

Abstract
A Boc-protected aminooxy end-functionalized poly(N-isopropylacrylamide) (pNIPAAm) was synthesized by reversible addition-fragmentation chain transfer (RAFT) polymerization. The monomer was polymerized in the presence of a Boc-protected aminooxy trithiocarbonate chain transfer agent (CTA) utilizing 2,2’-azobis(2-isobutyronitrile) (AIBN) as the initiator in DMF at 70 °C. The final polymer had a number-average molecular weight (Mn) of 4,200 Da as determined by 1H NMR spectroscopy and a narrow polydispersity index (1.14) by gel permeation chromatography (GPC). The Boc group was removed, and the polymer was then incubated with Nε-levulinyl lysine-modified bovine serum albumin (BSA). Gel electrophoresis confirmed that the conjugation was successful. The aminooxy end-functionalized pNIPAAm was also immobilized on a gold surface after reduction of the trithiocarbonate end-group. The pNIPAAm surface was then incubated with an aldehyde-modified heparin to yield the polysaccharide-functionalized surface. All surface modifications were monitored by FT-IR spectroscopy.
Introduction
Oxime chemistry is increasingly exploited to conjugate proteins, peptides and sugars to small molecules, polymers, and surfaces.1 There are several reasons for this trend. The first is that reaction of a hydroxylamine, also called an aminooxy, and an aldehyde or ketone is chemoselective.2 The reaction readily occurs, without side reactions with natural functionality found on biomolecules. This allows for site-selective conjugation, which is important for retention of bioactivity for most applications.3,4 Indeed, oxime chemistry has lead to protein-polymer conjugates whereby the protein, erythropoietin (Epo), was one hundred percent as active as the unmodified version;5 this is rarely the case for heterogeneous therapeutic protein-polymer conjugates.3,4 The second is that site-selective modification of biomolecules to incorporate aminooxy, aldehyde, or ketone moieties is straightforward. For proteins, this can be accomplished by modifying natural amino acids with levulinic acid or aminooxy acetic acid.5-7 This approach can be site selective if the targeted amino acid is rare or if native chemical ligation is utilized. Other methods include transamination reactions to modify N-termini with α-oxoamides,8-10 recombinant methods utilizing ketone modified non-natural amino acids,11 or by enzymatic N-terminal addition.12 Polysaccharides are easily oxidized to aldehydes at vicinal diols.13 In the study described herein, we exploited oxime chemistry to conjugate a protein and polysaccharide to a polymer. Importantly, we developed a straightforward synthetic approach to synthesize the partner end-functionalized aminooxy polymer for these conjugation reactions.
Polymers with either hydroxylamine or reactive carbonyl groups at the chain end or side chain have been synthesized by a variety of approaches. For example, hydroxylamines are readily introduced into PEG chain ends via a Mitsunobu reaction;9,14 this reaction is typically repeated twice to increase the yield. Free radical polymerization has been utilized to produce aminooxy15 or aldeyde16 groups side chain polymers. In these cases, the polymers have broad molecular weight distributions. However, there are several advantages to polymers with low polydispersities (PDIs) including uniform solubilities, toxicity profiles, and pharmacokinetics.17 This can be achieved utilizing controlled radical polymerizations (CRPs). Our group has synthesized aminooxy poly(styrene), poly(PEG acrylate), pNIPAAm, and poly(2-hydroxypropylmethacrylate) by atom transfer radical polymerization (ATRP).6,18 Others have reported aldehyde end-functionalized polymers by ATRP.19 Poly(methacrylates) with side chain aldehydes or ketones and have been prepared by both reversible addition-fragmentation chain transfer (RAFT) polymerization and ATRP.20-25 In some cases, these have been subsequently reacted with aminooxy-functionalized compounds, including peptides.21-23 In this manuscript, we report for the first time a synthetic method to produce aminooxy semi-telechlic polymers that exploits RAFT polymerization.
RAFT polymerization is widely used because it provides well-defined polymers with targeted molecular weights and narrow molecular weight distributions.26-28 The technique is robust and can be employed with success in the presence of a wide variety of functional groups. RAFT polymerization requires a chain transfer agent (CTA), which typically is a dithioester or trithiocarbonate compound. End-group functionality can be easily incorporated into the polymers by designing a CTA with functional “R” or “Z” groups resulting in semi-telechelic or telechelic polymers.29-37 In this manner, polymers have been prepared by RAFT polymerization with biotin, pyridyl disulfide, maleimide, azide, cysteine, glutathione, and protein end groups.29-32,35,37
Compared to our reported examples of utilizing ATRP to produce aminooxy-functionalized polymers, RAFT polymerization has some advantages. Foremost, by placing the hydroxylamine in the “R” group of the CTA, the “Z” group end of the polymer chain can be further manipulated. The dithioester or trithiocarbonate can be removed by heating the polymer in the presence of a radical initiator.34 This has been be utilized to introduce distinct chemical reactivity at the chain end.38 The “Z” moieties can also be reduced to thiol groups.39-41 This has been exploited to introduce fluorophores by reaction with the sulfhydryl, which is potentially useful to track biomolecules in vitro.42,43 The ease with which this particular reduction reaction occurs offers additional versatility; for example, the thiol can be used to chemisorb the polymer to gold or silver surfaces.41,44,45 Sumerlin and coworkers used this approach to fabricate surfaces modified with water soluble polymers.45 It was for these reasons that we exploited RAFT polymerization to produce aminooxy-end functionalized polymers. In this report, preparation of the CTA, synthesis of pNIPAAM hydroxylamine, and conjugation to ketone modified bovine serum albumin (BSA) are detailed. Immobilization of the polymer on gold surfaces and conjugation of the polysaccharide heparin to the pNIPAAm films are also disclosed.
