

Abstract
The total synthesis of (+)-lithospermic acid is reported, which exploits two successive C–H activation reactions as the key steps. Rh-catalyzed carbene C–H insertion reaction using Davies’ catalyst built the dihydrobenzofuran core, and a late-stage intermolecular C–H olefination coupled the olefin unit with the dihydrobenzofuran core to construct the molecule in a highly convergent manner.
Since its first isolation and characterization in 1975,1 lithospermic acid has been implicated as an active component in Danshen, one of the most popular traditional herbs used in the treatment of cardiovascular disorders, cerebrovascular diseases, various types of hepatitis, chronic renal failure and dysmenorrheal (Figure 1).2 Recent studies showed that (+)-lithospermic acid (1) has potent and nontoxic anti-HIV activity.3 Not surprisingly, the total synthesis of lithospermic acid and its derivatives has attracted significant interest.4 Preparation of racemic heptamethyl lithospermate was first reported by the Jacobson group in 1979,5 while the first total synthesis of (+)-lithospermic acid was accomplished by the Ellman and Bergman group, which elegantly showcased their C–H activation/hydroarylation reaction en route to the chiral dihydrobenzofuran core.6 Herein, we report a synthesis of (+)-lithospermic acid greatly facilitated by two key C–H functionalization reactions, including an intermolecular C–H olefination of arenes, through which the dihydrobenzofuran core and the olefin unit are coupled at a late stage of the synthesis to achieve the highest convergency.
Figure 1.

(+)-Lithospermic Acid (1)
Since demethylation of heptamethyl lithospermate through a two-step deprotection sequence to give lithospermic acid had already been demonstrated,6a we focused our efforts on the assembly of hexamethyl lithospermate 2 (Figure 2). Inspired by a number of total syntheses using C–H olefination of indoles and pyrroles,7 we envisioned 2 could be prepared by an intermolecular C–H olefination of acid 4 with acrylate 3, which would put to the test our recently developed carboxyl-directed C–H olefination reaction (Figure 2),8 especially in the presence of other potentially more reactive electron-rich arenes and two potentially racemizable chiral stereocenters (C20 and C21). This disconnection would also enable a highly convergent approach toward the synthesis of 2. Olefin 3 can be readily made in three steps from commercially available rosmarinic acid. We decided to construct the chiral dihydrobenzofuran 4 from diazo-intermediate 6 through another C–H functionalization, an intramolecular asymmetric C–H carbene insertion using the Davies’ Rh(II) catalyst9 and a chiral auxiliary developed by Fukuyama.10
Figure 2.

Retrosynthesis of (+)-Lithospermic Acid
Following the above-mentioned synthetic plan, we began our experimental efforts to prepare the diazo-intermediate 6. We initially attempted to use a 3,4-dimethoxybenzyl protecting group for the phenol of phenylacetic acid 7; unfortunately, the 3,4-methoxylbenzyl protecting group proved to be incompatible with the synthetic sequence leading to this phenylacetic acid skeleton (the 3,4-methoxylbenzyl moiety was cleaved under oxidative conditions). Thus, we instead prepared 7 from readily available O-eugenol in three steps (see supporting information).
Reacting 7 with (S)-N,N-tetramethylenelactamide under Mitsunobu conditions appended the lactamide-type chiral auxiliary 8 in 83% yield (Scheme 1).11 Removal of the benzyl group gave the corresponding phenol in quantitative yield, which was then protected with 3,4-dimethoxybenzyl bromide in the presence of K2CO3 to provide the cyclization precursor 9 in 86% yield. Treatment of 9 with a diazo transfer reagent in the presence of DBU at room temperature afforded diazo compound 6 in 82% yield,9d which was isolated and immediately treated with 0.5 mol% of Davies’ Rh2(S-DOSP)4 catalyst. Gratifyingly, the C–H insertion reaction proceeded smoothly at room temperature and provided the trans-dihydrobenzofuran core 10 in good yield and diastereoselectivity (85%, 8:1 d.r.). Both CH2Cl2 and hexanes were found to be effective solvents for the cyclization reaction. Basic hydrolysis conditions afforded the carboxylic acid 4 in 86% yield.
