

Abstract
The 14–18 amino acid kinesin neck linker domain links the core motor to the coiled-coil dimerization domain. One puzzle is that the neck linker appears too short for the 4 nm distance each linker must stretch to enable an 8 nm step – when modeled as an entropic spring, high inter-head forces are predicted when both heads are bound to the microtubule. We addressed this by analyzing the length of the neck linker across different kinesin families and using molecular dynamics simulations to model the extensibility of Kinesin-1 and Kinesin-2 neck linkers. The force-extension profile from molecular dynamics agrees with the Worm Like Chain (WLC) model for Kinesin-1 and supports the puzzling prediction that extending the neck linker 4 nm requires forces multiple times the motor stall force. Despite being 3 amino acids longer, simulations suggest that extending the Kinesin-2 neck linker by 4 nm requires similarly high forces. A possible resolution to this dilemma is that helix α-6 may unwind to enable the two-head bound state. Finally, simulations suggest that cis/trans isomerization of a conserved proline residue in Kinesin-2 accounts for the differing predictions of molecular dynamics and the WLC model, and may contribute to motor regulation in vivo.
Keywords: Molecular Biomechanics, Entropic Spring, Bioinformatics, Worm Like Chain, Molecular Motor, Microtubule
INTRODUCTION
Kinesin motor proteins, which transport intracellular cargo along microtubules, provide an excellent model for studying molecular biomechanics and investigating the interplay of mechanical forces and biochemistry at the level of a single protein molecule. Kinesins can be grouped into 14 different families based on sequence similarity.1,2 Within each family, both the overall structural organization and for the most part the cellular roles are consistent. However, motors in different families possess significantly different motor characteristics – they move at different speeds, in different directions and for varying distances along microtubules. In some cases, the structural basis of these differences have been worked out,3 but for the most part the relation of specific sequence differences to resulting motor function are poorly understood.
Kinesins exist as monomers, dimers, and tetramers, and the motor domain can be at the N-terminus, the C-terminus, or internal.1,2 Here, we focus on N-terminal kinesins, which walk to the plus-ends of microtubules, and include kinesin families 1, 2, 3, 5 and 7. All of these motors contain four major components: a motor domain (or head), a flexible neck linker, a coiled-coil stalk, and a cargo binding tail domain. Kinesin-1, also called conventional kinesin, was the first kinesin to be discovered and the one that has received the most intensive investigation. It is processive, meaning that it takes on the order of 100 steps per interaction with a microtubule,4,5 and this processivity has been shown to rely on coordination between the chemomechanical cycles of the two head domains, such that at least one head remains bound to the microtubule at all times.6 Because understanding the basic force-generating transition that underlies kinesin mechanics requires understanding the interplay between the two head domains, understanding interdomain coordination in kinesins has been an area of intense research.7
In studying kinesin’s force generating mechanism, the neck linker domain has emerged as a key structural feature. The neck linker is a region of roughly 14–18 amino acids that links α-6, the last alpha helix in the core motor domain to α-7, the first alpha helix of the coiled-coil dimerization domain (Figure 1). A body of data support the idea that the principal force-generating transition in kinesin is a transition of the neck linker from a disordered or random coil state to an ordered beta-sheet conformation stabilized by interactions with the core motor domain and the N-terminal extension.8–11 In addition to its role in the force-generating conformational change, an equally important role of the neck linker is to transmit mechanical tension between the two head domains. Virtually all mechanisms that have been put forward to explain how kinesin can take multiple steps along the microtubule without dissociating involve mechanical tension transmitted between the two head domains.7,12–16 For instance, the “rear-head gating” mechanism holds that as a kinesin walks along a microtubule, the trailing head remains bound until the leading head attaches and produces forward-directed strain that pulls the trailing head off of the microtubule.14,17 The “front-head gating” mechanism holds that when both kinesin heads are bound to a microtubule, rearward strain in the leading head prevents ATP from binding, pausing the motor’s hydrolysis cycle until the trailing head detaches from the microtubule.12,13 Consistent with these ideas, artificially extending the neck linker of Kinesin-1 by introducing extra amino acids into the C-terminus region of the neck linker leads to diminished processivity, slower velocities, or both.15,16,18
Figure 1. Kinesin neck linker structure and mechanism.