Materials and Methods
Materials
All chemicals were purchased from Aldrich or Acros and used as received, unless otherwise specified. Heparin sodium salt grade I-A, 50,000 units, from porcine intestinal mucosa, ~170 USP units/mg was purchased from Sigma Aldrich. N,N,N’N’-Tetramethyl-O-(1H-benzotriazol-1-yl)uranium hexafluorophosphate (HBTU) was bought from Novabiochem and 1-hydroxybenzotriazole (HOBT) was obtained from AnaSpec Incorporated. AIBN was recrystallized from cold ethanol and dried under vacuum. The trithiocarbonate acid 1 was synthesized according to published procedures.46,47 BSA was purchased from Fisher Scientific and modified to include ketones using the procedure outlined by Heredia et al.6
Analytical techniques
1H NMR spectra were recorded on a Bruker Avance500 spectrometer; all polymer spectra were taken with a minimum delay time of 10 seconds. GPC was conducted on a Shimadzu high performance liquid chromatography (HPLC) system equipped with a refractive index detector RID-10A and two Polymer Laboratories PLgel 5 μm mixed D columns (with guard column). LiBr (0.1 M) in DMF at 40 °C was used as a solvent (flow rate: 0.8 mL/min). Near-monodisperse poly(methyl methacrylate) standards (Polymer Laboratories) were employed for calibration. Chromatograms were processed with the EZStart 7.2 chromatography software. Preparatory reverse phase HPLC was carried out on a Shimadzu HPLC system equipped with a UV detector using a Luna 5u C18 (2) 100A column (preparatory: 5μm, 250 × 21.2 mm) with monitoring at λ =254 nm and 220 nm. A water: acetonitrile (55:45) solvent system was used as the mobile phase (flow rate: 22 mL/min). UV-Vis spectra were recorded on a ThermoSpectronic Biomate 5 Spectrometer. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was carried out using 4-20% precast gradient gels from Invitrogen. For reducing conditions, samples were dissolved in tris(hydroxymethyl)aminomethane (TRIS) buffer containing SDS, bromophenol blue and glycerol. Dithiothreitol (DTT) was freshly added, and the samples were heated at 95 °C for 4 minutes. Staining was accomplished using coomassie blue. Ellipsometry was performed using a Gaertner LSE ellipsometer equipped with a 633 nm HeNe laser fixed at a 70° incidence angle. Infrared spectra were obtained with a Perkin Elmer Spectrum One instrument equipped with a universal ATR accessory. Surface IR spectra were obtained using a Jasco 670 Plus FTIR spectrometer equipped with a MCT detector and a variable angle ATR-FTIR accessory (Harrick Scientific). The spectra were recorded over 1000 scans using a hemispherical germanium ATR crystal at an angle of 65° and a scan resolution of 4 cm-1. The UCLA Molecular Instrumentation Center (MIC) performed the mass spectrometry.
Synthesis of the trithiocarbonate alcohol 2
Trithiocarbonate acid 1 (0.35 g, 1.55 mmol, 1 equiv.), HOBT (0.25 g, 1.86 mmol, 1.2 equiv.) and HBTU (0.70 g, 1.86 mmol, 1.2 equiv.) were evacuated-argon refilled 3 times and dissolved in dry methylene chloride (4 mL), followed by addition of N,N-diisopropylethylamine (DIPEA, 0.61 mL, 3.72 mol, 2.4 equiv.). Twenty min later, 2-(2-aminoethoxy)ethanol (0.19 mL, 1.86 mmol, 1.2 equiv.) was added to the reaction mixture. After five hours, the mixture was concentrated, redissolved in chloroform, and washed 2 times with water and once with brine. The solution was then dried over magnesium sulfate, filtered, and concentrated to obtain the trithiocarbonate alcohol 2 in 96% yield. 1H NMR (CDCl3): δ 6.87 (s, 1H, NH), 3.66 (m, 2H, CH2OH), 3.48 (t, 2H, CH2O, J = 5.2 Hz), 3.47 (t, 2H, CH2CH2OH, J = 4.4 Hz), 3.43-3.37 (m, 2H, CONHCH2), 3.28-3.22 (q, 2H, CH3CH2, J = 7.4 Hz), 1.66 (s, 6H, CCH3CH3), 1.29 (t, 3H, CH3CH2, J = 7.4 Hz).
Synthesis of Boc-aminooxy CTA 3
The alcohol 2 (0.60 g, 1.92 mmol, 1 equiv.), Boc-aminooxy acetic acid (0.55g, 2.89 mmol, 1.5 equiv.), N,N’-dicyclohexylcarbodiimide (DCC, 0.60 g, 2.89 mmol, 1.5 equiv.) and 4-(dimethylamino)pyridine (DMAP, 0.03 g, 0.23 mmol, 0.12 equiv.) were evacuated-argon refilled 3 times, dissolved in dry methylene chloride (9 mL), and stirred for 12 hours at room temperature. The reaction was filtered, concentrated, and purified by column chromatography (diethyl ether: hexanes 7:3, Rf of the product was 0.76). The CTA 3 was then further purified by HPLC (55% acetonitrile: 45% water); the product was recovered by collecting the peak at 8 minutes. After lyophilization, the CTA 3 was obtained in 32% yield. 1H NMR (CDCl3): δ 7.8 (s, 1H, ONH), 6.84 (s, 1H, NH), 4.46 (s, 2H, COCH2O), 4.27 (m, 2H, CH2OCO), 3.60 (t, 2H, CH2CH2OCO, J = 4.7 Hz), 3.49 (m, 2H, CH2O), 3.43-3.37 (m, 2H, CONHCH2), 3.29-3.23 (q, 2H, CH3CH2, J = 7.4 Hz), 1.68 (s, 6H, SCCH3CH3), 1.47 (s, 9H, CCH3CH3CH3), 1.30 (t, 3H, CH3CH2, J = 7.4 Hz). Mass spectrum (MALDI) calcd: 507.13 m/z (M+Na+); observed: 507.05 m/z (M+Na+). UV-vis: λmax: 308 nm.
Typical Polymerization of NIPAAm from CTA 3
N-Isopropylacrylamide (NIPAAm, 0.64 g, 5.68 mmol, 100 equiv.), AIBN (0.009 g, 0.057 mmol, 1 equiv.) and CTA 3 (0.055 g, 0.11 mmol, 2 equiv.) were charged into a Schlenk tube, dissolved in DMF, (2.9 mL) and subjected to 3 freeze/pump/thaw cycles. The flask was refilled with Ar and immediately transferred to a 70 °C oil bath to initiate the polymerization. The polymerization was stopped after 2.5 hours by freezing the reaction in liquid N2. The DMF was removed and the mixture was then dissolved in THF and precipitated 3 times from cold hexanes. The polymer was recovered as a yellow powder, with a Mn (calculated by 1H NMR) of 4,200 Da and a PDI (calculated by GPC) of 1.14. 1H NMR (CDCl3, peaks are broad): δ 6.22 (1H, NH), 4.45 (end-group, CH2ON), 4.32 (end-group, CH2OCO), 3.98 (1H, NHCH), 3.67 (end-group, CH2CH2OCO), 3.53 (end-group, CH2O), 3.5-3.29 (end-group, CH2S, CONHCH2), 2.71-0.90 (CCH3CH3 and backbone). IR (cm-1): 3448, 3292, 3075, 2972, 2933, 1628, 1537, 1458, 1386, 1367, 1171, 1130, 928, 883. UV-vis: λmax: 307 nm.
Kinetic Studies of the Polymerization of NIPAAm from CTA 3
NIPAAM, 3, and AIBN in the same amounts as above were dissolved in DMF in a 25 mL Schlenk tube and subjected to 3 freeze/pump/thaw cycles. The tube was then transferred to a 70 °C oil bath to initiate the polymerization. Samples were taken from the mixture at defined time points. The DMF was removed, and the samples were diluted into CDCl3 for 1H NMR analysis and DMF with LiBr (0.1 M) for GPC analysis.