Scheme 1.

Synthesis of Dihydrobenzofuran Carboxylic Acid 4a
a Reagents and conditions: (a) pyrrolidinyl (S)-lactamide (1.2 equiv), PPh3 (1.2 equiv), DEAD (1.2 equiv), toluene, 0 to 23 °C, 2 h; (b) H2 (balloon), Pd/C (5 mol%), MeOH, 23 °C, 12 h; (c) 3,4-dimethoxybenzyl bromide (1.45 equiv), K2CO3 (2 equiv), THF (2 equiv), 80 °C, over night, 71% over three steps; (d) p-ABSA (2 equiv), DBU (2 equiv), MeCN, 0 to 23 °C, 24 h, 82%; (e) Rh2(S-DOSP)4 (0.5 mol%), CH2Cl2, 23 °C, 2 h, 85%, dr 8:1; (f) Ba(OH)2·8H2O (1.1 equiv), THF/MeOH (1:1), 0 to 23 °C, 4 h, 86%.
Next, we prepared the acrylate coupling partner 3 (Scheme 2). Alcohol 5 was synthesized by pentamethylation of rosmarinic acid and subsequent hydrolysis.6a By treating alcohol 5 with acrylic acid in the presence of EDC and DMAP at room temperature, acrylate 3 was obtained in 91% yield. Compound 3 is stable when stored at 0 °C.
Scheme 2.

Synthesis of Acrylate 3a
a Reagents and conditions: (a) Me2SO4 (10 equiv), K2CO3 (10 equiv), acetone, reflux, 10 h, 97%; (b) NaOMe (1 equiv), MeOH, 23 °C, 2 h, 88%; (c) acrylic acid (1.5 equiv), EDC-HCl (2 equiv), DMAP (2 equiv), CH2Cl2, 0 to 23 °C, 91%.
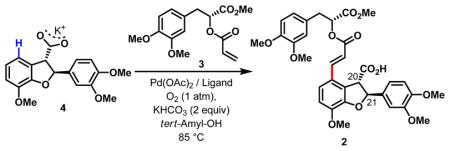
With carboxylic acid 4 and acrylate 3 in hand, we were ready to test the key C-H olefination step. While this strategy promised to assemble the lithospermic acid backbone at a late stage in a highly convergent manner, several potential complications could jeopardize such an approach. First, the presence of multiple electron-rich and hence reactive arenes could cause non-selective C–H olefination reactions. Second, the two chiral centers adjacent to the esters (C20 and C21) could be prone to racemization under the reaction conditions. Third, intermolecular C–H olefination of two relatively large fragments involves considerable entropy costs compared to previous intramolecular C–H olefinations used in synthesis.7 Finally, the carboxyl directing group is pointing away from the target C–H bonds with an approximate dihedral angle of 53° (estimated using ChemBio 3D). Nonetheless, despite these disadvantages, we hoped that combination of the directing power of the carbonyl group in carboxyl potassium salt and ligand acceleration8 could deliver this late-stage C–H olefination process.
Consistent with our hypothesis, olefination of 4 with 3, when carried out in the presence of 5 mol% Pd(OAc)2, 5 mol% Ac-Ile-OH and 2 equiv KHCO3 in a solution of tert-amyl alcohol at 85 °C for 2 h under 1 atm O2, afforded the desired product 2 in 93% isolated yield (Table 1, entry 3). Catalyst loading can be lowered to 2 mol% with prolonged reaction time (entry 4). In the absence of Ac-Ile-OH ligand, olefination of 4 under the optimized conditions also proceeded, albeit giving significantly lower yields (entries 1, 2). This late-stage coupling of two structurally complex substrates represents the most sophisticated application of arene C–H olefination to date, and demonstrates the broad applicability of this transformation to natural product synthesis. Mechanistically, these results further showcase the directing power of a weak coordination of Pd(II) with the carbonyl group in the carboxyl potassium salt formed in situ.