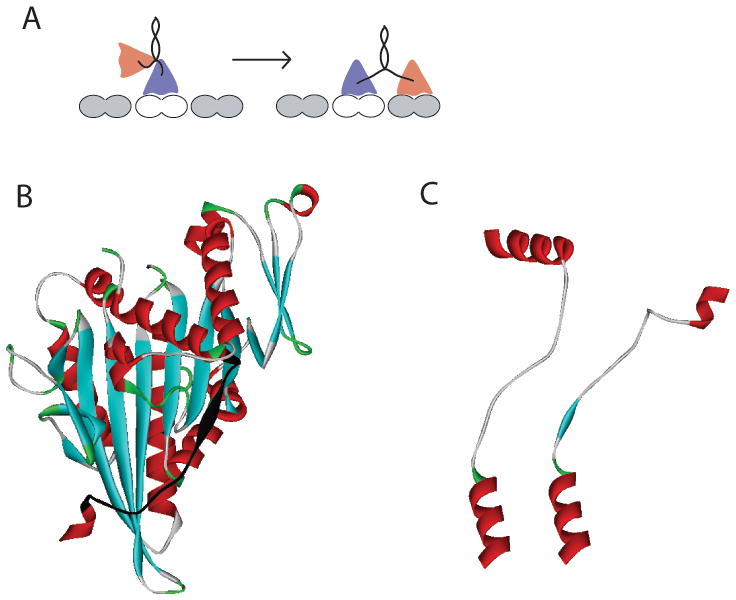
A: Schematic of kinesin stepping cycle. As the motor walks along the microtubule, the neck linker domains that connect each head to their shared coiled-coil transition between disordered states when only one head is bound, to ordered and stretched conformations when both heads are bound to the microtubule. B: Kinesin head structure (from rat kinesin dimer PDB:3KIN) showing the neck linker domain highlighted in black. C: Comparison of docked neck linkers from Kinesin-1 (rat kinesin dimer PDB:3KIN) and Kinesin-2 (human KIF3B, PDB:3B6U) crystal structures. In Kinesin-2, a proline at position 13 in the neck linker causes a kink. Structures were aligned using α-6 at bottom (last helix in the core head domain).
One surprising feature of the kinesin neck linker is that it is so short (14 amino acids in Kinesin-19,18). Kinesin heads bind to sequential tubulin subunits along a microtubule, and therefore their spacing matches the 8 nm tubulin dimer spacing.19,20 Structural investigations of microtubule-bound kinesin heads suggest that there are only subtle conformational changes in the core motor domain in different nucleotide states.21,22 This means that when both heads of kinesin are bound to the microtubule the neck linkers most likely emanate from each bound kinesin head at approximately the same point. Finally, cysteine crosslinking experiments show that preventing unwinding of the coiled-coil domain does not block motility,23 arguing that the α-7 coiled-coil remains dimerized when both heads are bound to the microtubule. Hence, when both kinesin heads are bound to the microtubule 8 nm apart, each 14-amino acid neck linker needs to stretch a distance of 4 nm. Using molecular modeling, it was shown that it is possible for the neck linker sequence to stretch that distance.24 However, by modeling the unstructured neck linker as an entropic spring using the equation for a Worm Like Chain (WLC), it has been argued that the force required to stretch the neck linkers to that degree requires forces of between two and six times the ~6 pN stall force of conventional kinesin.18,25 These high forces seem improbable, but are impossible to rule out as there are no experiments to date that have measured the force-extension properties of the kinesin neck linker.
The first goal of the current work is to compare neck linker sequences between different kinesin families and correlate the neck linker length with motor processivity. We recently compared the processivity of Drosophila Kinesin-1 to mouse KIF3A/B, a member of the Kinesin-2 family.18 Kinesin-2 motors have a longer neck linker domain (17 amino acids compared to 14 in Kinesin-1), and were found to take fourfold fewer steps along the microtubule per interaction. This reduced processivity suggests that inter-head tension is crucial for the coordination between the heads and that extending the neck linker, which results in greater compliance between the two heads, disrupts this communication. Consistent with this idea, a Kinesin-1 mutant containing a neck linker domain that was extended by three amino acids was found to be five-fold less processive than conventional kinesin.18 This neck linker extension result is consistent with results from Hackney15 but it contrasts with similar experiments by Yildiz et al.16
The second goal of the current work is to investigate the mechanical characteristics of the kinesin neck linker using molecular dynamics simulations. The WLC model has been shown to faithfully fit the force-extension characteristics of unfolded proteins like titin in atomic force microscopy experiments.26–28 The assumption has been that it also applies to shorter polypeptides and that the force-extension characteristics are independent of sequence, but these assumptions have not been tested. Molecular dynamics simulations provide a valuable window into these questions and a good test of the validity of the WLC model for short peptide sequences. Here we find that the force-extension profile from molecular dynamics simulations is consistent with the WLC model for Kinesin-1, strengthening the prediction that extending the Kinesin-1 neck linker 4 nm requires forces multiple times the motor stall force. Furthermore, the force-extension properties are sequence dependent and a conserved proline in the Kinesin-2 neck linker effectively shortens the neck linker in that motor, necessitating similary high forces to extend the linker.
METHODS
Bioinformatics Tools
The bioinformatics approach used to identify the kinesin neck linker domain across the superfamily was broken down into three stages. First, using the AlignX feature of the VectorNTI® Advance 10.3 Suite, primary structures were aligned according to the conserved LAGSE, FAYGQT, and α-6 regions of each motor. The primary sequences used were obtained from the RCSB Protein Data Bank in .pdb format and motor proteins of a given family were compared in a single alignment. The second step of the alignment was to define α-7, the start of the coiled-coil domain, using the coiled-coil prediction programs PCOILS, Marcoil, PairCoil2, and PSIPRED. All four of these programs, which are freely available online, predict the tendency for a given amino acid sequence to take on a coiled-coil conformation. Because each program has been shown to exhibit certain biases in their assignment of coiled coils,29 sequences were analyzed with all four approaches.