Chain extension studies
NIPAAm (0.30 g, 2.65 mmol, 200 equiv.), pNIPAAm 4 (macro CTA, Mn by 1H NMR: 3,800 Mn by GPC: 9,100, PDI: 1.08; 0.10 g, 0.027 mmol, 2 equiv.) and AIBN (0.002 g, 0.013 mmol, 1 equiv.) in DMF (0.8 mL) were charged into a Schlenk tube, subjected to 3 freeze/pump/thaw cycles and heated to 70 °C. The polymerization was stopped after 3 hours by freezing the reaction in liquid N2. The DMF was removed under vacuum and the polymer purified by dissolving in THF and precipitating 3 times from cold hexanes. The polymer was recovered as a yellow powder, with a Mn (calculated by 1H NMR) of 14,100. The Mn calculated by GPC was 26,800 with a PDI of 1.16.
Deprotection and Conjugation
Boc-protected pNIPAAm (10 mg, Mn = 8,100 Da determined by 1H NMR) was dissolved in 1 mL of a 50% v/v TFA/CHCl3 solution. After 30 min the solvent was removed by bubbling with argon, and the product dried under vacuum. Nε-Levulinyl lysine-modified BSA (1 mg) and deprotected pNIPAAm (5 mg) were dissolved in 300 μL of 50/50 v/v acetonitrile/Milli-Q H2O. After 30 min the solvent was removed by lyophilization. The protein was then redissolved in 200 μL of H2O. To isolate the pNIPAAm–BSA conjugate, 150 μL of this solution was heated above 32 °C to precipitate the polymer and then centrifuged at 15,000 rpm to isolate the solids. The pellet was redissolved in 50 μL of water for SDS-PAGE analysis.
pNIPAAm modified surface
Aminooxy-pNIPAAm 5 (0.10 g, 0.0063 mmol, 1 equiv.), n-butylamine (0.006 mL, 0.063 mmol, 10 equiv.) and tris(2-carboxyethyl)phosphine (TCEP, 0.001 mmol, 1 equiv.) were added to a 2-necked round bottom flask and evacuated-argon refilled 3 times. Degassed THF (2 mL) was added, and after one hour the reaction was stopped. The polymer was purified by precipitation from cold hexanes and dried under vacuum for 1 hour. The polymer (6) was recovered as a white powder. The polymer was then immediately dissolved in dry methanol (1 mL) and incubated with a piranha-cleaned (4:1 sulfuric acid: 30% hydrogen peroxide, CAUTION) gold surface for 12 hours to form an aminooxy end-functionalized pNIPAAm modified surface. The surface was rinsed with methanol and dried with a stream of argon. Ellipsometry indicated a surface thickness of 1.17 ± 0.04 nm. IR (cm-1): 3289, 2980, 2973, 2919, 1661, 1456, 1367, 1277, 1201, 899.
Heparin modified surface
Heparin oxidation to install aldehyde moieties was achieved following a literature procedure.13 Briefly, heparin was dissolved in 0.5 M sodium periodate (NaIO4) in 0.05 M acetate buffer (2 mL, pH 5). After quenching with glycerol, the polysaccharide was subjected to centriprep purification (3000 MWCO). Modification of heparin was confirmed by reaction with Purpald using a reported procedure.48 The reaction was also confirmed by IR (cm-1): 3435, 1747 1656, 1227, 1176, 1038, 1001, 893. The aldehyde-modified heparin was then immobilized by incubating the solution with a pNIPAAm modified gold surface for 12 hours at 4 °C. The surface was then rinsed with water and dried with a stream of argon. IR (cm-1): 3284, 3017, 2944, 2940, 2925, 2853, 1736, 1644, 1534, 1457, 1364, 1277, 1219, 1152, 1138, 1070, 936.
Heparin modified surface control
pNIPAAm 4 was reduced and immobilized on a gold surface as described above. Aldehyde-modified heparin was then incubated for 12 hours at 4 °C. The surface was then rinsed with water and dried with a stream of argon. IR (cm-1): 3246, 2942, 2923, 2852, 1639, 1536, 1400, 1366, 1048, 909.
Results and Discussion
Synthesis of the N-Boc protected aminooxy pNIPAAm
A trithiocarbonate CTA was designed with a protected hydroxylamine as the “R” group for several reasons. An aminooxy group was chosen due to its favorable reactivity with ketones and aldehyde groups. This makes it an ideal chemoselective reactive group for biomolecules such as proteins and saccharides. The “R” group was targeted so that the aminooxy would be stable to hydrolysis in the final polymer and that the trithiocarbonate “Z” group would be available for further modification.
The trithiocarbonate acid 1 was synthesized in one step from acetone, ethanethiol, and carbon disulfide following a literature procedure.46 After purification by recrystallization, the trithiocarbonate acid was added to 2-(2-aminoethoxy)ethanol in the presence of HOBt, HBTU, and DIPEA to yield trithiocarbonate alcohol 2 (Scheme 1) in 96% yield. Alcohol 2 was then coupled with Boc-aminooxy acetic acid in methylene chloride. After 12 hours the reaction mixture was filtered and concentrated. However, attempts to purify the target molecule by both flash chromatography and extraction were unsuccessful, as an unidentified (13%) impurity persisted. Purification of the Boc-aminooxy functionalized CTA 3 was achieved by reverse phase HPLC, utilizing an acetonitrile: water (55:45) mixture as the mobile phase. The CTA was recovered as a yellow oil in 32% yield. CTA 3 was characterized by 1H NMR (Figure 1) and MALDI MS, which showed that the pure compound had been made. The presence of the trithiocarbonate group was confirmed by UV-vis which exhibited a maximum absorbance at 308 nm corresponding to a π–π* transition of the thiocarbonyl moiety.
Scheme 1.
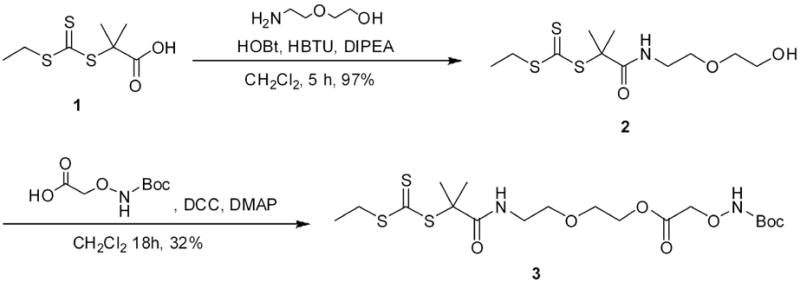
Synthesis of CTA.
Figure 1.