Table 1.
Synthesis of Hexamethyl Lithospermate 2
 | ||||
|---|---|---|---|---|
| Entry | Pd(OAc)2 | Time | Ligand | Yield |
| 1 | 5 mol% | 12 h | 5 mol% BQ | 31 % |
| 2 | 5 mol% | 24 h | 5 mol% BQ | 50 % |
| 3 | 5 mol% | 2 h | 10 mol% Ac-Ile-OH | 93 % |
| 4 | 2 mol% | 10 h | 4 mol% Ac-Ile-OH | 91 % |
To confirm the structure of 2, we converted 2 into two previously characterized derivatives, dimethyl ester 11 and diacid 12 (Scheme 3). Treatment of monomethyl ester 2 with diazomethane gave dimethyl ester 11 in 96% yield. Treatment of 2 or 11 under Nicolaou’s hydrolysis conditions afforded diacid 12 in 91% yield.12 1H and 13C NMR spectra of 11 and 12 were in good agreement with those reported by Ellman and Bergman (see supporting information).6a
Scheme 3.

Completion of the Synthesis of (+)-Lithospermic Acida
a Reagents and conditions: (a) Me3SnOH (3 equiv), 1,2-dichloroethane, 80 °C, 24 h, 91%; (b) 1-trimethylsilylquinolinium iodide (40 equiv), neat, 130 °C, 3 h, 31%; (c) CH2N2, Et2O, 0 °C, 10 min, 96%.
These results encouraged us to perform the two-step demethylation procedure that would complete the synthesis of the natural product. Conversion of mono acid 2 to diacid 12 under Nicolaou’s conditions12 and subsequent treatment of diacid 12 with TMSI–quinoline6a gave the natural product (+)-lithospermic acid (1) in 31% yield (Scheme 3).
In summary, total synthesis of (+)-lithospermic acid was accomplished in 12 steps and 11% overall yield from eugenol. The highly convergent strategy employed was made possible by two C–H functionalization reactions, including a late-stage Pd-catalyzed intermolecular C–H olefination reaction accelerated by the amino acid ligand Ac-Ile-OH, the most sophisticated application of arene C–H olefination to the synthesis of complex natural products.
Supplementary Material
Acknowledgments
We gratefully acknowledge the National Institutes of Health (NIGMS, 1 R01 GM084019-01A1) and National Science Foundation under the Center of Chemical Innovation in Stereoselective C–H Functionalization (CHE-0943980), and Amgen for financial support. We are thankful for predoctoral fellowships from the China Scholarship Council (D.-H.W.). We thank Prof. P. S. Baran, Prof. H. M. L. Davies, Dr. T. Newhouse and D. Sarlah for valuable discussions, and thank Dr. Shun Su for helping with the isolation of final product on prep. HPLC.
Footnotes
Supporting Information Available: Experimental procedures and characterization of all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kelley CJ, Mahajan JR, Brooks LC, Neubert LA, Breneman WR, Carmack M. J Org Chem. 1975;40:1804.The name of lithospermic acid was given in 1963, see: Johnson G, Sunderwirth SG, Gibian H, Coulter AW, Gassner FX. Phytochemistry. 1963;2:145.