In PCOILS,30–32 Position Specific Iterated BLAST runs were used to quantify the coiled-coil probability of specific sequences. The MTIDK matrix was employed, which contains a subset of data drawn from known crystal structures, along with a weighting scheme to avoid biases from charge-rich sequences. Marcoil uses a windowless prediction method that incorporates a probability distribution function.33 Posterior probabilities were calculated at each amino acid and those probabilities calculated to be above the default threshold were identified as a being in a coiled-coil domain. Thresholds of 1 %, 10 %, 50 %, 90 %, and 99 % stringency were used, resulting in a range of liberal to conservative coiled-coil predictions. PairCoil234,35 predicts the parallel coiled-coil folds from primary protein sequences using pair-wise residue probabilities with the Paircoil algorithm, and an updated coiled-coil database. Probabilities, or p-scores, are assigned to each residue, with a lower p-score signifying a greater likelihood of coiled-coil formation. The default p-score of 0.025 and a 21 amino acid window were used. Finally, the PSIPRED secondary structure prediction method incorporates two feed-forward neural networks that analyze outputs obtained from Position Specific Iterated BLAST runs.36–40 To reduce the false positive rate, the option of masking low complexity regions was enabled.
Results from this suite of bioinformatics tools were compared to identify the start of the α-7 coiled-coil. In general, PCOILS provided the most liberal estimates of coiled-coil formation, Multicoil was the most conservative and Marcoil and PSPIRED were intermediate in their predictions. To resolve differences between the software predictions and to most precisely identify the start of the α-7 coiled-coil, the final stage of sequence analysis was to manually identify the start of the first heptad repeat. This was done by identifying the hydrophobic a and d residues in the adjacent heptad repeats (a–g) and defining the first a or d residue in the first heptad repeat as the start of the coiled-coil dimerization domain.
Molecular Dynamics Tools
Neck linker force-extension curves were constructed using the constraint and force clamp pulling modes in GROMACS 3.3.3 & 4.0.3, respectively. The simulations all used 2 fs timesteps, and were run using both a single dual-core processing node on a computer cluster and a dual-core desktop computer. A water/peptide system was constructed using the spc216 flexible water model, and a minimum peptide-to-edge distance of 1.5 nm and periodic boundary conditions were used to limit edge effects. Counter-ions were added to achieve a charge-neutral system. A preliminary solvation simulation of 10 ps was run, incorporating both Berendsen temperature-coupling and Parrinello-Rahman pressure-coupling schemes. Following solvation, a 10 ps equilibration simulation was run. After energy minimization, a set of neck linker structures at different end-to-end lengths was produced by first pushing the ends together as close as possible without steric collisions, and then pulling the ends out to their full contour length. These structures were then used for the constraint mode simulations.
In the constraint mode, the distance between the first and last alpha carbons of the peptide remained fixed within a tolerance of 0.001 nm, and the force required to restrain the peptide in that conformation was calculated for each time step over the 160 ps simulation. At each end-to-end distance, an average force was calculated from the 80,000 time steps. This procedure was repeated for the same peptide constrained at varying end-to-end distances to generate a corresponding force-extension profile. For the force clamp simulations, instead of fixing the distance between the terminal alpha-carbons, these atoms were permitted to fluctuate and a force was applied in a direction along the vector between the two terminal alpha carbons. An end-to-end distance was calculated for each time step of the 160 ps simulation and averaged, and the procedure was repeated with forces ranging from a 50 pN compressive force to a 200 pN force pulling force. Both the constraint and force-clamp methods used the Berendsen temperature-coupling and Parrinello-Rahman pressure-coupling schemes. Force-extension curves were generated using OriginPro 8.
RESULTS AND DISCUSSION
Analysis of neck linker lengths
Despite the importance of the kinesin neck linker in force production and head-head coordination, there has not been a systematic analysis of the neck linker lengths across different kinesin families. Our first goal was to define the number of amino acids in the Kinesin-1 neck linker domain by using existing crystal structures to define the end of helix α-6 and the start of helix α-7. We then used sequence alignments and secondary structure predictions to predict the neck linker length in kinesin families 2, 3, 5 and 7. Sequence alignments for all five families are presented in Figure 2, and the consensus neck linker lengths and sequences are presented in Table 1.
Figure 2. Sequence alignment for five N-terminal kinesin families.

Predicted neck linker regions and predicted heptad repeats are shown. The last three motors in the Kinesin-1 family are fungal kinesins that contain a proline as the first residue of the coiled-coil. Because prolines are known to be found at the N-termini of alpha helicies and act as “caps” to stabilize the helix,62 we predict the neck linker to be 14 amino acids in these fungal Kinesin-1 motors.