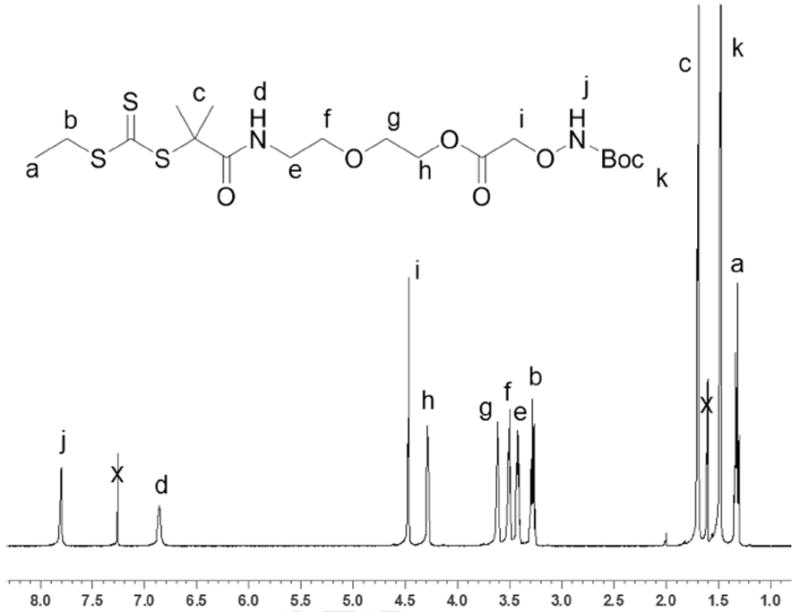
1H NMR (500 MHz) of CTA 3 in CDCl3.
Following purification and characterization of the CTA, NIPAAm was polymerized by RAFT. A variety of CTA to initiator ratios and concentrations were surveyed, and the optimal CTA concentration was 0.04 M with a 3 to AIBN ratio of 2:1. NIPAAm was polymerized in anhydrous DMF at 70 °C, with a ratio of NIPAAm to 3 of 50:1 to obtain pNIPAAm 4 (Scheme 2). Kinetic studies revealed that the reaction reached 92% conversion after 2 hours and there was no delay period (Figure 2a). The Mn of pNIPAAm also increased linearly with conversion, and the PDI remained below 1.15 throughout the polymerization (Figure 2.2b), indicating a controlled polymerization. The polymerization was stopped, and the polymer was purified by precipitation from hexanes to recover 4 as a yellow solid.
Scheme 2.
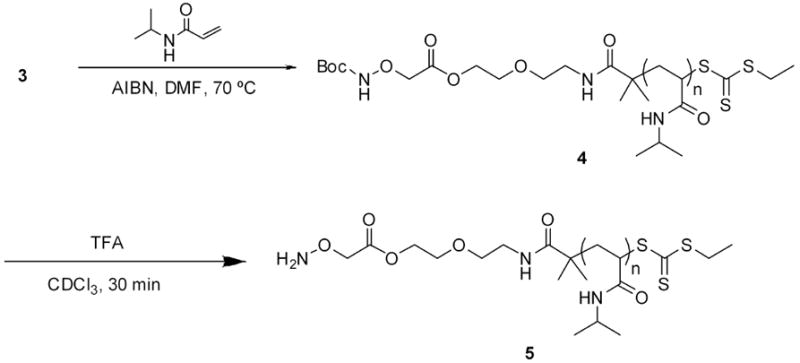
Polymerization of NIPAAm from CTA 3 and end-group deprotection
Figure 2.
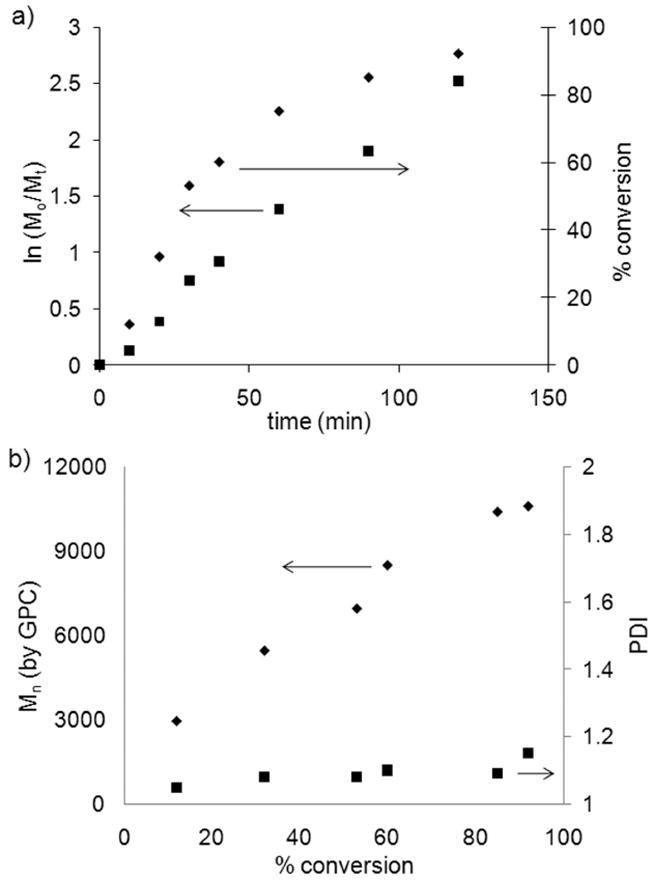
Polymerization of NIPAAm from CTA 3 in DMF at 70 °C [NIPAAm]:[3]:[AIBN] = 100:2:1. a) kinetic plot; b) experimental Mn (from GPC) and PDI vs. conversion.
The polymer was characterized by 1H NMR, GPC, IR and UV-vis spectroscopies. The Mn determined by 1H NMR was 4,200 Da, which was in reasonably close agreement with the expected Mn (5,300 Da). The molecular weight of the polymer was calculated by comparing the integrations of the CH peak of pNIPAAm at 3.98 ppm with the COOCH2 end-group peak at 4.32 (j and b respectively in Figure 3). The molecular weight distribution by GPC was narrow (PDI 1.15). In the IR spectrum the N-H stretch of the amide (3292 cm-1), the C-H stretch of the sp3 carbons (~2972 cm-1), the C=O stretch of the amide (1630 cm-1) and the CNH vibration (1540 cm-1) were observed (Figure 7a). The presence of the trithiocarbonate end-group was confirmed by UV-Vis, which showed a maximum absorbance at 307 nm; the same wavelength observed in the UV-Vis spectrum of the CTA 3.
Figure 3.
