibition in the mouse SARS-CoV replication model. A hybrid interferon, interferon alpha (IFN-α) B/D, and a mismatched double-stranded (ds) RNA interferon inducer, Ampligen (poly I:poly C124), were shown to potently inhibit virus titers in the lungs of infected mice (Barnard et al., 2006a). We also demonstrated that interferon alfacon-1 inhibited SARS-CoV infection in human bronchial epithelial Calu-3 cells (Kumaki et al., 2008).
Stinging nettle lectin Urtica dioica agglutinin (UDA), which is a small (8.7 kDa) plant monomeric lectin with a N-acetylglucosamine specificity (Beintema and Peumans, 1992, Van Damme et al., 1988), has been demonstrated to inhibit Nidovirales in vitro with some selectivity (van der Meer et al., 2007). Keyaerts et al. (2007) showed that UDA was a potent and selective inhibitor of SARS-CoV (Frankfurt 1 strain) with an EC50 = 1.3 ± 0.1 μM and an IC50 of >100 μM, resulting in an SI > 76.9. In addition, another lectin, the mannose specific lectin Hippeastrum hybrid agglutinin (HHA), likely inhibited SARS-CoV attachment to the cells or acted to inhibit the virus at the end of the infectious virus cycle (Keyaerts et al., 2007).
To evaluate the prophylactic potential of antivirals directed against SARS-CoV infection, new lethal animal models for SARS are needed to facilitate antiviral research. We previously adapted and characterized a new strain of SARS-CoV (strain v2163) that was highly lethal in 5–6-week-old BALB/c mice (Day et al., 2009). A number of compounds were tested for efficacy in SARS-CoV-infected BALB/c mice, including UDA. We earlier reported that UDA was partially protective in SARS-CoV-infected mice (Day et al., 2009). UDA at 5 mg/kg/day was shown to significantly delay the mean day of death compared to the average onset of death for animals receiving physiological sterile saline (PSS) (6.2 ± 1.7 days, p < 0.01). However, only 50% of the UDA-treated animals survived. Nevertheless, visual lung scores for both day 3 and day 6 for all lectin treatments in that experiment were lower than those observed in the lungs of placebo-treated mice. In addition, weight reduction changes from day 0 to day 4 (difference between day 0 and day 4 weights) were also less for lectin-treated animals than for PSS-treated mice. These data suggest that UDA treatment of mice did ameliorate the lethality and some of the associated lung pathogenesis in mice infected with the mouse-adapted SARS-CoV. Furthermore, a higher concentration of UDA was tested to determine its efficacy in reducing lethality and ameliorating the pathogenesis in mice infected with a lethal dose of mouse-adapted SARS-CoV. Our data showed that increasing the concentration of UDA treatment to 15 mg/kg/day did not increase the efficacy of UDA compared to UDA used at 5 mg/kg/day. Because of the positive results obtained from in vitro assays and the initial studies showing the efficacy of UDA treatment in significantly reducing mortality in the lethal SARS-CoV mouse model, UDA was further evaluated in the lethal mouse model for SARS-CoV to see if efficacy could be improved. We optimized the dosage regimen to increase the effectiveness of UDA against SARS-CoV in BALB/c mouse model in terms of survival. We also further investigated the mode of action of UDA in vitro.
2. Materials and methods
2.1. Cells
Vero 76 cells, which were obtained from American Type Culture Collection (ATCC, Manassas, VA), were routinely grown in minimal essential medium (MEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Thermo Fisher Scientific Co., Logan, UT). For in vitro antiviral assays, the serum was reduced to 2% in Vero 76 cells and gentamicin was added to the medium at a final concentration of 50 μg/ml. The human primary embryonic kidney cells (293T), which express ACE2 (293T-ACE2), were obtained from the ATCC, and were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated FBS.
2.2. Viruses
SARS-CoV, strain Urbani (200300592), was obtained from the Centers for Disease Control (CDC, Atlanta, GA, USA). The Frankfurt strain was kindly provided by Jindrich Cinatl (Klinikum der J.W. Goethe Universitat, Frankfurt Am Main, Germany). The Toronto-2 strain was supplied by Heinz Feldman (National Microbiology Laboratory, Winnipeg, Manitoba, Canada). The CHUK-W1 strain was received from Paul K.S. Chan (The Chinese University of Hong Kong, China). All strains were propagated and titrated in Vero 76 cells. Personnel entering the facility wore powered air-purifying respirators (3M HEPA Air-Mate; 3M, Saint Paul, MN) and full body protection Tyvek suits.
2.3. Development of mouse-adapted virus
To adapt the human clinical isolate strain Urbani to mice, mice were infected intranasally with Urbani strain using a 1:5 dilution of cell-cultured amplified virus. Three or five days after infection, the lungs were removed and homogenized and then a 1:5 dilution of the clarified lung homogenate was used to re-infect a subsequent group of mice. This infection step continued for 25 times through BALB/c mice lungs. The virus was then plaque-purified 3 times and yielded a virus causing severe lung disease and mortality in infected mice. The virus was verified as SARS-CoV by ELISA, and PCR (Day et al., 2009). In the mouse model, animals infected with three LD50 of virus die between day 4 and day 8, with 90–100% mortality achieved by day 8. The lungs are severely inflamed and exhibit extreme lung histopathology. Weight loss is excessive: >25% of the total initial body weight. The occasional surviving animal may lose 25% or more weight but seems to regain the weight by day 9 or day 10 and lives for at least 21 days or more. Virus titers in the lungs often exceed 107/ml, with the titers peaking at days 3–4. Virus lung titers persist at least until day 7 in mice that survive that long.
2.4. Plasmid
Plasmids encoding spike (S) glycoprotein from SARS-CoV and human ACE2 have been described previously (Simmons et al., 2004, Simmons et al., 2005) and provided by the Simmon's laboratory. Plasmid pNL4-3 Luc-R-E- (pNL-luc) encodes a replication-incompetent variant of the human immunodeficiency virus type 1 (HIV-1) molecular clone NL4-3, in which the nef gene has been replaced by a firefly luciferase (luc) reporter, and the env and vpr genes were inactivated, as previously described (Connor et al., 1995).
2.5. Compounds
The N-acetylglucosamine-specific stinging nettle lectin (U. dioica agglutinin, UDA) was purchased from EY Laboratories Inc. (San Mateo, CA). Stock solutions of UDA were prepared at a concentration of 5 mg/ml in distilled water and stored at −20 °C. UDA was subsequently diluted in MEM for in vitro experiments. For in vivo studies, UDA was prepared in PSS solution. Stock solutions of interferon alfacon 1 (IFN-alfacon 1) (InterMune, Inc., Brisbane, CA) were provided at a concentration of 30 μg/ml and stored at −20 °C. IFN-alfacon 1 was solubilized in MEM for in vitro experiments at a final concentration of 3.0 ng/ml. The interferon inducer, poly IC:LC was obtained from Ribopharm Corporation (Bethesda, MD). N-Acetyl-d-glucosamine was obtained from the Sigma–Aldrich Group (Sigma–Aldrich, St. Louis, MO).
2.6. Cytopathic effect (CPE) inhibition assay
A modified protocol of Barnard et al. (2004a) was used for the in vitro evaluation of antiviral efficacy of the inhibitors of SARS-CoV replication. Vero 76 cells were seeded onto 96-well tissue culture plates. Test compound and virus were added in equal volumes to near-confluent cell monolayers in 96-well tissue culture plates the next day. The multiplicity of infection (MOI) used ranged from 0.01 to 0.025 in order to produce complete virus cytopathic effects (CPE) in 100% of the cells in the virus control wells within 3–4 days. The plates were incubated at 37 °C until the cells in the virus control wells showed complete viral CPE as observed by light microscopy. Each concentration of drug was assayed for virus inhibition of viral CPE in triplicate and for cytotoxicity in duplicate. Six wells per plate were set aside as uninfected, untreated cell controls and six wells per plate received virus only and represented controls for virus replication. IFN alfacon-1 (infergen™) was titrated as the positive control drug for each set of compounds tested.
Morphological changes resulting from cytotoxicity of test compound or virus CPE were graded on a scale of 0–5, with 5 defined as the appearance of complete cytotoxicity or CPE involving the entire monolayer as observed by light microscopy. The values obtained were then converted to percentages of untreated, uninfected controls. The 50% cell cytotoxic concentrations (CC50) and 50% virus inhibitory concentrations (IC50), representing the putative concentration at which 50% of the monolayers would show compound cytotoxicity or virus CPE, respectively, were estimated by regression analysis. A selectivity index (SI) was calculated using the formula SI = CC50/IC50. The activity in the CPE assay was then verified spectrophotometrically by neutral red (NR) uptake assay on the same plate.