Table 1.
| Family | Characteristic Length (# of Amino Acids) | Characteristic Sequence | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Kinesin 1 | 14 | α6 | → | K | T | I | K | N | T | V | S | V | N | L | E | L | T | → | α7 | ||||
| Kinesin 2 | 17 | α6 | → | K | N | I | K | N | K | P | R | V | N | E | D | P | K | D | A | L | → | α7 | |
| Kinesin 3 | 17 | α6 | → | K | Q | I | _ | C | N | A | V | I | N | E | D | P | N | A | K | L | → | α7 | |
| Kinesin 5 | 18 | α6 | → | K | N | I | _ | N | K | P | _ | V | N | Q | K | L | _ | K | K | _ | L | → | α7 |
| Kinesin 7 | 18 | α6 | → | K | _ | _ | _ | N | _ | P | _ | V | N | E | _ | _ | T | D | _ | A | L | → | α7 |
Although a number of Kinesin-1 crystal structures have been solved, there is not universal agreement in the literature regarding where helix α-6, the last helix in the core motor domain ends and where the flexible neck linker domain begins. As seen in Figure 2, there is a conserved arginine-alanine-lysine (RAK) sequence at this interface. In the human Kinesin-1 monomer structure in ADP (PDB:1BG2),41 α-6 ends just before the R, while in the rat Kinesin-1 monomer structure in ADP (PDB:2KIN)42 and the Neurospora kinesin monomer in ADP (NcKin PDB:1GOJ),43 α-6 ends two residues later, after the A. Confusingly, in the dimeric rat kinesin crystal structure (PDB:3KIN), α-6 ends after the A in one head and after the subsequent K in the second head.44
To help resolve this uncertainty regarding the start of the neck linker domain, we examined existing crystal structures from other kinesin families. In the Kinesin-2 family, α-6 of both human KIF3B (PDB:3B6U) and Giardia KIF3A (PDB:GiKIN2a)45 end in RA, and in the Kinesin-5 family, α-6 of human Eg5 (PDB:1II6)46 also ends in RA. The Kinesin-3 family appears to be the exception – α-6 of KIF1A ends with R in the ADP structure (PDB:1I5S) and includes RAK and the subsequent Q in the AMPPCP structure (PDB:1I6I).21 However, this KIF1A construct is actually a chimaera in which most of the native Kinesin-3 neck linker is replaced by the corresponding sequence from Kinesin-1, and it is possible that this result is sequence dependent. Hence, based on the majority of the existing kinesin crystal structures, we define the end of the α-6 helix as the conserved RA and the start of the neck linker domain as the conserved K in the RAK tripeptide (Figure 2). Across different kinesin families, α-6 appears to take on a similar structure, suggesting that the start of the neck linker is conserved across kinesin families, but it cannot be ruled out that the end of α-6 unfolds either in different nucleotide states or under tension.
The next step in defining the neck linker length was to define the start of the α-7 coiled-coil domain. Because this dimerization domain is present in the rat dimeric Kinesin-1 crystal structure, and because the predicted heptad repeats in α-7 are well conserved across the Kinesin-1 family, the start of α-7 can be assigned with high confidence. From this analysis, the Kinesin-1 neck linker is 14 amino acids (Figure 2 and Table 1). Much of the secondary structure analysis we carried out involved defining the start of the α-7 coiled-coil domain in the other N-terminal kinesin families. Motors in the Kinesin-2 family transport cargo along axonemal microtubules in cilia and flagella, and they also carry out a number of transport tasks along cytoplasmic microtubules.47 In our previous work, we compared crystal structures of Kinesin-1 and Kinesin-2 and concluded that the Kinesin-2 neck linker domain is 17 amino acids, making it three residues longer than most Kinesin-1 motors.18 Interestingly, most Kinesin-2 motors contain two proline residues in their neck domain. In the crystal structure of human KIF3B (PDB:3B6U), which includes the entire neck linker domain, the first proline is in the “straight” trans conformation, while the second proline is in the “kinked” cis conformation (see Figure 1C). In the molecular dynamics simulations below, we compare the mechanical properties of Kinesin-1 and Kinesin-2 neck linker domains and analyze the dynamics of this proline residue.
Kinesin-3 motors are often referred to as monomeric kinesins48 and so defining the α-7 coiled-coil domain is debatable, but as they have been shown to move as dimers in vivo49 α-7 is clearly a dimerization domain. The Kinesin-3 neck linker has a consensus length of 17 amino acids, and like Kinesin-2 and fungal Kinesin-1 motors also includes a proline residue, but because it is not present in any existing Kinesin-3 crystal structures, its conformation is unknown.
The Kinesin-5 and Kinesin-8 families are predicted to have the longest neck linker domains at 18 amino acids. Motors in the Kinesin-5 family, which are responsible for sliding antiparallel microtubules during mitosis,50 have been shown to be minimally processive in vitro.51,52 This minimal processivity is consistent with the hypothesis that longer neck linkers enhance the mechanical compliance between the two motor domains and diminish head-head coordination. However, the consensus neck linker of Kinesin-7 (CENP-E) is also 18 amino acids and these mitotic motors have been shown to have comparable processivity to Kinesin-1 motors in vitro.53,54 Interestingly, though, the first two heptad repeats in the Kinesin-7 coiled-coil are highly positively charged, and it has been shown that positive charge in this region can enhance processivity due to electrostatic tethering.55 Consistent with this, Yardimci et al. showed that single-molecule movement of Kinesin-7 along microtubules has diffusive components in addition to slow plus-end directed motility.54 This result suggests that Kinesin-7 processivity is enhanced by mechanisms beyond interdomain coordination. Hence, while there is not a clear rule, there is clearly a trend that kinesin families with longer neck linkers are less processive.