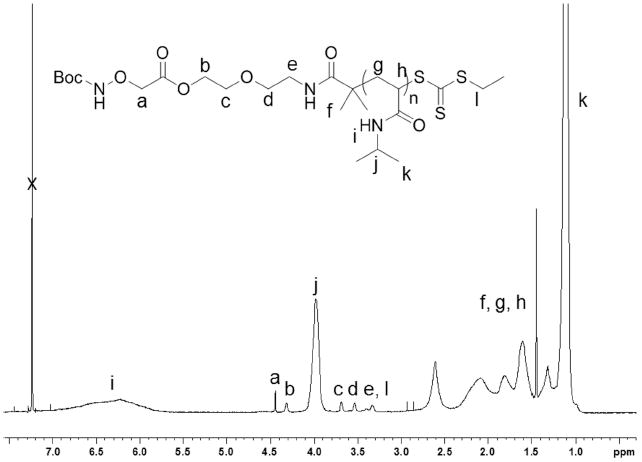
1H NMR (500 MHz) of pNIPAAm 4 in CDCl3.
Figure 7.
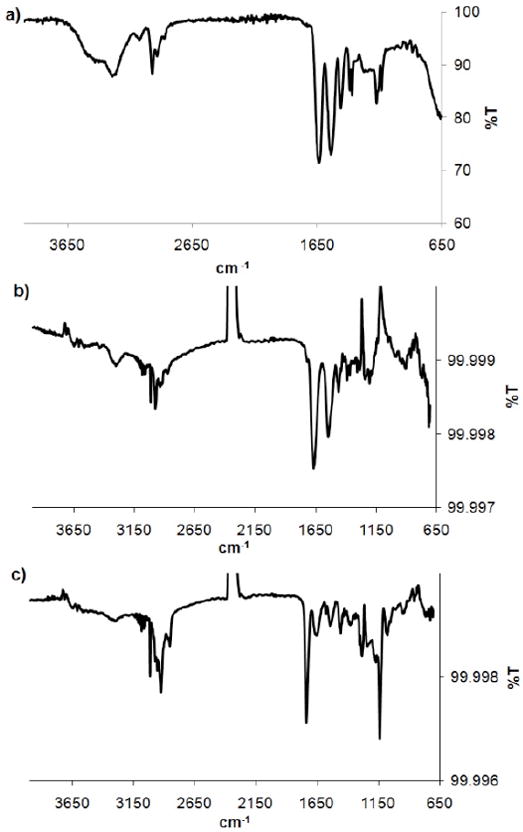
FT-IR spectra of (a) pNIPAAm 4 powder, (b) pNIPAAm 7 immobilized on a gold surface, (c) heparin immobilized on 7 on gold.
Chain extension studies
Chain extension studies were performed to confirm that 4 contained the trithiocarbonate end-group. A macro CTA 4 with a Mn determined by 1H NMR of 3,800 Da and 9,100 Da by GPC with a PDI of 1.08 was chosen for this study. NIPAAm was polymerized in the presence of the Boc-aminooxy macro CTA 4, utilizing recrystallized AIBN as the initiator in anhydrous DMF at 70 °C ([NIPAAm]: [4]: [AIBN] = 200:2:1) with a macro CTA concentration of 0.03 M. The polymerization was stopped after 3 hours (98% conversion) and the crude mixture was analyzed by GPC. The GPC trace of the final polymer displayed a shift to lower retention time when compared to macro CTA 4 (Figure 2.4). The Mn determined by GPC was 26,800 Da, which, as expected, was three times the original molecular weight by GPC (9,100 Da). The Mn of the final polymer determined by 1H NMR was 14,100 Da, which was in close agreement with the expected Mn (14,900 Da), confirming CTA efficiency. The GPC trace of the extended polymer did show residual unmodified macro CTA. Yet, this was minimal, which confirmed the presence of the trithiocarbonate moiety in the majority of the polymer chains. The polymer was recovered as a pale yellow solid after purification by precipitation from cold hexanes.
Protein Conjugation
Protein-polymer conjugates have significant advantages over unmodified proteins.1,3,49 These include increased stability and solubility, the ability to modulate protein activity, and increased bioavailability. pNIPAAm is a stimuli responsive polymer that undergoes phase separation from water at 32 °C.50 It is well known that pNIPAAm and other “smart” polymers are capable of maintaining this phase behavior characteristic when conjugated to biomolecules, while the biomolecules retain their bioactivity.49-52 pNIPAAm conjugates are useful for biotechnology applications where temperature can be utilized to “turn off” active sites by sterically hindering the binding of analyte or to isolate proteins. Therefore, we explored whether or not this polymer synthesized by RAFT could be utilized to make protein-polymer conjugations.
The polymer was deprotected following the procedure of Heredia et al. (Scheme 2).6 Briefly, the polymer was incubated with a 1:1 mixture of trifluoroacetic acid (TFA)/chloroform for 30 min. After the solvent was removed, the deprotection was confirmed by 1H NMR spectroscopy (Figure 5). The shift of the peak arising from the protons on the methylene α to the aminooxy at 4.45 ppm to 4.73 ppm indicated that the deprotection was successful. Comparison of the integration of the signals from the end group with the methyne proton of the isopropyl group demonstrated that the molecular weight remained unchanged after deprotection, which was important because it showed that the end group was not cleaved during the reaction.
Figure 5.
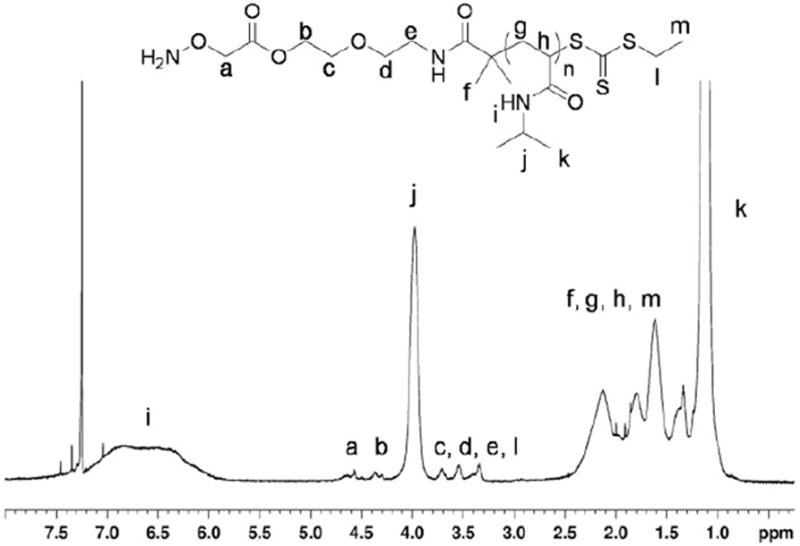
1H NMR (500 MHz) of deprotected polymer 5.