2.7. Neutral red (NR) uptake assay for determination of antiviral efficacy and cytotoxicity of test compound
This assay was done for each CPE inhibition test plate described above to verify the inhibitory activity and the cytotoxicity detected by visual observation. In our experience, the usual correlation between visual and neutral red (NR) uptake assays in our hands has been greater than 95%. The neutral red (NR) uptake assay was performed using a modified method of Cavanaugh et al. (1990) as described by Barnard et al. (2004b). Briefly, medium was removed from each well of a plate, 0.011% neutral red (NR) was added to each well of the plate, and the plate was incubated for 2 h at 37 °C in the dark. The neutral red (NR) solution was removed from the wells, the wells were rinsed and any remaining dye was extracted using Sörenson's citrate buffered ethanol. Absorbances at 540 nm/405 nm were read with a microplate reader (Opsys MR™, Dynex Technologies, Chantilly, VA). Absorbance values were expressed as percentages of untreated controls and IC50, CC50, and SI values were calculated as described above.
2.8. Virus yield reduction assay
Virus yield reduction assay was used to confirm the results of the CPE inhibition/NR uptake assays. Infectious virus yield from the CPE inhibition assay was determined on the supernatant from the test well as previously described (Barnard et al., 2004b). After the CPE was scored as described above, each plate was frozen at −80 °C and then thawed. Sample wells at the concentrations of each test compounds were pooled and titered in Vero 76 cells for infectious virus by CPE assay as previously described by Barnard et al. (2004a). A 90% reduction in virus yield (IC90) was then calculated by linear regression analysis. This value represented a one-log10 inhibition in titer when compared to untreated virus controls.
2.9. Pseudotype virus production
Pseudotyped viruses were generated by cotransfecting 293T cells with 30 μg of Env-encoding plasmid and 10 μg of pNL-luc reporter backbone per 10-cm dish in the presence of calcium phosphate. Forty hours after transfection, the supernatant was filtered through a 0.45-μm-pore-size screen, and then purified by ultracentrifugation (28,000 rpm in an SW28 rotor, Beckman, Coulter Inc., Brea, CA) over a 20% sucrose cushion and stored at −80 °C as aliquots (Zhou et al., 2010). The amount of virus was assessed with a p24 antigen capture enzyme-linked immunosorbent assay (p24 ELISA, Aalto Bio Reagents Ltd., Dublin, Ireland).
2.10. Pseudotype virus assay
293T cells (293T-ACE2) were seeded to the 96-well white Nunclon surface tissue culture plates (Nalgene Nunc., Rochester, NY) at 1 × 104 cells/well and grown overnight. For pre-treatment assay, 293T-ACE2 cells were pre-incubated with UDA at different concentrations at 37 °C for 1 h and, infected with SARS-CoV pseudotyped virus. For post-treatment assay, 293T-ACE2 cells were infected with SARS-CoV pseudotyped virus for 1, 4, or 24 h and, treated with UDA at different concentrations. In addition, SARS-CoV pseudotyped virus was pre-incubated with UDA at different concentrations at 37 °C for 1 or 0 h before addition to 293T-ACE2 cells. Then, plates were incubated for 2 days at 37 °C and firefly luciferase reporter expression was determined with reagents from Promega (Madison, WI), and the percentage of infection calculated. For cell viability assay, 293T-ACE2 cells were seeded and treated with UDA. The quantity of the ATP present in metabolically active cells was determined with CellTiter-Glo® Luminescent Cell Viability Assay Systems (Promega, Madison, WI).
2.11. Animals
Specific pathogen-free female 14–18 g BALB/c mice were obtained from Charles River Laboratories (Wilmington, MA) for this study. They were maintained on standard mouse chow and tap water ad libitum. The BALB/c mice were quarantined for 24 h prior to use. The animal studies were carried out in an approved bio-safety level 3+ animal facility.
2.12. Animal experimental design
The BALB/c mice were anesthetized with a 0.1 ml intraperitoneal injection of 20 mg/kg of Ketamine® and SARS-CoV was administered intranasally (i.n.) in a volume of 0.05 ml. Groups of 20 mice were administered UDA intraperitoneally (i.p.) twice a day for 4 days (bid × 5) beginning 4 h after virus exposure. Poly IC:LC was administered intranasally at 24 h before virus exposure and 8 h after exposure to virus and served as a positive control for controlling the virus infection. Thirty mice were treated i.p. with PSS at 24 h prior to virus exposure and 4 h after virus exposure and then twice a day for three more days. Mice in this group represented the placebo controls. Animal deaths were recorded for up to 21 days post virus exposure. Following intranasal administration of SARS-CoV (1 LD90), five mice from each group were sacrificed on day 3 and day 6. SARS-CoV-infected and mock-infected mice were weighed every day and clinical signs of disease were observed. Weight loss was also determined in all infected and uninfected groups of mice. Animals that lost greater than 30% of their initial body weight were humanely euthanized by CO2 asphyxiation, and the day of euthanization was designated as the day of death due to infection.
2.13. Compound toxicity determination
For UDA, a dose range finding experiment was done to determine the maximum tolerated concentration. Three mice were used per treatment group. Toxicity was evaluated in terms of weight change and adverse events. Adverse events for which observations were made included ruffling of fur, lethargy, paralysis, incontinence, repetitive circular motion, and aggression. Mice were also weighed every day from 24 h prior to virus infection to day 11 and day 15 post virus exposure.
2.14. Lung score/lung weight determinations
Samples from each mouse lung lobe were weighed and placed in a petri dish. Lungs were scored based on surface appearance of lungs. Lungs were then assigned a score ranging from 0 to 4, with 0 indicating that the lungs looked normal and 4 denoting that the entire surface area of the lung was inflamed and showed plum colored lung discoloration (Sidwell et al., 1995). Significant differences in lung scores were determined by Kruskal–Wallis test followed by Dunn's pairwise comparison post tests. Analysis of variance (ANOVA) was used to determine significant differences in lung weights. Pairwise comparisons were made by Newman–Keuls post tests.
2.15. Lung virus titer determination
Lung virus titers were analyzed from mice sacrificed on day 3 and day 6 post virus exposure. A lobe from each mouse lung was homogenized in MEM supplemented with 10% FBS and the tissue fragments were allowed to settle. The varying dilutions of the supernatant fluids were assayed in triplicate for infectious virus in Vero 76 cells by CPE assay. The titers (CCID50 values) were calculated using the Reed–Muench method. Significant differences were detected by ANOVA. Pairwise comparisons were made by Newman–Keuls post tests.
2.16. Statistical analysis of death and survival
Mice were weighed in groups prior to treatment and then every day thereafter to determine the average weight change for all animals in each treatment group. Weights were expressed as group averages for each day and evaluated by two-way analysis of variance for significant differences among treatment groups followed by pairwise comparisons using Bonferroni post tests.
Survival analysis was done using the Kaplan–Meier graphical method and a Logrank test. The analysis revealed significant differences among the treatment groups. Therefore, pairwise comparisons of survivor curves (PSS versus any treatment) were analyzed by the Gehan–Breslow–Wilcoxon pairwise comparisons test, and the relative significance was adjusted to a Bonferroni-corrected significance threshold for the number of treatment comparisons done. Mean day of death was calculated and analyzed by the Kruskal–Wallis test, followed by Dunn's post tests for evaluating the significant pairwise comparisons. Live numbers of mice per total mice were totaled and treatment group differences were evaluated by contingency table analysis. Fisher's exact tests were used to make pairwise comparisons to placebo-treated mice.
2.17. Cytokine microarray analysis
At day 3 and day 6 post-infection, lung samples were taken from the mice in each treatment group. Lung samples from each time point were homogenized in a volume adjusted for lung weights, immediately frozen, and stored prior to analysis. Cytokine levels in the supernatant fluids, adjusted for total protein, were measured on the same day using the Quansys Q-Plex™ Mouse Cytokine Array (Quansys Biosciences, Logan, UT). This platform was chosen for its ability to simultaneously measure 16 different mouse cytokines in a small sample volume (30 μl). Cytokines contained interleukin-1 alpha (IL-1α), interleukin-1 beta (IL-1β), interleukin-2 (IL-2), interleukin-3 (IL-3), interleukin-4 (IL-4), interleukin-5 (IL-5), interleukin-6 (IL-6), interleukin-10 (IL-10), interleukin-12 (IL-12), interleukin-17 (IL-17), monocyte chemotactic protein-1 (MCP-1), interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), macrophage inflammatory protein-1-alpha (MIP-1α), granulocyte-macrophage colony stimulating factor (GM-CSF), and RANTES. Total protein in each sample was determined using a bicinchoninic acid (BCA™) assay. The total protein values were then used to adjust the measured cytokine levels in proportion to the amount of protein present in each sample. Significant differences were detected by ANOVA and Newman–Keuls post tests.