Modeling the neck linker force-extension curve
While sequence analysis can help define the length of the kinesin neck linker domain across different families, it does not address how sequence differences may alter the mechanical properties of these flexible domains. From experiments on Kinesin-1 motors there is evidence for structural changes in the neck linker in different nucleotide states9 and in different microtubule binding states.14 but to date experimental insights into kinesin neck linker properties have needed to be extrapolated from the behavior of the entire head domain such as in optical trapping studies.16 Molecular dynamics (MD) provides an approach for simulating, at high resolution, the force-extension properties of the kinesin neck linker domain, and so we modeled the force-extension properties of Kinesin-1 and Kinesin-2 neck linker domains.
The flexibility of polypeptide chains derives from the minimally constrained rotation around the N-Cα (phi angle) and Cα-C (psi angle) bonds in each amino acid.56 Most peptides are in the trans conformation, defined as the relationship of the alpha carbons in adjacent amino acids around the axis of their shared peptide bond. Proline is unique in that it is highly constrained due to its cyclic structure and can readily take on either a cis (kinked) or trans (straight) conformation.56 Unstructured polypeptide chains, like DNA and RNA, are often described as “entropic springs” – flexible chains that can take on many different conformations.57 Their elasticity derives from the fact that pulling them taut reduces the possible number of conformational states (reducing disorder) and thus requires energy input. Of the two models most widely used to describe biopolymers, the Freely Jointed Chain (FJC) and the Worm Like Chain (WLC), the WLC is the most widely used model for predicting the force-extension properties of polypeptide chains.26–28 The WLC model describes the force (F) required to extend a polymer with a given contour length (Lc) and persistence length (Lp) a given end-to-end distance (x):
Here kBT is Boltzman’s constant times the absolute temperature (=4.1 pN-nm). The contour length of a polypeptide is equal to the number of amino acids multiplied by the distance along the chain per amino acid. The distance per amino acid is usually taken as 0.38 nm, as this is the length of one amino acid from early crystal structures.56 However, because each amino acid is linked to each nearest neighbor at an angle,58 the length of the extended chain will be necessarily be shorter than a sum of the subunit lengths, and arguably a distance of 0.364 nm per amino acid should be used. For consistency with previous work and because this correction alters the results by less than 5%, we use 0.38 nm per amino acid. The persistence length of polypeptide chains, a measure of their flexibility, has been estimated by fitting the WLC equation to force-extension profiles of unfolded titin, and found to be 0.4 nm28 or between 0.5 and 2.4 nm, depending on sequence and ionic strength.59 We choose a persistence length of 0.5 nm for our fitting.
One of the weaknesses of using the WLC model to fit polypeptides is that the persistence length is equal to the length of ~2 amino acids, and it is known that the flexibility of polypeptides results from rotations around reasonably stiff bonds and not from continuous bending of a homogeneous chain. Because the WLC model fits force-extension data well for long polypeptides like titin domains, these weaknesses have been ignored, but for short polypeptides (<20 amino acids) they are cause for concern. Hence, to test the validity of the WLC model for short polypeptides, we compared this model to results from MD simulations.
In theory, molecular dynamics can be performed on a peptide structure that is assembled de novo, but by beginning the molecular dynamics simulations from known crystal structures, uncertainties regarding local energetic minima can be avoided. Hence, for this study we compared neck linkers from a Kinesin-1 (rat dimeric kinesin, PDB: 3KIN) and a Kinesin-2 motor (human KIF3B, PDB: 3B6U). We recently compared the velocity and processivity of Kinesin-1 and Kinesin-2 and found that Kinesin-2 is four-fold less processive, and so these MD simulations serve as a complement to this published experimental work.18 One amino acid was added to each end of the neck linker and was used in the simulations as a constraint point for defining the end-to-end distance or as a point for applying defined forces. The polypeptide was shortened from its initial (crystal structure) length down to 0.5 nm on the short end and stretched to approximately 0.5 nm beyond its contour length on the long end. An output file for this pulling simulation was generated consisting of successive .pdb files corresponding to the full range of end-to-end distances. Figure 3 shows the structure of the Kinesin-1 neck linker at a range of end-to-end distances. In principle, it is possible to construct a force-extension curve using this ramp approach by simply extracting the force at various lengths during the 300 ps pull. However, considerable force fluctuations were observed over short time windows, and we also wanted to exclude the possibility that the pulling rate influences the calculated force. Hence, we calculated the force-extension profile using two approaches – the constraint mode and the force clamp mode.
Figure 3. Neck linker structures.

In the constraint mode, the Kinesin-2 neck linker was pulled from its crystal structure length to its maximal contour length. The 17 residue neck linker contains one extra amino acid on each end and end-to-end distances are measured from the first to the last Cα. Five different neck structures at end-to-end distances of 3.54, 4.54, 5.54, 6.54 and 7.20 nm are shown.
The “constraint method” in the GROMACS v3.3.3 package involves clamping the position of one atom and recording the force necessary to maintain that position. We used the constraint mode to clamp the alpha carbon of the first and last amino acids in our given structure and recorded the average force required to maintain that end-to-end distance over a 160 ps window. For each end-to-end distance, the initial structure was taken from the collection of pdb files generated from the ramp pull. While there were significant force fluctuations (>100 pN) over the 160 ps simulation, the data consisted of 80,000 points, which enabled a reliable mean force value to be calculated for each length. The force-extension curve for the Kinesin-1 and Kinesin-2 neck linkers obtained using the constraint mode are plotted in Figure 4A and B.