Bioconjugation was subsequently performed by mixing deprotected aminooxy pNIPAAm with the Nε-levulinyl-modified BSA in a 50/50 v/v solution of acetonitrile/water (Scheme 3). The conjugate was purified and covalent attachment of the protein to the polymer was verified by thermal precipitation. The polymer was lyophilized, taken up water and heated to 32 °C to induce a phase transition of the polymer. SDS-PAGE analysis of the resulting conjugate displayed a clear shift in molecular weight following conjugation with pNIPAAm (Figure 6, Lane 1 vs. Lane 2). These data demonstrate the ability to utilize this polymer for conjugation to a protein modified to contain ketones. Further, this shows that “smart” polymer conjugates that respond to temperature can be prepared by this approach.
Scheme 3.
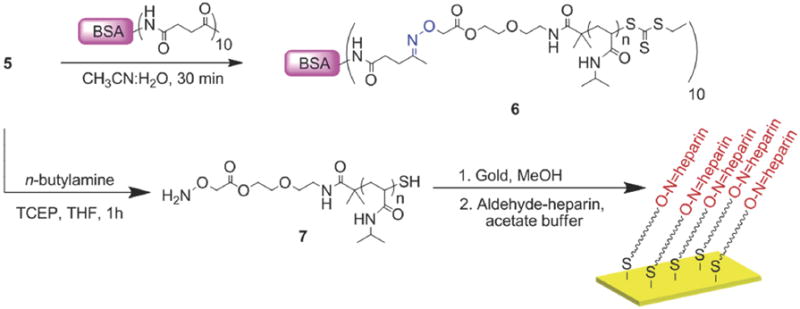
Bioconjugation and surface modification
Figure 6.
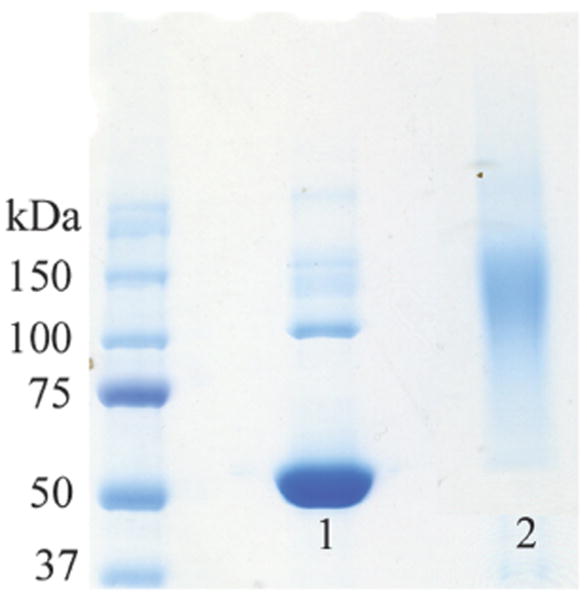
SDS-PAGE of pNIPAAm-BSA conjugate. Lane 1: Unmodified BSA, Lane 2: BSA-pNIPAAm following thermal precipitation.
Surface Modification with pNIPAAm 5
We next proceeded to prepare surfaces of pNIPAAm. “Smart” polymers retain thermosensitivity when immobilized on surfaces. This is important for a variety of applications. For example, control of cell-surface interactions and absorption and desorption of biomolecules with “smart” surfaces is known.53,54 To determine if the aminooxy pNIPAAm could be immobilized on surfaces, 4 was treated with TFA to reveal the aminooxy group following the same procedure described above. The solvent was removed and pNIPAAm 5 was redissolved in THF and treated with an excess of n-butylamine to remove the trithiocarbonate end-group. This was performed in the presence of TCEP to prevent disulfide formation (Scheme 3). The reaction was stopped after one hour, and pNIPAAm 7 was recovered as a white solid after precipitation from cold hexanes. The polymer was redissolved in dry methanol and incubated with a freshly cleaned gold surface. Thiols covalently attach to gold and the omega thiol resulting from reduction of the trithiocarbonate served as an anchoring point. The surface was characterized by IR spectroscopy (Figure 7b). A small band was observed around 3289 cm-1 which was attributed to the N-H stretch of the amide. The C-H stretches (~2973 cm-1) of the sp3 carbons, the C=O stretch (1659 cm-1), and CNH vibration (1540 cm-1) of the amide were also observed (Figure 7b). The IR of the surface-bound polymer was similar to the IR of the free polymer confirming immobilization of pNIPAAm 7 (Figure 7a vs. 7b). Ellipsometry measurements indicated in a thickness of 1.17 ± 0.04 nm, which suggested a low surface coverage. This is not unexpected with a “grafted to” approach.
Heparin surfaces
We next functionalized the gold surfaces with heparin (Scheme 3). Heparin is a sulfated polysaccharide involved in many biological processes. It is a well known anticoagulant drug that is immobilized on surfaces to prevent blood clotting in biomedical devices.55,56 Due to its interaction with a variety of proteins, this polysaccharide has also been studied as a drug delivery system and for protein storage and stabilization; it can also be used to immobilize growth factors for cell adhesion on surfaces.57-59 Heparin is a heterogeneous mixture of sulfated and unsulfated disaccharide units.56 Thus, to bind heparin to the pNIPAAm film on gold, the polysaccharide was incubated with NaIO4. Sodium periodate was utilized to oxidize the vicinal unsulfated diols to aldehydes for reaction with the aminooxy group of the polymer using a literature procedure.13 Confirmation of the reaction was obtained utilizing Purpald.48 The dye reacts only with aldehydes to produce a characteristic purple color, which was observed both by eye and by UV-vis (λmax = 554 nm). To corroborate these results, the IR spectrum of unmodified and modified heparin were compared. The former had characteristic peaks corresponding to the C=O stretch of the amide of the glucosamine unit (1621 cm-1), the S=O stretch of the sulfate groups (1230 cm-1), and the C-O vibrations of the alcohols (1062 cm-1). The IR spectrum of the modified heparin was similar but revealed a new peak at 1747 cm-1 corresponding to the C=O stretch of the aldehyde. These data confirmed that the heparin had been modified as desired.
Modified heparin was placed on a pNIPAAm surface, and following incubation for 12 hours, characterized by IR (Figure 7c). The pNIPAAm bands were still visible with the N-H stretch of the amide (a weak band at 3284 cm-1), the C=O stretch of the amide (1644 cm-1) which overlapped with the amide of the heparin, and the CNH vibration at 1534 cm-1. In addition, the aldehyde group vibration (1736 cm-1) of the modified heparin, sulfate stretch (1219 cm-1) and CO stretch at 1138 cm-1 were visible. It is likely that the C=O stretch of the aldehyde was observed following conjugation because there were a large excess of aldehydes per heparin chain compared to a single aminooxy group on each polymer chain. The C=N stretch (around 1650 cm-1) of the oxime bond was not observed; it should overlap with the C=O stretch of the carbonyl groups and is also typically weak. In addition, the low content of aminooxy group (one per polymer chain) would result in a weak, undetectable intensity.