- 2.(a) Cheng TO. Int J Cardiol. 2007;121:9. doi: 10.1016/j.ijcard.2007.01.004. [DOI] [PubMed] [Google Scholar]; (b) Jiang RW, Lau KM, Hon PM, Mak TCW, Woo KS, Fung KP. Curr Med Chem. 2005;12:237. doi: 10.2174/0929867053363397. [DOI] [PubMed] [Google Scholar]; (c) Zhou L, Zuo Z, Chow MSS. J of Clin Pharmacol. 2005;45:1345. doi: 10.1177/0091270005282630. [DOI] [PubMed] [Google Scholar]; (d) Lu Y, Foo LY. Phytochemistry. 2002;59:117. doi: 10.1016/s0031-9422(01)00415-0. [DOI] [PubMed] [Google Scholar]; (e) Watzke A, O’Malley SJ, Bergman RG, Ellman JA. J Nat Prod. 2006;69:1231. doi: 10.1021/np060136w. [DOI] [PubMed] [Google Scholar]
- 3.(a) Abd-Elazem IS, Chen HS, Bates RB, Huang RCC. Antiviral Res. 2002;55:91. doi: 10.1016/s0166-3542(02)00011-6. [DOI] [PubMed] [Google Scholar]; (b) Shigematsu T, Tajima S, Nishikawa T, Murad S, Pinnell SR, Nishioka I. Biochim Biophys Acta, Gen Subj. 1994;1200:79. doi: 10.1016/0304-4165(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 4.For recent syntheses of structually related members of rosmarinic acid family, see: Snyder SA, Kontes F. J Am Chem Soc. 2009;131:1745. doi: 10.1021/ja806865u.
- 5.Jacobson RM, Raths RA. J Org Chem. 1979;44:4013. [Google Scholar]
- 6.(a) O’Malley SJ, Tan KL, Watzke A, Bergman RG, Ellman JA. J Am Chem Soc. 2005;127:13496. doi: 10.1021/ja052680h. [DOI] [PubMed] [Google Scholar]; (b) Colby DA, Bergman RG, Ellman JA. Chem Rev. 2010;110:624. doi: 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Trost BM, Godleski SA, Genêt JP. J Am Chem Soc. 1978;100:3930. [Google Scholar]; (b) Cushing TD, Sanz-Cervera JF, Williams RM. J Am Chem Soc. 1993;115:9323. [Google Scholar]; (c) Baran PS, Corey EJ. J Am Chem Soc. 2002;124:7904. doi: 10.1021/ja026663t. [DOI] [PubMed] [Google Scholar]; (d) Garg NK, Caspi DD, Stoltz BM. J Am Chem Soc. 2004;126:9552. doi: 10.1021/ja046695b. [DOI] [PubMed] [Google Scholar]; (e) Beck EM, Hatley R, Gaunt MJ. Angew Chem, Int Ed. 2008;47:3004. doi: 10.1002/anie.200705005. [DOI] [PubMed] [Google Scholar]; (f) Bowie AL, Jr, Trauner D. J Org Chem. 2009;74:1581. doi: 10.1021/jo801791j. [DOI] [PubMed] [Google Scholar]
- 8.(a) Wang DH, Engle KM, Shi BF, Yu JQ. Science. 2010;327:315. doi: 10.1126/science.1182512. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Engle KM, Wang DH, Yu JQ. Angew Chem, Int Ed. 2010;49:6169. doi: 10.1002/anie.201002077. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Engle KM, Wang DH, Yu JQ. J Am Chem Soc. 2010;132:14137. doi: 10.1021/ja105044s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Davies HML, Hansen T. J Am Chem Soc. 1997;119:9075. [Google Scholar]; (b) Davies HML, Hansen T, Churchill MR. J Am Chem Soc. 2000;122:3063. [Google Scholar]; (c) Davies HML, Antoulinakis EG. J Organomet Chem. 2001;617:47. [Google Scholar]; (d) Davies HML, Cantrell WR, Jr, Romines KR, Baum JS. Org Synth. 1992;70:93. [Google Scholar]
- 10.(a) Kurosawa W, Kan T, Fukuyama T. Synlett. 2003:1028. doi: 10.1021/ja036011k. [DOI] [PubMed] [Google Scholar]; (b) Kurosawa W, Kan T, Fukuyama T. J Am Chem Soc. 2003;125:8112. doi: 10.1021/ja036011k. [DOI] [PubMed] [Google Scholar]
- 11.Ito Y, Kobayashi Y, Kawabata T, Takase M, Terashima S. Tetrahedron. 1989;45:5767. [Google Scholar]
- 12.Nicolaou KC, Estrada AA, Zak M, Lee SH, Safina BS. Angew Chem, Int Ed. 2005;44:1378. doi: 10.1002/anie.200462207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.