Figure 4. Neck linker force-extension.

A and B: Force-extension profiles of Kinesin-1 and Kinesin-2 neck linkers using constraint mode simulations. Each point represents mean ± SEM from a 160 ps simulation at each end-to-end distance. Solid curves are predictions from the WLC model for a 15 and 18 residue peptide (measured from Cα to Cα of flanking residues) using 0.5 nm persistence length and 0.38 nm per residue contour length. C and D: Force extension profiles of Kinesin-1 and Kinesin-2 neck linkers using force clamp mode. WLC curves are same as in A and B.
Despite averaging the forces over many time points, there was still significant variability in the data resulting form the significant force fluctuations inherent in the constraint mode. Hence, we used a second approach for simulating the force-extension curve and employed the force clamp mode provided in the newly released GROMACS v4.0.3 package. In this method, a force is applied between two atoms along a vector connecting them, and the resulting fluctuations in their interatomic distance are calculated over time. This approach is a complement to the constraint mode and it provides a way to confirm the consistency of the constraint data. Each force clamp simulation started with an initial structure taken from the “constraint mode” results. A specific force was defined between the alpha carbons of the first and last residue in the structure, and the mean end-to-end distance averaged over the 160 ps simulation (Figure 4C and D). It should be noted that the direction of the force is on a line connecting the two alpha carbons, though because the structure can rotate about those carbons, the clamp is equivalent to a free pivot.
We compared the simulated force-extension properties of Kinesin-1 and Kinesin-2 to predictions from the WLC model (Figure 4). Overall, the four force-extension profiles agree reasonably well with predictions from the WLC model, which lends support to the validity of the WLC model for approximating the force-extension profile of short polypeptide chains. Importantly, the relationship of the MD simulations to the WLC predictions differs for the two neck linker sequences analyzed, indicating that the mechanical properties of these short peptide domains are sequence specific to some degree. One region of the curves where all of the simulations diverge from the WLC predictions is at short end-to-end distances. From the WLC equation, it is clear that force is positive for all end-to-end distances, but in the MD simulations negative forces (pushing forces) were required to shorten the neck linkers below approximately 2 nm (Figure 4). These resisting forces result from steric clashes of the amino acid side chains, features that are absent in the theoretical WLC paradigm. From the force clamp simulations at zero load (Figure 4C and D), the mean end-to-end distance of the free neck linker peptide in solution can be obtained. The mean length is 2.0 nm for the Kinesin-1 neck linker, which has a 5.3 nm contour length, and 2.2 nm for the Kinesin-2 neck linker, which has a 6.5 nm contour length. In principle, it is possible to derive this same end-to-end distance at zero force from the constraint mode data (Figure 4A and B), but the variability in the data precludes a reliable estimate.
The most relevant region of the force-extension curves is around 4 nm, the distance that each neck linker must stretch when both heads are bound to the microtubule. For Kinesin-1, the mean force in the range of 3.8 – 4.2 nm is predicted to be 28 pN from the WLC model, 15 pN from the constraint mode MD simulations and 35 pN from force clamp MD simulations (Figure 4A and C). Hence, the MD simulation values bracket the WLC prediction, and all three values are considerably higher than the ~6 pN stall force of Kinesin-1 motors. Due to the longer contour length of the Kinesin-2 neck linker (17 amino acids compared to 14 amino acids for Kinesin-1), the force at 4 nm predicted from the WLC is 15 pN or roughly half that of Kinesin-1. Interestingly, however, the predicted forces in the 3.8 to 4.2 nm range for Kinesin-2 are 32 pN and 35 pN from the constraint and force clamp simulations, respectively. Hence, even though the contour length of the Kinesin-2 neck linker is considerably longer, the force required to stretch the Kinesin-2 neck linker to 4 pN is quite similar to predictions for Kinesin-1.
At the highest forces, the Kinesin-1 MD force-extension profile agrees favorably with the WLC predictions. As the flexible domains are pulled to near their contour length, they stiffen considerably and very high forces are seen when the end-to-end distance approaches and exceeds 5 nm. For Kinesin-2 there was considerably less agreement between the MD results and the WLC predictions. For both the constraint and force clamp simulations, the forces in the 4.0 – 5.5 nm range were consistently higher than predicted from the WLC. To understand the Kinesin-2 neck linker properties at high forces, we investigated conformations of conserved proline residues.