3. Results
3.1. Effects of UDA on SARS-CoV infection in vitro
We first examined the effects of different concentrations of UDA on the replication of SARS-CoV Urbani strain in Vero 76 cells at 37 °C. UDA potently inhibited virus replication (Table 1 ). UDA was shown to have an IC50 of 0.9 ± 0.3 μg/ml determined by visual assay (data not shown) and, to have an IC50 of 2.6 ± 3.7 μg/ml determined by neutral red (NR) uptake assay. We then examined whether UDA affected the replication of other SARS-CoV strains. UDA inhibited the other SARS-CoV isolates with SI values ranging from 5.5 to 54.2 as determined by neutral red uptake assay (Table 1).
Table 1.
UDA inhibition of SARS-CoV replication in Vero 76 cells.
| Virus strains | Neutral red (NR) assay |
Virus yield reduction assay |
|||
|---|---|---|---|---|---|
| IC50a (μg/ml) | CC50b (μg/ml) | SIc | IC90d (μg/ml) | Virus yielde | |
| Urbani | 2.6 ± 3.7 | 12.5 ± 4.4 | 10.2 ± 5.6 | 1.1 ± 0.4 | 4.8 ± 0.4 |
| Mouse-adapted virus | 0.6 ± 0.6 | 9.9 ± 0.2 | 42.8 ± 47.5 | 0.7 ± 0.7 | 5.3 ± 0.4 |
| Frankfurt v1940 | 2.0 ± 1.1 | 9.2 ± 1.0 | 5.5 ± 2.0 | 1.7 ± 1.1 | 5.1 ± 0.1 |
| Hong Kong v2157 | 1.7 ± 0.2 | 14.3 ± 0.6 | 8.6 ± 1.1 | 1.3 ± 0.2 | 5.4 ± 0.3 |
| Toronto-2 v2147 | 0.9 ± 1.2 | 9.5 ± 0.9 | 54.2 ± 52.5 | 0.9 ± 0.9 | 5.2 ± 0.4 |
50% virus inhibitory concentration.
50% cell cytotoxic concentration of drug.
Selective index: SI = CC50/IC50.
90% virus inhibitory concentration.
Average virus titers (log10 CCID50 per 0.18 ml).
The activity of UDA was confirmed in a virus yield reduction assay. UDA reduced virus yields of the Urbani strain by 90% at 1.1 ± 0.4 μg/ml in Vero 76 cells, which correlated well with the potent activity detected by neutral red (NR) uptake assay. UDA also blocked the replication of other SARS-CoV strains, reducing virus yields of these viruses by 90%, with IC90 values ranging from 0.7 ± 0.7 to 1.7 ± 1.1 μg/ml (Table 1).
3.2. Effects of UDA on weight change of BALB/c mice
Given the potent activity detected in vitro, efficacy was also evaluated in a lethal SARS-CoV mouse model. Most mice randomly assigned to the toxicity control groups gained weight at rates nearly equal to mice receiving physiological sterile saline (PSS) (Fig. 1 ). The mice treated with poly IC:LC were the only group of mice to lose noticeable amounts of weight, this occurred at day 2. However, they still gained back all the lost weight after the nadir of weight loss at day 2 and the mice in this group managed to have gained enough weight by the end of the experiment that their gain was statistically equal to the other groups of mice. No other adverse events were observed for any of the toxicity control mice used in the experiment.
Fig. 1.
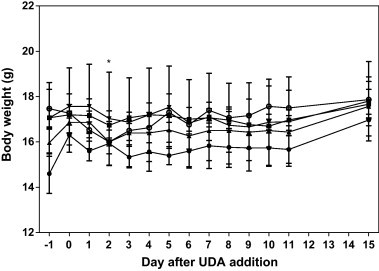
Effects of UDA and poly IC:LC on weight change of un-infected BALB/c mice. * p < 0.05 versus PSS. The mock-infected BALB/c mice were treated with PSS (●), UDA at 20 mg/kg/day (■), 10 mg/kg/day (▴), 5 mg/kg/day (▾), or poly IC:LC at 1.0 mg/kg/day (○).
3.3. Effects of UDA on weight change and death of BALB/c mice infected with a lethal dose of mouse-adapted SARS-CoV
Infected mice receiving any concentration of UDA and poly IC:LC were protected against the serious weight loss suffered by infected mice receiving the placebo, PSS (Table 2 ). This protection against weight loss was significant for all UDA treatment groups by day 12, at which time at least 50% of the mice survived in all of the treatment groups (Fig. 2 ). All mice receiving UDA or poly IC:LC went on to gain weight by the end of the experiment that was equal to or exceeded the original body weight recorded at the initiation of the study (Table 2). For this experiment, 90% of the mice receiving the calculated LD90 dose died as expected (Fig. 2). The most significantly efficacious dose of UDA used for treating infected mice was UDA at 5 mg/kg/day. At this dose, mice were significantly protected against weight loss by day 6 (Table 2, p < 0.05) and thereafter, all mice were protected against death due to virus infection (p < 0.001, Fig. 2). Further analysis of death revealed that this same dose of UDA significantly enhanced the ratio of live mice to total mice in the treatment group (p < 0.01) and obviously increased the mean day of death (p < 0.05, Table 3 ). Interestingly, the 10 mg/kg/day dose of UDA also afforded significant protection against death when evaluating all parameters of death, and also protected against weight loss (Fig. 2 and Table 2, Table 3). In contrast, the higher dose of 20 mg/kg/day was not nearly as protective against death, was slightly toxic in uninfected mice at day 2 and day 3 after treatment (Fig. 1), and did not ameliorate weight loss in infected, treated mice during the critical days of infection (days 3–9) (Table 2 and Fig. 2). However, this dose still prolonged the survival of mice as evidenced by the fact that the median day of death (the calculated day on which most mice died) was 16 days. In addition, even when mice treated with UDA at 20 mg/kg/day, they were almost 3-times less likely to die as rapidly as mice treated with placebo (see hazard ratio, Table 3). This suggests that the 10 mg/kg/day dose of UDA might be the maximum tolerated efficacious dose.
Table 2.
Effects of UDA and poly IC:LC on weight averages of female BALB/c mice infected with a lethal dose of mouse-adapted SARS-CoV.
| Day post virus exposure | Average group weight (g) |
||||
|---|---|---|---|---|---|
| PSS | UDA (20 mg/kg/day) | UDA (10 mg/kg/day) | UDA (5.0 mg/kg/day) | Poly IC:LC (1.0 mg/kg/day) | |
| −1 | 17.0 ± 0.8 | 17.3 ± 0.9 | 16.8 ± 0.6 | 17.0 ± 0.7 | 16.8 ± 0.8 |
| 0 | 17.0 ± 0.7 | 17.4 ± 1.1 | 16.8 ± 0.7 | 16.7 ± 0.7 | 16.1 ± 0.6 |
| 1 | 17.2 ± 0.8 | 17.1 ± 0.8 | 16.7 ± 0.7 | 17.1 ± 0.7 | 16.0 ± 0.7 |
| 2 | 16.3 ± 0.8 | 16.1 ± 0.7 | 16.0 ± 0.7 | 16.8 ± 0.7 | 16.5 ± 0.7 |
| 3 | 14.7 ± 0.7 | 15.4 ± 0.8 | 14.9 ± 0.6 | 15.2 ± 0.8 | 17.1 ± 0.6*** |
| 4 | 13.8 ± 0.7 | 14.7 ± 0.7 | 14.0 ± 0.8 | 14.5 ± 0.8 | 17.2 ± 0.5*** |
| 5 | 13.1 ± 0.6 | 14.3 ± 1.1 | 13.8 ± 1.1 | 14.5 ± 1.4 | 17.5 ± 0.5*** |
| 6 | 12.4 ± 0.5 | 13.7 ± 0.7 | 13.6 ± 1.5 | 14.2 ± 1.6* | 17.5 ± 0.6*** |
| 7 | 12.2 ± 0.5 | 13.4 ± 0.9 | 13.7 ± 1.5 | 14.7 ± 2.0*** | 17.5 ± 0.5*** |
| 8 | 11.9 ± 0.5 | 13.2 ± 0.9 | 14.3 ± 1.6** | 15.2 ± 2.4*** | 17.7 ± 0.6*** |
| 9 | 11.5 ± 0.4 | 13.0 ± 1.0 | 14.9 ± 1.5*** | 15.6 ± 2.4*** | 17.8 ± 0.6*** |
| 10 | 11.8 ± 0.3 | 13.8 ± 1.6 | 15.5 ± 1.4** | 15.7 ± 2.5*** | 17.8 ± 0.7*** |
| 11 | 11.9 ± 0.6 | 14.7 ± 1.2 | 16.5 ± 1.4*** | 15.8 ± 2.5*** | 17.8 ± 0.7*** |
| 12 | 12.1 ± 0.6 | 15.6 ± 1.6* | 16.3 ± 1.3*** | 16.0 ± 2.5*** | 18.1 ± 0.8*** |
| 13 | 12.2 ± 0.3 | 15.8 ± 1.5** | 16.5 ± 1.1*** | 16.0 ± 2.6** | 18.2 ± 0.7*** |
| 14 | 12.1 ± 0.7 | 16.3 ± 1.4*** | 16.6 ± 1.0*** | 16.1 ± 2.6*** | 18.3 ± 0.8*** |
| 15 | 12.1 ± 0.7 | 16.3 ± 1.4*** | 16.6 ± 1.0*** | 16.1 ± 2.6*** | 18.3 ± 0.8*** |
| 21 | 13.2 ± 1.8 | 17.0 ± 1.1** | 17.9 ± 0.7*** | 17.5 ± 2.4*** | 19.4 ± 1.0*** |
p < 0.05 versus PSS.