To verify that heparin was immobilized on the surface through a covalent bond instead of surface adsorption, a control study was performed. The polymer surface was fabricated by treatment of Boc-protected aminooxy polymer 4 with n-butylamine followed by incubation with a gold surface following the same conditions described above. After the addition of aldehyde-heparin, the mixture was placed on the surface, and the IR spectrum was taken. As expected, the N-H (3246 cm-1) and C=O (1639 cm-1) stretches of pNIPAAm were observed, but the aldehyde C=O stretch was not observed. This confirmed that the heparin does not physically adsorb to the surface, but requires free aminooxy groups for reaction. This also suggests that heparin modification is via oxime bonds. Together these results demonstrated that functional pNIPAAm surfaces are obtained and that heparin is readily immobilized on the surface by oxime chemistry. We anticipate that a wide variety of polysaccharides and polymers will be amenable to this methodology.
Conclusions
A Boc-protected aminooxy trithiocarbonate CTA was synthesized for the fabrication of end-group functionalized polymers. pNIPAAm was prepared by RAFT polymerization and the polymerization was controlled. UV-vis and chain extension studies confirmed the presence of the trithiocarbonate at the omega chain of the polymers. Conjugation to a levulinyl lysine modified BSA demonstrated that protein-polymer conjugates could be prepared using this procedure. The ability to thermally precipitate the polymer while attached to the protein displayed that the thermosensitivity of the polymer was conferred to the protein. This polymer could therefore be used to form thermo-responsive bioconjugates with other proteins modified with aldehydes or ketones. Modification of a gold surface with aminooxy-pNIPAAm was shown, and immobilization of aldehyde-heparin on the pNIPAAm film proved to be successful. This technique could be applied to the stabilization and storage of growth factors on surfaces or used to coat medical devices were the anticoagulant activity of heparin is needed such as for renal dialysis instruments.
Figure 4.

Chain extensions studies ([NIPAAm]:[4]:[AIBN] = 200:2:1). GPC chromatogram of 4 (—) and chain extension pNIPAAm product (- -).
Acknowledgments
This work was supported by the NSF (CAREER CHE-0695793). VVD thanks the National Institutes of Health (Grant # 31 GM077086-02) for a predoctoral fellowship. ZPT thanks the NIH-sponsored Biotechnology Training in Biomedical Sciences and Engineering Program for funding. HDM thanks the Alfred P. Sloan foundation for additional funding. The authors appreciate Dr. Karina L. Heredia for the synthesis of the trithiocarbonate alcohol 2, and Caitlin Stevens for help with the Purpald assay. The authors are grateful to Professor Bruce Dunn for granting access to his surface IR and ellipsometer, and Steven Jonas for help with the instruments.
References
- 1.Heredia KL, Maynard HD. Org Biomol Chem. 2007;5:45–53. doi: 10.1039/b612355d. [DOI] [PubMed] [Google Scholar]
- 2.Lemieux GA, Bertozzi CR. Trends Biotechnol. 1998;16:506–13. doi: 10.1016/s0167-7799(98)01230-x. [DOI] [PubMed] [Google Scholar]
- 3.Duncan R. Nat Rev Drug Discov. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 4.Duncan R. Nat Rev Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- 5.Kochendoerfer GG, Chen SY, Mao F, Cressman S, Traviglia S, Shao HY, Hunter CL, Low DW, Cagle EN, Carnevali M, Gueriguian V, Keogh PJ, Porter H, Stratton SM, Wiedeke MC, Wilken J, Tang J, Levy JJ, Miranda LP, Crnogorac MM, Kalbag S, Botti P, Schindler-Horvat J, Savatski L, Adamson JW, Kung A, Kent SBH, Bradburne JA. Science. 2003;299:884–887. doi: 10.1126/science.1079085. [DOI] [PubMed] [Google Scholar]
- 6.Heredia KL, Tolstyka ZP, Maynard HD. Macromolecules. 2007;40:4772–4779. [Google Scholar]
- 7.Shao H, Crnogorac MM, Kong T, Chen SY, Williams JM, Tack JM, Gueriguian V, Cagle EN, Carnevali M, Tumelty D, Paliard X, Miranda LP, Bradburne JA, Kochendoerfer GG. J Am Chem Soc. 2005;127:1350–1. doi: 10.1021/ja043096w. [DOI] [PubMed] [Google Scholar]
- 8.Dixon HBF. Biochem J. 1964;92:661–&. doi: 10.1042/bj0920661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilmore JM, Scheck RA, Esser-Kahn AP, Joshi NS, Francis MB. Angew Chem Int Ed. 2006;45:5307–5311. doi: 10.1002/anie.200600368. [DOI] [PubMed] [Google Scholar]
- 10.Wu PG, Brand L. Methods Enzymol. 1997;278:321–330. doi: 10.1016/s0076-6879(97)78017-0. [DOI] [PubMed] [Google Scholar]
- 11.Chin JW, Cropp TA, Anderson JC, Mukherji M, Zhang ZW, Schultz PG. Science. 2003;301:964–967. doi: 10.1126/science.1084772. [DOI] [PubMed] [Google Scholar]
- 12.Connor RE, Piatkov K, Varshavsky A, Tirrell DA. Chembiochem. 2008;9:366–9. doi: 10.1002/cbic.200700605. [DOI] [PubMed] [Google Scholar]
- 13.Yoshitomi Y, Nakanishi H, Kusano Y, Munesue S, Oguri K, Tatematsu M, Yamashina I, Okayama M. Cancer Lett. 2004;207:165–174. doi: 10.1016/j.canlet.2003.11.037. [DOI] [PubMed] [Google Scholar]
- 14.Christman KL, Requa MV, Enriquez-Rios VD, Ward SC, Bradley KA, Turner KL, Maynard HD. Langmuir. 2006;22:7444–7450. doi: 10.1021/la0608213. [DOI] [PubMed] [Google Scholar]
- 15.Esser-Kahn AP, Francis MB. Angew Chem Int Ed. 2008;47:3751–4. doi: 10.1002/anie.200705564. [DOI] [PubMed] [Google Scholar]