The consensus Kinesin-2 neck linker contains a proline residue at the 7th position that is in the trans (straight) conformation and a proline at the 13th position that is in the kinked cis conformation in the crystal structure (see kink in Figure 1). As prolines reduce the flexibility of disordered protein domains, it seems surprising that the C-terminal half of the Kinesin-2 neck linker would contain a proline in the cis conformation, and it clearly causes a structural divergence between the Kinesin-1 and Kinesin-2 neck linker domains (Figure 1C). One possibility is that, although the Kinesin-2 neck linker is 3 amino acids longer than Kinesin-1, the kink caused by this proline means that their effective lengths are comparable. To investigate whether this proline isomerizes during the mechanical perturbations, we investigated its conformation in the Kinesin-2 constraint MD simulations. At end-to-end distances below ~4 nm, the proline remained in its kinked cis conformation (Figure 5A). However at the longest distances examined, the proline was found to be in the straight trans conformation, which results in an effective extension of the neck linker domain (Figure 5B). Interestingly, when we examined the neck linker structures at intermediate lengths, we found that the proline isomerized between the cis and trans conformations multiple times during the 160 ps simulations (Figure 5C). Hence, our interpretation of the shallower Kinesin-2 force-extension curve is that at low forces the proline at the 13th position in the neck linker is in a cis conformation, which effectively shortens the neck linker domain, while at higher forces it isomerizes to a trans conformation.
Figure 5. Kinesin-2 proline conformations at different force levels.

A: Representative image of proline in cis conformation at 4.0 nm extension. B: Representative image of proline in trans conformation at 6.9 nm extension. C: Images of proline isomerizing from cis to trans and back to cis conformation during 160 ps simulation at 5.0 nm extension.
Proline isomerizations have been studied in the context of protein folding, where it is thought that the slow isomerization can slow the kinetics of folding and even act as a molecular timer for biomolecular interactions.60 Although neither the cis or trans state is generally favored by more than a few kcal/mol, the activation barrier between them is high and in solution the transitions are thought to occur on the timescale of ~103 seconds,60 although intracellular proline isomerases are expected to accelerate these rates. AFM experiments on elastin found that forces in the 200–300 pN range strongly favored proline isomerization to the trans conformation,61 consistent with our MD simulations. However, even at these high forces, the estimated isomerization rates were in the microsecond range, while ours are in the sub-nanosecond range. Furthermore, the reversal of isomerization back to the cis conformation against such high forces is still predicted to be in the ~103 sec range or slower, so it is very surprising that we see multiple reversals in our 160 ps simulations.
Proline isomerization in the Kinesin-2 neck linker may represent a novel and previously unrecognized feature of kinesin motility. The fact that all of the Kinesin-2 and most of the Kinesin-3 motors contain a proline in the C-terminal half of the neck linker suggests that this residue is important. For these structural changes to occur on timescales relevant for motor regulation (10s of seconds) or motor stepping (~10 msec), the isomerization would need to be catalyzed. In theory, adjacent sequences could influence the isomerization kinetics or the core motor domain itself (particularly when the neck linker is in the docked conformation) could act as an isomerase. One puzzling experimental observation that may be explained by this proline isomerization mechanism is the tendency, when a Kinesin-2-coated bead is held next to a microtubule during optical trapping experiments, for the motors to wait for a period of seconds to tens of seconds before engaging with the microtubule and walking (S. Shastry, J. Andreasson and W.O. Hancock, unpublished observations). Kinesin-1 motors display no such delay, and following the pause the Kinesin-2 motors walk normally. It is possible that this pause is caused by proline isomerization that converts the motor from an inactive to an active state. In theory, this isomerization could act as a regulatory switch in cells, or it could play a role in directional switching of cargo in intraflagellar transport. Intraflagellar transport particles that are transported to the ends of cilia and flagella by Kinesin-2 are transported back to the cell body by dynein motors, but the switch that determines which motor type is active not understood.
An obvious experiment to test these ideas is to substitute the proline in the 13th position of the Kinesin-2 neck linker with an alanine or even a flexible glycine residue and measure changes in the motor run length (a measure of head-head coordination). If the proline is normally in the kinked cis conformation during motor stepping as seen in the crystal structure, then substituting it would be predicted to effectively extend the neck liker, which would be analogous to inserting one or two amino acids at the C-terminus end of the neck linker. Alternatively, if the crystal structure does not represent the normal conformation and instead the proline is normally in the trans conformation during motor stepping, then substituting it should not measurably alter the motor processivity. This experiment is currently under way in our laboratory.
CONCLUSIONS
Based on the 8 nm spacing between adjacent tubulins in a microtubule and the relatively short length of the Kinesin-1 neck linker domain, it has been proposed that there is considerable inter-head tension when both kinesin heads are bound to the microtubule. If the flexible neck linker domain is treated as an entropic spring that has a very low probability of being in the fully extended state, then diffusion of the tethered head to the next binding site may even be a kinetic limitation in the overall kinesin stepping cycle. Using a suite of bioinformatics tools, we find that among N-terminal kinesin families, the 14 amino acid Kinesin-1 neck linker is the shortest, and neck linkers in the Kinesin-2, Kinesin-3, Kinesin-5 and Kinesin-7 families are either 17 or 18 amino acids.