p < 0.01 versus PSS.
p < 0.001 versus PSS.
Fig. 2.
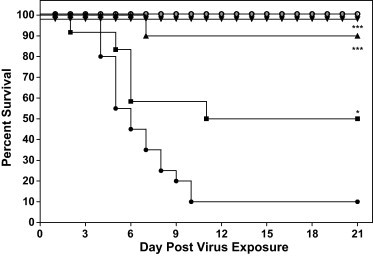
Effects of UDA on the survival of BALB/c mice infected with a lethal dose of mouse-adapted SARS-CoV. * p < 0.05 versus PSS, *** p < 0.0001 versus PSS. SARS-CoV-infected BALB/c mice were treated with PSS (●), UDA at 20 mg/kg/day (■), 10 mg/kg/day (▴), 5 mg/kg/day (▾), or poly IC:LC at 1.0 mg/kg/day (○).
Table 3.
Effects of UDA on various survival parameters measured for BALB/c mice infected with a lethal dose of SARS-CoV.
| Treatment | Live/total | Mean day of death | Hazard ratioa |
|---|---|---|---|
| PSS | 3/20 | 6.0 | – |
| UDA (20 mg/kg/day) | 6/10 | 16.0 | 2.79 (1.16–6.72) |
| UDA (10 mg/kg/day) | 9/10* | Undefined | 7.74 (2.95–20.31) |
| UDA (5 mg/kg/day) | 10/10** | Undefined | 32.32 (11.39–91.76) |
| Poly IC:LC (1 mg/kg) | 10/10** | Undefined | 32.32 (11.39–91.76) |
Hazard ratios are relative to the PSS control.
p < 0.05 versus PSS control.
p < 0.01 versus PSS control.
3.4. Effects of UDA on lung scores, lung weights, and virus lung titers of BALB/c mice infected with a lethal dose of mouse-adapted SARS-CoV at day 3 and day 6 post virus exposure
The pathology of the lungs of SARS-CoV-infected mice worsened in placebo-treated mice due to a combination of viral damage to alveolar cells causing necrosis of the capillary walls leading to lung hemorrhage, as well as to late-occurring vascular phenomena resulting from immune response to infection (pathology not shown). The gross pathology of the lungs was also severe in placebo-treated mice on both days it was evaluated (Table 4, Table 5 ). At day 3, the 10 and 20 mg/kg/day doses of UDA afforded significant protection against the surface hemorrhaging and inflammation noted for lungs from infected, untreated mice (p < 0.05, Table 4). At day 6, protection provided by the 10 mg/kg/day dose was not as significant, but the protection was still significantly greater than that observed in mice treated with 20 mg/kg/day UDA or those mice receiving PSS (p < 0.05, Table 5). The lungs of mice receiving the 5 mg/kg/day dose of UDA displayed significantly little surface pathology at both day 3 (Table 4, p < 0.05) and day 6 (Table 5, p < 0.05). This dose was, again, the most effective dose of UDA for ameliorating the surface hemorrhage and inflammation due to the virus infection.
Table 4.
Effects of UDA treatment on various measured lung parameters at day 3 post virus exposure for BALB/c mice infected with a lethal dose of SARS-CoV.
| Treatment | Day 3 lung scores (g) ± SD | Day 3 lung weights (g) ± SD | Day 3 virus lung titers/g tissue ± SD |
|---|---|---|---|
| PSS | 1.70 ± 0.57 | 0.18 ± 0.03 | 6.31 ± 0.13 |
| UDA (20 mg/kg/day) | 0.50 ± 0.35* | 0.18 ± 0.03 | 6.75 ± 0.35* |
| UDA (10 mg/kg/day) | 0.30 ± 0.27** | 0.17 ± 0.01 | 6.75 ± 0.00* |
| UDA (5 mg/kg/day) | 0.20 ± 0.27 | 0.18 ± 0.01 | 6.50 ± 0.20 |
| Poly IC:LC (1 mg/kg) | 0.60 ± 0.22 | 0.17 ± 0.01 | 4.69 ± 0.13*** |
p < 0.05 versus PSS control.
p < 0.01 versus PSS control.
p < 0.001 versus PSS control.
Table 5.
Effects of UDA treatment on various measured lung parameters at day 6 post virus exposure for BALB/c mice infected with a lethal dose of SARS-CoV.
| Treatment | Day 6 lung scores (g) ± SD | Day 6 lung weights (g) ± SD | Day 6 virus lung titers/g tissue ± SD |
|---|---|---|---|
| PSS | 2.50 ± 0.71 | 0.36 ± 0.02 | 5.50 ± 0.00 |
| UDA (20 mg/kg/day) | 1.60 ± 0.55 | 0.20 ± 0.04 | 5.56 ± 0.24 |
| UDA (10 mg/kg/day) | 1.20 ± 0.67* | 0.20 ± 0.02 | 5.19 ± 0.47 |
| UDA (5 mg/kg/day) | 0.40 ± 0.22* | 0.20 ± 0.03 | 5.25 ± 0.41 |
| Poly IC:LC (1 mg/kg) | 0.20 ± 0.27* | 0.18 ± 0.03** | 2.94 ± 0.55*** |
p < 0.05 versus PSS control.
p < 0.01 versus PSS control.
p < 0.001 versus PSS control.
When evaluating other pathological effects such as edema for each treatment group, which was measured indirectly by lung weights, the lungs of mice treated with 10 mg/kg/day of UDA at day 3 after virus exposure weighed slightly less than did the lungs of mice in the other groups (Table 4). At day 6 after virus exposure, all groups of mice receiving UDA or poly IC:LC had much lower lung weights than did the placebo-treated mice (Table 5), although the lower lung weight was only significant for mice receiving poly IC:LC (p < 0.001). This reduction in lung weights at day 6 (probably due to reduction in edema and lung consolidation) was significant for mice receiving poly IC:LC (p < 0.001).
The pattern of dosage efficacy described above was not detected when evaluating virus lung titers. At day 3 after virus exposure, mice treated with UDA did not have lower virus lung titers compared to placebo treated mice except mice receiving UDA at 10 mg/kg/day (Table 4). This pattern held true at day 6 post virus exposure, where the lung virus titers in the group of mice receiving UDA at 20 and 5 mg/kg/day were nearly equal to those recorded for placebo-treated mice, but mice receiving UDA at 10 mg/kg/day had what appeared to be virus lung titers lower than virus titers from placebo-treated mice. As expected, the lung virus titers for mice receiving poly IC:LC were significantly reduced at day 3 and day 6 post virus exposure (Table 4, Table 5, p < 0.001).
3.5. Effects of treatment on cytokines and chemokines
In a previous experiment, we demonstrated that the mouse-adapted strain of SARS-CoV elicited expression of significant amount of cytokines IL-1α, IL-6, and chemokines MCP-1, MIP-1α and RANTES in the mouse lungs on day 3 after infection as measured with the multiplex ELISA assay (Day et al., 2009). In the current study, levels of IL-2, IL-3, IL-4, IL-5, IL-10, IL-12, IL-17, IFN-γ, TNF-α, and GM-CSF were below the minimum detection level for all test groups at day 3 post virus exposure. High levels of IL-1β were detected in the lungs of mice infected with mouse-adapted strain (Fig. 3A). Levels of IL-1β, IL-6, MCP-1 and MIP-1α dropped when BALB/c mice were treated with UDA (5 mg/kg/day) or poly IC:LC (1 mg/kg/day) (Fig. 3A). Although there was a recognizable decrease in the cytokines, this decrease was not statistically significant. In contrast, at day 3 the lungs of infected mice treated with UDA (5 mg/kg/day) or poly IC:LC (1 mg/kg/day) had much higher RANTES levels compared to the day 3 values for lungs from infected, untreated mice (Fig. 3B).