- 16.Zabransky J, Houska M, Plichta Z, Kalal J. Makromol Chem. 1985;186:231–245. [Google Scholar]
- 17.Godwin A, Hartenstein M, Muller AHE, Brocchini S. Angew Chem Int Ed. 2001;40:594–597. doi: 10.1002/1521-3773(20010202)40:3<594::AID-ANIE594>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 18.Kopping JT, Tolstyka ZP, Maynard HD. Macromolecules. 2007;40:8593–8599. [Google Scholar]
- 19.Tao L, Mantovani G, Lecolley F, Haddleton DM. J Am Chem Soc. 2004;126:13220–13221. doi: 10.1021/ja0456454. [DOI] [PubMed] [Google Scholar]
- 20.Cheng C, Sun G, Khoshdel E, Wooley KL. J Am Chem Soc. 2007;129:10086–10087. doi: 10.1021/ja073541y. [DOI] [PubMed] [Google Scholar]
- 21.Hwang JY, Li RC, Maynard HD. J Controlled Release. 2007;122:279–286. doi: 10.1016/j.jconrel.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 22.Li RC, Broyer RM, Maynard HD. J Polym Sci Part A: Polym Chem. 2006;44:5004–5013. [Google Scholar]
- 23.Li RC, Hwang J, Maynard HD. Chem Commun. 2007:3631–3633. doi: 10.1039/b709304g. [DOI] [PubMed] [Google Scholar]
- 24.Mittal A, Sivaram S, Baskaran D. Macromolecules. 2006;39:5555–5558. [Google Scholar]
- 25.Sun GR, Cheng C, Wooley KL. Macromolecules. 2007;40:793–795. doi: 10.1021/ma062592x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moad G, Rizzardo E, Thang SH. Aust J Chem. 2006;59:669–692. [Google Scholar]
- 27.Barner L, Davis TP, Stenzel MH, Barner-Kowollik C. Macromol Rapid Commun. 2007;28:539–559. [Google Scholar]
- 28.Perrier S, Takolpuckdee P. J Polym Sci Part A: Polym Chem. 2005;43:5347–5393. [Google Scholar]
- 29.Liu J, Bulmus V, Barner-Kowollik C, Stenzel MH, Davis TP. Macromol Rapid Commun. 2007;28:305–314. [Google Scholar]
- 30.Bathfield M, D’Agosto F, Spitz R, Charreyre MT, Delair T. J Am Chem Soc. 2006;128:2546–2547. doi: 10.1021/ja057481c. [DOI] [PubMed] [Google Scholar]
- 31.Zhao YL, Perrier S. Chem Commun. 2007:4294–4296. doi: 10.1039/b708293b. [DOI] [PubMed] [Google Scholar]
- 32.Hong CY, Pan CY. Macromolecules. 2006;39:3517–3524. [Google Scholar]
- 33.You YZ, Manickam DS, Zhou QH, Oupicky D. Biomacromolecules. 2007;8:2038–2044. doi: 10.1021/bm0702049. [DOI] [PubMed] [Google Scholar]
- 34.Perrier S, Takolpuckdee P, Mars CA. Macromolecules. 2005;38:2033–2036. [Google Scholar]
- 35.Boyer C, Liu J, Bulmus V, Davis TP, Barner-Kowollik C, Stenzel MH. Macromolecules. 2008;41:5641–5650. [Google Scholar]
- 36.Heredia KL, Grover GN, Tao L, Maynard HD. Macromolecules. 2009;42:2360–2367. doi: 10.1021/ma8022712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heredia KL, Nguyen TH, Chang CW, Bulmus V, Davis TP, Maynard HD. Chem Commun. 2008:3245–3247. doi: 10.1039/b804812f. [DOI] [PubMed] [Google Scholar]
- 38.Tao L, Kaddis CS, Loo RRO, Grover GN, Loo JA, Maynard HD. Chem Commun. 2009:2148–2150. doi: 10.1039/b822799c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qiu XP, Winnik FM. Macromol Rapid Commun. 2006;27:1648–1653. [Google Scholar]
- 40.Narain R, Gonzales M, Hoffman AS, Stayton PS, Krishnan KM. Langmuir. 2007;23:6299–6304. doi: 10.1021/la700268g. [DOI] [PubMed] [Google Scholar]
- 41.Roth PJ, Kessler D, Zentel R, Theato P. Macromolecules. 2008;41:8316–8319. [Google Scholar]
- 42.Henry SM, Convertine AJ, Benoit DSW, Hoffman AS, Stayton PS. Bioconjugate Chem. 2009;20:1122–1128. doi: 10.1021/bc800426d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.York AW, Scales CW, Huang FQ, McCormick CL. Biomacromolecules. 2007;8:2337–2341. doi: 10.1021/bm700514q. [DOI] [PubMed] [Google Scholar]
- 44.Christman KL, Vazquez-Dorbatt V, Schopf E, Kolodziej CM, Li RC, Broyer RM, Chen Y, Maynard HD. J Am Chem Soc. 2008;130:16585–16591. doi: 10.1021/ja803676r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sumerlin BS, Lowe AB, Stroud PA, Zhang P, Urban MW, McCormick CL. Langmuir. 2003;19:5559–5562. [Google Scholar]
- 46.Lai JT, Filla D, Shea R. Macromolecules. 2002;35:6754–6756. [Google Scholar]
- 47.Convertine AJ, Lokitz BS, Vasileva Y, Myrick LJ, Scales CW, Lowe AB, McCormick CL. Macromolecules. 2006;39:1724–1730. [Google Scholar]
- 48.Jacobsen NW, Dickinson RG. Anal Chem. 1974;46:298–299. [Google Scholar]
- 49.Hoffman AS, Stayton PS. Macromol Symp. 2004;207:139–151. [Google Scholar]
- 50.Alarcon CDH, Pennadam S, Alexander C. Chem Soc Rev. 2005;34:276–285. doi: 10.1039/b406727d. [DOI] [PubMed] [Google Scholar]
- 51.Hoffman AS. Clin Chem. 2000;46:1478–1486. [PubMed] [Google Scholar]
- 52.Heredia KL, Bontempo D, Ly T, Byers JT, Halstenberg S, Maynard HD. J Am Chem Soc. 2005;127:16955–16960. doi: 10.1021/ja054482w. [DOI] [PubMed] [Google Scholar]
- 53.Mendes P. Chem Soc Rev. 20008;37:2412–2429. [Google Scholar]
- 54.Da Silva RMP, Mano JF, L RR. Trends Biotechnol. 2007;25:577–583. doi: 10.1016/j.tibtech.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 55.Murugesan S, Xie J, Linhardt RJ. Curr Top Med Chem. 2008;8:80–100. doi: 10.2174/156802608783378891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Capila I, Linhardt RJ. Angew Chem Int Ed. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 57.Edelman ER, Mathiowitz E, Langer R, Klagsbrun M. Biomaterials. 1991;12:619–626. doi: 10.1016/0142-9612(91)90107-l. [DOI] [PubMed] [Google Scholar]
- 58.Schroeder-Tefft JA, Bentz H, Estridge TD. J Controlled Release. 1997;49:291–298. [Google Scholar]
- 59.Sakiyama-Elbert SE, Hubbell JA. J Controlled Release. 2000;65:389–402. doi: 10.1016/s0168-3659(99)00221-7. [DOI] [PubMed] [Google Scholar]