Fig. 3.
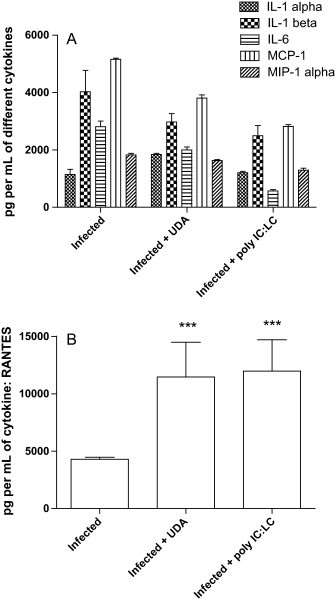
(A) Average levels of the cytokines detected in BALB/c mice infected with a lethal dose of mouse-adapted SARS-CoV on day 3. (B) Average levels of the cytokine RANTES detected in BALB/c mice infected with a lethal dose of mouse-adapted SARS-CoV on day 3. *** p < 0.001, each compound versus PSS.
3.6. Mode of action of UDA in vitro
To investigate how UDA might inhibit SARS-CoV infection, we further studied its mode of action in vitro. UDA was added to cells just before (pre-treatment) the addition of virus to cells or after adsorption of the virus (post-treatment) to cells. For UDA pre-treatment, Vero 76 cells were treated with UDA for 1 h, and then infected with virus. For UDA post-treatment, Vero 76 cells were infected with SARS-CoV for 1 h, and then treated with UDA. Marked inhibitory effects on SARS-CoV replication were observed when the treatment occurred just before virus exposure to cells, but the inhibition was three times less when the treatment occurred after adsorption (Table 6 ). This effect was even more pronounced when comparing IC90 values in virus yield reduction assay, pre-treating cells with UDA resulted in an IC50 of 0.05 μg/ml. We also tested whether UDA would affect SARS-CoV replication 24 h after infection. Treatment with UDA 24 h after infection did not inhibit SARS-CoV replication. In order to examine the possibility that the inhibitory effect was due to direct binding of UDA to SARS-CoV, UDA was added to the virus for 1 h before inoculation. Marked inhibition of SARS-CoV replication was observed comparable to UDA pre-treatment of cells, suggesting UDA might react directly with SARS-CoV (Table 6).
Table 6.
Antiviral activity of UDA against SARS-CoV replication in Vero 76 cells when added to cells at various times before and after virus exposure to cells.
| Treatment | Neutral red (NR) uptake assay |
Virus yield reduction assay |
|||
|---|---|---|---|---|---|
| IC50a (μg/ml) | CC50b (μg/ml) | SIc | IC90d (μg/ml) | Virus yield | |
| Pre-treatment (1 h) | 0.51 ± 0.09 | 5.30 ± 0.40 | 10.63 ± 2.12 | 0.05 ± 0 | 4.6 ± 0.4 |
| Post-treatment (1 h) | 1.80 ± 0.35 | 6.70 ± 0.66 | 3.87 ± 1.02 | 2.4 ± 0.7 | 4.2 ± 0.5 |
| Post-treatment (4 h) | 1.43 ± 0.15 | >10.0 ± 0 | 7.03 ± 0.7 | ND | ND |
| Post-treatment (24 h) | >10.0 ± 0 | >10.0 ± 0 | 0 ± 0 | ND | ND |
| Virus-treatment (1 h) | 0.53 ± 0.24 | 5.80 ± 0.36 | 12.20 ± 4.26 | 0.4 ± 0 | 4.7 ± 0.2 |
50% virus inhibitory concentration.
50% cell cytotoxic concentration of drug.
Selective index: SI = CC50/IC50.
90% virus inhibitory concentration.
To further investigate whether UDA plays a role in the early steps of viral infection, we next examined the effect of UDA on SARS-CoV pseudotyped virus infection in 293T-ACE2 target cells. Confirming what was described above, pre-treatment with UDA, but not post-treatment, in 293T-ACE2 cells resulted in a significant dose-dependent reduction of infectivity (Fig. 4 ). In addition, pre-incubation of SARS-CoV pseudotyped virus with UDA at 37 °C for 1 or 0 h in 293T-ACE2 cells also resulted in significant dose-dependent reduction of infectivity (Fig. 4); UDA inhibition was almost 50-fold more potent compared to treating with UDA 1 h after virus adsorption. Thus, the inhibition of live SARS-CoV and SARS-CoV pseudotyped virus infections indicated that UDA acted at the early steps of viral infection.
Fig. 4.
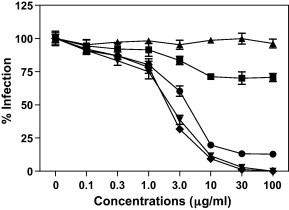
Effect of infectivity of retroviral particles pseudotyped virus with SARS-CoV spike (S) protein by UDA. 293T-ACE2 cells were infected with pseudotyped virus and then, treated with UDA 1 (●), 4 (■) or 24 h (▴) after infection; 293T-ACE2 cells were treated with UDA for 1 h and then, infected with pseudotyped virus (▾); SARS-CoV was exposed to UDA for 1 h (♦).
3.7. Effect of addition of UDA on a single cycle of virus replication
To better determine the target step of UDA in SARS-CoV life cycle, the time-of-addition assay was performed according to the method previously described (Keyaerts et al., 2005). Vero 76 cells were infected with mouse-adapted SARS-CoV at a multiplicity of infection of 1.0 to ensure almost all the cells were exposed to virus in order to study a single cycle virus replication. After 1 h of adsorption, the cells were washed with the medium and UDA at 3.2 μg/ml, which is almost over 5 times the IC50, was added at various time points after infection. Simultaneously, UDA was found to inhibit the early phase (1, 2, 4, 6, 8 or 12 h after infection), but not the late phase (20 or 24 h after infection), of SARS-CoV life cycle as previously shown above (Table 7 ).
Table 7.
Effects of UDA treatment on infection with mouse-adapted SARS-CoV in Vero 76 cells at various time points post virus exposure.
| Treatmenta | 1 h | 2 h | 4 h | 6 h | 8 h | 12 h | 20 h | 24 h |
|---|---|---|---|---|---|---|---|---|
| UDA (3.2 μg/ml) | – | – | – | – | – | – | + | + |
| UDA (1.0 μg/ml) | + | + | + | + | + | + | + | + |
| UDA (0.32 μg/ml) | + | + | + | + | + | + | + | + |
+: CPE was observed.
UDA was cytotoxic to Vero 76 cells at 10 μg/ml.
3.8. Pre-exposure of Vero 76 cells to UDA prior to infection
Since UDA binds to N-acetylglucosamine, we further examined whether UDA would affect the binding site to cell-surface sugar chains in Vero 76 cells. Vero 76 cells were incubated with UDA for 1 h and UDA was removed. Then, the cells were washed with the medium, aspirated to near dryness, and, infected with SARS-CoV Urbani strain. The incubation was continued for 3 days and the antiviral activity was determined as described above. In this experiment, treatment of the cells with UDA before infection did not inhibit SARS-CoV replication of Vero 76 cells, suggesting that UDA did not affect the binding site to cell-surface sugar chains in Vero 76 cells (data not shown). Similar results were obtained when 293T-ACE2 cells were treated with UDA for 1 h, washed with the medium and then, infected with SARS-CoV pseudotyped virus (data not shown).
3.9. Pre-exposure of cell-free SARS-CoV to UDA prior to infection
In this experiment, a concentrated cell-free SARS-CoV Urbani isolate was exposed to UDA at various concentrations for 1 h at 37 °C. Then, the UDA-exposed virus suspensions were titrated in Vero 76 cells in 96-well plate by endpoint dilution as described previously (Barnard et al., 2004a). All the assays were done in triplicate. At day 4 or day 5 post infection, the viral titers in Vero 76 cells were quantified as described above. Pre-exposure of SARS-CoV to UDA was shown to block the viral replication in a dose-dependent manner (Fig. 5A). In doing these titrations, UDA was diluted out to well below its EC50 concentration (see Table 1), indicating that the antiviral effect was the result of incubation of UDA with virus and not from carryover of the compound during the titration. These data indicated that UDA neutralizes the virus infectivity, presumably by binding to the SARS-CoV spike (S) glycoprotein.
Fig. 5.
(A) Effect of live SARS-CoV infection by UDA (virus titers: log10 CCID50 per 0.18 ml). SARS-CoV was exposed to UDA for 1 h and the UDA-exposed virus suspension was titrated in Vero 76 cells. (B) Effect of live SARS-CoV infection by N-acetylglucosamine (virus titers: log10 CCID50 per 0.18 ml). N-Acetylglucosamine was exposed to UDA at 37 °C for 1 h and the N-acetylglucosamine-exposed UDA was incubated with a concentrated cell-free SARS-CoV Urbani isolate at 37 °C for another 1 h.
3.10. UDA might bind directly to the SARS-CoV spike glycoprotein
To determine if the activity of UDA against SARS-CoV was due to specific interaction with the spike glycoprotein, additional experiments utilizing N-acetylglucosamine were evaluated for their antiviral activity in vitro. N-Acetylglucosamine was incubated with UDA at 37 °C for 1 h and the N-acetylglucosamine-exposed UDA was incubated with a concentrated cell-free SARS-CoV Urbani isolate at 37 °C for 1 h. Then, the virus was titrated in Vero 76 cells. These studies showed that the N-acetylglucosamine-exposed UDA did not prevent the SARS-CoV infectivity, whereas UDA, not treated with N-acetylglucosamine, reduced viral titers by about 100-fold (Fig. 5B). These data indicate that UDA neutralized virus infectivity by binding to a moiety such as N-acetylglucosamine presumably found on the virus protein responsible for adsorption.
4. Discussion
We have demonstrated the potent inhibition of SARS-CoV replication in cell culture by neutral red uptake assay and virus yield reduction assay. We have also shown that UDA inhibited retroviral particles pseudotyped with SARS-CoV spike (S) infection in vitro, suggesting that a function of SARS-CoV spike (S) protein might be targeted by UDA. This in vitro inhibitory activity also translated into inhibition of virus-induced mortality in a lethal SARS-CoV challenge model of mice, but did not reduce the virus lung titers. One possibility to explain the efficacy of UDA in spite of not significantly reducing virus titers may be attributed to the putative mechanism of action supported by the data previously described. It is likely that UDA inhibits attachment and thus limits spread of virus from infected cells to uninfected cells by binding to released virus to prevent attachment to uninfected cells. Since in the current study entire lungs were harvested, homogenized, and titrated for virus, there would be no way of differentiating intracellular infectious virus from virus released from infected cells. If bronchial fluids from lung lavages had been assayed for virus, there may have well been a decrease in titers of released extracellular virus in response to UDA treatment, especially at day 3 and day 4 when maximal virus titers from lung homogenates are achieved.
On day 3 post challenge, multiple cytokines and chemokines such as IL-1α, IL-1β, IL-6, MCP-1, MIP-1α and RANTES were significantly elevated in the lungs of BALB/c mice infected with mouse-adapted SARS-CoV. Similar findings have also been reported (Chen and Subbarao, 2007). Yen et al. (2006) showed that human monocytic THP-1 cell line was susceptible to SARS-CoV. In that study, SARS-CoV induced THP-1 cells to express MCP-1, IL-8, MIP-1α, IP-10, MIP-1β, and RANTES. Li et al. (2005a) reported that RANTES inhibited SARS-CoV replication in THP-1 cells when added into the culture at the same time with the SARS-CoV or before, but not after, infection. In this report, significantly high levels of mouse RANTES in the lungs of BALB/c mice were detected when SARS-CoV-infected mice were treated with UDA or poly IC:LC, indicating that elevated RANTES inhibits the disease progression. In addition, IL-1β, IL-6 and MCP-1 were more strongly expressed in SARS-CoV-infected mice. IL-6 and MCP-1 have also been observed in human SARS patients (Chen and Subbarao, 2007). Jiang et al. (2005) demonstrated that superinfection after immune injury and acute respiratory distress as well as lung failure are the main causes of death. The rapid increase of IL-6, IL-8 and MCP-1 is a sign of superinfection, indicating a high risk of death. Conversely, we observed that detectable levels of IL-1β, IL-6, MCP-1 and MIP-1α dropped when BALB/c mice were treated with UDA or poly IC:LC. Therefore, cytokine/chemokine are likely good early indicators of disease outcome in mice.
The angiotensin-converting enzyme 2 (ACE2) has been identified as a functional receptor for SARS-CoV (Li et al., 2003). CD209L (also called L-SIGN, DC-SIGNR, and DC-SIGN2), a different human cellular glycoprotein, has been demonstrated as an alternative receptor for SARS-CoV (Jeffers et al., 2004). Plant lectins are sugar-binding proteins with high affinity for their specific sugar residues and play important roles in biological recognition phenomena involving cells and proteins. Some viruses use lectins to attach themselves to the host cells to initiate infection (Jeffers et al., 2004). SARS-CoV antigens have been detected in alveolar epithelial cells (primarily type II pneumocytes), bronchial epithelial cells, and alveolar macrophages of the respiratory tract (Ding et al., 2004, Ye et al., 2007). All of these cell types have ACE2 (Li et al., 2003). CD209L is expressed in human lung on type II alveolar cells and can mediate infection by SARS-CoV (Jeffers et al., 2004). SARS-CoV spike (S) glycoprotein attaches to use both ACE2 and CD209L in the early steps of viral infection. Because previous studies have shown that SARS-CoV spike (S) glycoprotein binds to the cell surface lectins DC-SIGN and DC-SIGNR (Marzi et al., 2004, Yang et al., 2004), it needs to be further determined whether UDA blocks binding of SARS-CoV to DC-SIGN and DC-SIGNR. The in vitro data suggest that the mode of inhibition of the lectin UDA might be binding to the virion to prevent adsorption, perhaps by UDA binding to the epitope found on the SARS-CoV spike protein that binds to DC-SIGN and DC-SIGNR.
Many enveloped viruses have glycoproteins on their surface. These glycoproteins are mediators of the receptor binding and membrane fusion of the virion with the host cells. Therefore, they are attractive therapeutic targets for novel antiviral therapies. When van der Meer et al. (2007) used an immunoperoxidase staining assay and a luciferase-based assay to evaluate the antiviral activity of UDA, they showed that UDA inhibited Nidovirales in vitro. These assays are based on the number of infected cells and expression of viral proteins. UDA binds to the viral glycoproteins to inhibit virus entry into the target cells (Balzarini, 2007). O’Keefe et al. (2010) recently reported that a potent antiviral lectin griffithsin (GRST), a 12.7 kDa protein originally isolated from red alga Griffithsia sp., inhibited SARS-CoV infection both in vitro and in vivo. GRST is known to bind not only to N-acetylglucosamine, but also to mannose in a multivalent manner via its three independent carbohydrate-binding domains (Ziolkowska et al., 2006). GRST specifically bound to SARS-CoV spike glycoprotein to inhibit viral entry likely because SARS-CoV spike protein is heavily glycosylated and contains 23 putative N-glycosylation sites (Krokhin et al., 2003, Rota et al., 2003). We also confirmed that the inhibitory effects of SARS-CoV infection were observed when UDA was added to the live SARS-CoV Urbani strain or SARS-CoV pseudotyped virus for 1 h before inoculation, suggesting UDA might react directly with SARS-CoV. Based on these observations, we further examined whether UDA would affect the binding site to cell-surface sugar chains in Vero 76 or 293T-ACE2 cells. UDA had been removed after 1 h treatment and both cells had been washed with the medium before infection. No reduction in viral CPE was observed. Our data suggest that UDA did not affect the binding site to cell-surface sugar chains at all. Furthermore, we demonstrated that UDA neutralized the virus infectivity, presumably by binding to the SARS-CoV spike (S) glycoprotein.
In support of this, Ritchie et al. (2010) identified the N-glycans associated with SARS-CoV spike protein. They demonstrated that the majority (30% of total glycans), N-glycans were high-mannose hybrid (28%) and bi-, tri-, and tetra-antennary complex glycans (42%). Many of the terminal residues of the antennary couples appeared to be N-acetylglucosamine, the glycan to which UDA preferentially binds. However, the authors did not map the positions of these residues within the S protein or determine whether this clustering of N-glycans complexes in S1 attachment portion of the S protein or the S2 fusion portion of the protein. Nevertheless, the data support our conclusion that UDA likely binds to the S protein via N-acetylglucosamine residues of the SARS-CoV S protein to inhibit attachment to cells, thereby preventing virus infection of cells.
Although the in vitro data suggest that one mode of inhibition might be the binding of UDA to the virion to prevent adsorption, another possibility that needs to be further investigated is whether UDA might protect mice from virus challenge by regulating immune responses in addition to or in lieu of inhibition of SARS-CoV directly. For example, it has been found that UDA might function as a superantigenic lectin in activating T-cell responses and cytokine production (Galelli and Truffa-Bachi, 1993). The mechanism of immune modulation is also supported by the significant increase in RANTES levels in infected mice receiving UDA treatment compared to RANTES levels detected in un-treated, infected mice. Given the demonstrated in vitro and in vivo inhibition of SARS-CoV replication, UDA or an analog might qualify as a potential therapy for treating SARS-CoV infections.
Acknowledgments
We thank Jason R. Madsen, Michael A. Morrey, Meeyeon Ahn for kindly providing technical assistance; Craig W. Day and Gregory J. Podgorski for professional consultation; and Heather L. Greenstone for scientific discussion. We are grateful to other colleagues for their support. This work was supported by contracts NO1-A1-30048, NO1-AI-15435 and HHSN272201000039/HHSN27200002/A14 from the Virology Branch, National Institute of Allergic and Infectious Diseases (NIAID), National Institutes of Health (NIH) and a grant R01AI074986 from the NIAID, NIH (to G.S.). Joseph K.-K. Li was also supported in part by AES Project 538.
References
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Balzarini J. Carbohydrate-binding agents: a potential future cornerstone for the chemotherapy of enveloped viruses? Antivir. Chem. Chemother. 2007;18(1):1–11. doi: 10.1177/095632020701800101. [DOI] [PubMed] [Google Scholar]
- Barnard D.L., Day C.W., Bailey K., Heiner M., Montgomery R., Lauridsen L., Chan P.K., Sidwell R.W. Evaluation of immunomodulators, interferons and known in vitro SARS-coV inhibitors for inhibition of SARS-coV replication in BALB/c mice. Antivir. Chem. Chemother. 2006;17(5):275–284. doi: 10.1177/095632020601700505. [DOI] [PubMed] [Google Scholar]
- Barnard D.L., Day C.W., Bailey K., Heiner M., Montgomery R., Lauridsen L., Winslow S., Hoopes J., Li J.K., Lee J., Carson D.A., Cottam H.B., Sidwell R.W. Enhancement of the infectivity of SARS-CoV in BALB/c mice by IMP dehydrogenase inhibitors, including ribavirin. Antiviral Res. 2006;71(1):53–63. doi: 10.1016/j.antiviral.2006.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard D.L., Hubbard V.D., Burton J., Smee D.F., Morrey J.D., Otto M.J., Sidwell R.W. Inhibition of severe acute respiratory syndrome-associated coronavirus (SARSCoV) by calpain inhibitors and beta-d-N4-hydroxycytidine. Antivir. Chem. Chemother. 2004;15(1):15–22. doi: 10.1177/095632020401500102. [DOI] [PubMed] [Google Scholar]
- Barnard D.L., Hubbard V.D., Smee D.F., Sidwell R.W., Watson K.G., Tucker S.P., Reece P.A. In vitro activity of expanded-spectrum pyridazinyl oxime ethers related to pirodavir: novel capsid-binding inhibitors with potent antipicornavirus activity. Antimicrob. Agents Chemother. 2004;48(5):1766–1772. doi: 10.1128/AAC.48.5.1766-1772.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard D.L., Kumaki Y. Development in the search for the small-molecule inhibitors for treatment of severe acute respiratory syndrome coronavirus. In: LaFemina R.L., editor. Antiviral Research: Strategies in Antiviral Drug Discovery. ASM Press; Washington, DC: 2009. pp. 209–222. [Google Scholar]
- Beintema J.J., Peumans W.J. The primary structure of stinging nettle (Urtica dioica) agglutinin. A two-domain member of the hevein family. FEBS Lett. 1992;299(2):131–134. doi: 10.1016/0014-5793(92)80231-5. [DOI] [PubMed] [Google Scholar]
- Booth C.M., Matukas L.M., Tomlinson G.A., Rachlis A.R., Rose D.B., Dwosh H.A., Walmsley S.L., Mazzulli T., Avendano M., Derkach P., Ephtimios I.E., Kitai I., Mederski B.D., Shadowitz S.B., Gold W.L., Hawryluck L.A., Rea E., Chenkin J.S., Cescon D.W., Poutanen S.M., Detsky A.S. Clinical features and short-term outcomes of 144 patients with SARS in the greater Toronto area. JAMA. 2003;289(21):2801–2809. doi: 10.1001/jama.289.21.JOC30885. [DOI] [PubMed] [Google Scholar]
- Bosch B.J., Martina B.E., Van Der Zee R., Lepault J., Haijema B.J., Versluis C., Heck A.J., De Groot R., Osterhaus A.D., Rottier P.J. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl. Acad. Sci. U.S.A. 2004;101(22):8455–8460. doi: 10.1073/pnas.0400576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh P.F., Jr., Moskwa P.S., Donish W.H., Pera P.J., Richardson D., Andrese A.P. A semi-automated neutral red based chemosensitivity assay for drug screening. Invest. New Drugs. 1990;8(4):347–354. doi: 10.1007/BF00198590. [DOI] [PubMed] [Google Scholar]
- Chen J., Subbarao K. The immunobiology of SARS. Annu. Rev. Immunol. 2007;25:443–472. doi: 10.1146/annurev.immunol.25.022106.141706. [DOI] [PubMed] [Google Scholar]
- Cinatl J., Morgenstern B., Bauer G., Chandra P., Rabenau H., Doerr H.W. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet. 2003;361(9374):2045–2046. doi: 10.1016/S0140-6736(03)13615-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor R.I., Chen B.K., Choe S., Landau N.R. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206(2):935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
- Day C.W., Baric R., Cai S.X., Frieman M., Kumaki Y., Morrey J.D., Smee D.F., Barnard D.L. A new mouse-adapted strain of SARS-CoV as a lethal model for evaluating antiviral agents in vitro and in vivo. Virology. 2009;395(2):210–222. doi: 10.1016/j.virol.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y., He L., Zhang Q., Huang Z., Che X., Hou J., Wang H., Shen H., Qiu L., Li Z., Geng J., Cai J., Han H., Li X., Kang W., Weng D., Liang P., Jiang S. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. J. Pathol. 2004;203(2):622–630. doi: 10.1002/path.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348(20):1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- Galelli A., Truffa-Bachi P. Urtica dioica agglutinin. A superantigenic lectin from stinging nettle rhizome. J. Immunol. 1993;151(4):1821–1831. [PubMed] [Google Scholar]
- Groneberg D.A., Fischer A., Chung K.F., Daniel H. Molecular mechanisms of pulmonary peptidomimetic drug and peptide transport. Am. J. Respir. Cell Mol. Biol. 2004;30(3):251–260. doi: 10.1165/rcmb.2003-0315TR. [DOI] [PubMed] [Google Scholar]
- Groneberg D.A., Witt C., Wagner U., Chung K.F., Fischer A. Fundamentals of pulmonary drug delivery. Respir. Med. 2003;97(4):382–387. doi: 10.1053/rmed.2002.1457. [DOI] [PubMed] [Google Scholar]
- Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L., Luo S.W., Li P.H., Zhang L.J., Guan Y.J., Butt K.M., Wong K.L., Chan K.W., Lim W., Shortridge K.F., Yuen K.Y., Peiris J.S., Poon L.L. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302(5643):276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- Ho J.C., Ooi G.C., Mok T.Y., Chan J.W., Hung I., Lam B., Wong P.C., Li P.C., Ho P.L., Lam W.K., Ng C.K., Ip M.S., Lai K.N., Chan-Yeung M., Tsang K.W. High-dose pulse versus nonpulse corticosteroid regimens in severe acute respiratory syndrome. Am. J. Respir. Crit. Care Med. 2003;168(12):1449–1456. doi: 10.1164/rccm.200306-766OC. [DOI] [PubMed] [Google Scholar]
- Ho T.Y., Wu S.L., Chen J.C., Wei Y.C., Cheng S.E., Chang Y.H., Liu H.J., Hsiang C.Y. Design and biological activities of novel inhibitory peptides for SARS-CoV spike protein and angiotensin-converting enzyme 2 interaction. Antiviral Res. 2006;69(2):70–76. doi: 10.1016/j.antiviral.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers S.A., Tusell S.M., Gillim-Ross L., Hemmila E.M., Achenbach J.E., Babcock G.J., Thomas W.D., Jr., Thackray L.B., Young M.D., Mason R.J., Ambrosino D.M., Wentworth D.E., Demartini J.C., Holmes K.V. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. U.S.A. 2004;101(44):15748–15753. doi: 10.1073/pnas.0403812101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Xu J., Zhou C., Wu Z., Zhong S., Liu J., Luo W., Chen T., Qin Q., Deng P. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am. J. Respir. Crit. Care Med. 2005;171(8):850–857. doi: 10.1164/rccm.200407-857OC. [DOI] [PubMed] [Google Scholar]
- Keyaerts E., Vijgen L., Maes P., Neyts J., Van Ranst M. Growth kinetics of SARS-coronavirus in Vero E6 cells. Biochem. Biophys. Res. Commun. 2005;329(3):1147–1151. doi: 10.1016/j.bbrc.2005.02.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyaerts E., Vijgen L., Pannecouque C., Van Damme E., Peumans W., Egberink H., Balzarini J., Van Ranst M. Plant lectins are potent inhibitors of coronaviruses by interfering with two targets in the viral replication cycle. Antiviral Res. 2007;75(3):179–187. doi: 10.1016/j.antiviral.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokhin O., Li Y., Andonov A., Feldmann H., Flick R., Jones S., Stroeher U., Bastien N., Dasuri K.V., Cheng K., Simonsen J.N., Perreault H., Wilkins J., Ens W., Plummer F., Standing K.G. Mass spectrometric characterization of proteins from the SARS virus: a preliminary report. Mol. Cell. Proteomics. 2003;2(5):346–356. doi: 10.1074/mcp.M300048-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348(20):1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Kumaki Y., Day C.W., Wandersee M.K., Schow B.P., Madsen J.S., Grant D., Roth J.P., Smee D.F., Blatt L.M., Barnard D.L. Interferon alfacon 1 inhibits SARS-CoV infection in human bronchial epithelial Calu-3 cells. Biochem. Biophys. Res. Commun. 2008;371(1):110–113. doi: 10.1016/j.bbrc.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S.K., Woo P.C., Li K.S., Huang Y., Tsoi H.W., Wong B.H., Wong S.S., Leung S.Y., Chan K.H., Yuen K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U.S.A. 2005;102(39):14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard J.N., Schaffer D.V. Antiviral RNAi therapy: emerging approaches for hitting a moving target. Gene Ther. 2006;13(6):532–540. doi: 10.1038/sj.gt.3302645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D., Wu N., Yao H., Bader A., Brockmeyer N.H., Altmeyer P. Association of RANTES with the replication of severe acute respiratory syndrome coronavirus in THP-1 cells. Eur. J. Med. Res. 2005;10(3):117–120. [PubMed] [Google Scholar]
- Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Shi Z., Yu M., Ren W., Smith C., Epstein J.H., Wang H., Crameri G., Hu Z., Zhang H., Zhang J., McEachern J., Field H., Daszak P., Eaton B.T., Zhang S., Wang L.F. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- Marzi A., Gramberg T., Simmons G., Moller P., Rennekamp A.J., Krumbiegel M., Geier M., Eisemann J., Turza N., Saunier B., Steinkasserer A., Becker S., Bates P., Hofmann H., Pohlmann S. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 2004;78(21):12090–12095. doi: 10.1128/JVI.78.21.12090-12095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe B.R., Giomarelli B., Barnard D.L., Shenoy S.R., Chan P.K., McMahon J.B., Palmer K.E., Barnett B.W., Meyerholz D.K., Wohlford-Lenane C.L., McCray P.B., Jr. Broad-spectrum in vitro activity and in vivo efficacy of the antiviral protein griffithsin against emerging viruses of the family Coronaviridae. J. Virol. 2010;84(5):2511–2521. doi: 10.1128/JVI.02322-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S., Chu C.M., Cheng V.C., Chan K.S., Hung I.F., Poon L.L., Law K.I., Tang B.S., Hon T.Y., Chan C.S., Chan K.H., Ng J.S., Zheng B.J., Ng W.L., Lai R.W., Guan Y., Yuen K.Y. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S., Lai S.T., Poon L.L., Guan Y., Yam L.Y., Lim W., Nicholls J., Yee W.K., Yan W.W., Cheung M.T., Cheng V.C., Chan K.H., Tsang D.N., Yung R.W., Ng T.K., Yuen K.Y. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie G., Harvey D.J., Feldmann F., Stroeher U., Feldmann H., Royle L., Dwek R.A., Rudd P.M. Identification of N-linked carbohydrates from severe acute respiratory syndrome (SARS) spike glycoprotein. Virology. 2010;399(2):257–269. doi: 10.1016/j.virol.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300(5624):1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Sidwell R.W., Bailey K.W., Wong M.H., Huffman J.H. In vitro and in vivo sensitivity of a non-mouse-adapted influenza A (Beijing) virus infection to amantadine and ribavirin. Chemotherapy. 1995;41(6):455–461. doi: 10.1159/000239382. [DOI] [PubMed] [Google Scholar]
- Simmons G., Gosalia D.N., Rennekamp A.J., Reeves J.D., Diamond S.L., Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U.S.A. 2005;102(33):11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G., Reeves J.D., Rennekamp A.J., Amberg S.M., Piefer A.J., Bates P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. U.S.A. 2004;101(12):4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui J., Li W., Murakami A., Tamin A., Matthews L.J., Wong S.K., Moore M.J., Tallarico A.S., Olurinde M., Choe H., Anderson L.J., Bellini W.J., Farzan M., Marasco W.A. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl. Acad. Sci. U.S.A. 2004;101(8):2536–2541. doi: 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang K., Seto W.H. Severe acute respiratory syndrome: scientific and anecdotal evidence for drug treatment. Curr. Opin. Invest. Drugs. 2004;5(2):179–185. [PubMed] [Google Scholar]
- Tsang K.W., Ho P.L., Ooi G.C., Yee W.K., Wang T., Chan-Yeung M., Lam W.K., Seto W.H., Yam L.Y., Cheung T.M., Wong P.C., Lam B., Ip M.S., Chan J., Yuen K.Y., Lai K.N. A cluster of cases of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003;348(20):1977–1985. doi: 10.1056/NEJMoa030666. [DOI] [PubMed] [Google Scholar]
- Tsui P.T., Kwok M.L., Yuen H., Lai S.T. Severe acute respiratory syndrome: clinical outcome and prognostic correlates. Emerg. Infect. Dis. 2003;9(9):1064–1069. doi: 10.3201/eid0909.030362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme E.J., Broekaert W.F., Peumans W.J. The Urtica dioica agglutinin is a complex mixture of isolectins. Plant Physiol. 1988;86(2):598–601. doi: 10.1104/pp.86.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Meer F.J., de Haan C.A., Schuurman N.M., Haijema B.J., Peumans W.J., Van Damme E.J., Delputte P.L., Balzarini J., Egberink H.F. Antiviral activity of carbohydrate-binding agents against Nidovirales in cell culture. Antiviral Res. 2007;76(1):21–29. doi: 10.1016/j.antiviral.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U.S.A. 2003;100(23):13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.Y., Huang Y., Ganesh L., Leung K., Kong W.P., Schwartz O., Subbarao K., Nabel G.J. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 2004;78(11):5642–5650. doi: 10.1128/JVI.78.11.5642-5650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J., Zhang B., Xu J., Chang Q., McNutt M.A., Korteweg C., Gong E., Gu J. Molecular pathology in the lungs of severe acute respiratory syndrome patients. Am. J. Pathol. 2007;170(2):538–545. doi: 10.2353/ajpath.2007.060469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen Y.T., Liao F., Hsiao C.H., Kao C.L., Chen Y.C., Wu-Hsieh B.A. Modeling the early events of severe acute respiratory syndrome coronavirus infection in vitro. J. Virol. 2006;80(6):2684–2693. doi: 10.1128/JVI.80.6.2684-2693.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Lu K., Pfefferle S., Bertram S., Glowacka I., Drosten C., Pohlmann S., Simmons G. A single asparagine-linked glycosylation site of the severe acute respiratory syndrome coronavirus spike glycoprotein facilitates inhibition by mannose-binding lectin through multiple mechanisms. J. Virol. 2010;84(17):8753–8764. doi: 10.1128/JVI.00554-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziolkowska N.E., O’Keefe B.R., Mori T., Zhu C., Giomarelli B., Vojdani F., Palmer K.E., McMahon J.B., Wlodawer A. Domain-swapped structure of the potent antiviral protein griffithsin and its mode of carbohydrate binding. Structure. 2006;14(7):1127–1135. doi: 10.1016/j.str.2006.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]