Molecular dynamics simulations of the Kinesin-1 and Kinesin-2 neck linkers generally support predictions from the Worm Like Chain model. In particular, they suggest that extending the neck linker the 4 nm necessary to enable both heads to bind to the microtubule requires forces equal to three to six times the 6 pN kinesin stall force. It seems unlikely that the inter-head tension would be so much greater than the stall force, and diffusion of the tethered head to the next binding site against tensions of these magnitudes may be expected to be the kinetic limitation in the kinesin stepping cycle, so this puzzle is unresolved. One possible resolution of this puzzle is tension-dependent melting of the α-7 coiled-coil. However, because cysteine crosslinking at the first residue of α-7 has no effect on velocity and only a moderate effect on processivity,23 this possibility is discounted. A more attractive possibility is that α-6 may unwind in particular nucleotide states or under tension. Because of the difficulty in crystallizing motors in different nucleotide states and the complexity of observing the force dependence of this conformational change using other techniques, this is an open possibility, and focused experiments on this question are needed.
Finally, molecular dynamics simulations of the Kinesin-2 neck linker domain suggest that a proline in the cis (kinked) conformation at the 13th position effectively shortens the Kinesin-2 neck linker such that in the low force regime its 17 residue force-extension profile is similar to that of the 14 residue Kinesin-1 motor. At high forces, our simulations suggest that the proline isomerizes to the straighter trans conformation, leading to a shallower overall force-extension profile. This cis/trans proline isomerization may play a role in regulation of the motor or in motor stepping characteristics.
Figure 6. Comparison of Kinesin-2 results to WLC models.

Molecular dynamics constraint mode (open circles) and force clamp (filled squares) results superimposed on WLC model fits for 15 (dashed line) and 18 (solid line) amino acid neck linkers. Due to cis/trans proline isomerization at high forces, the force-extension plot transitions from the 15 to the 18 residue WLC predictions.
Acknowledgments
The authors thank Matthew L. Kutys and John Fricks for helpful discussions. This work was supported by NIH grant R01-GM076476 (to W.O.H.) and NSF DMS-0714939 (J. Fricks, PI).
References
- 1.Lawrence CJ, Dawe RK, Christie KR, Cleveland DW, Dawson SC, Endow SA, Goldstein LS, Goodson HV, Hirokawa N, Howard J, Malmberg RL, McIntosh JR, Miki H, Mitchison TJ, Okada Y, Reddy AS, Saxton WM, Schliwa M, Scholey JM, Vale RD, Walczak CE, Wordeman L. A standardized kinesin nomenclature. J Cell Biol. 2004;167:19–22. doi: 10.1083/jcb.200408113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miki H, Setou M, Kaneshiro K, Hirokawa N. All kinesin superfamily protein, KIF, genes in mouse and human. Proc Natl Acad Sci U S A. 2001;98:7004–11. doi: 10.1073/pnas.111145398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Endow SA. Determinants of molecular motor directionality. Nat Cell Biol. 1999;1:E163–7. doi: 10.1038/14113. [DOI] [PubMed] [Google Scholar]
- 4.Block SM, Goldstein LS, Schnapp BJ. Bead movement by single kinesin molecules studied with optical tweezers. Nature. 1990;348:348–52. doi: 10.1038/348348a0. [DOI] [PubMed] [Google Scholar]
- 5.Howard J, Hudspeth AJ, Vale RD. Movement of microtubules by single kinesin molecules. Nature. 1989;342:154–8. doi: 10.1038/342154a0. [DOI] [PubMed] [Google Scholar]
- 6.Hancock WO, Howard J. Processivity of the motor protein kinesin requires two heads. J Cell Biol. 1998;140:1395–405. doi: 10.1083/jcb.140.6.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Block SM. Kinesin motor mechanics: binding, stepping, tracking, gating, and limping. Biophys J. 2007;92:2986–95. doi: 10.1529/biophysj.106.100677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Case RB, Rice S, Hart CL, Ly B, Vale RD. Role of the kinesin neck linker and catalytic core in microtubule-based motility. Curr Biol. 2000;10:157–60. doi: 10.1016/s0960-9822(00)00316-x. [DOI] [PubMed] [Google Scholar]
- 9.Rice S, Lin AW, Safer D, Hart CL, Naber N, Carragher BO, Cain SM, Pechatnikova E, Wilson-Kubalek EM, Whittaker M, Pate E, Cooke R, Taylor EW, Milligan RA, Vale RD. A structural change in the kinesin motor protein that drives motility. Nature. 1999;402:778–84. doi: 10.1038/45483. [DOI] [PubMed] [Google Scholar]
- 10.Khalil AS, Appleyard DC, Labno AK, Georges A, Karplus M, Belcher AM, Hwang W, Lang MJ. Kinesin’s cover-neck bundle folds forward to generate force. Proc Natl Acad Sci U S A. 2008;105:19247–52. doi: 10.1073/pnas.0805147105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenfeld SS, Jefferson GM, King PH. ATP reorients the neck linker of kinesin in two sequential steps. J Biol Chem. 2001;276:40167–74. doi: 10.1074/jbc.M103899200. [DOI] [PubMed] [Google Scholar]
- 12.Hancock WO, Howard J. Kinesin’s processivity results from mechanical and chemical coordination between the ATP hydrolysis cycles of the two motor domains. Proc Natl Acad Sci U S A. 1999;96:13147–52. doi: 10.1073/pnas.96.23.13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hancock WO, Howard J. Kinesin: Processivity and chemomechanical coupling. In: Schliwa M, editor. Molecular Motors. Wiley-VCH; Weinheim, Germany: 2003. pp. 243–269. [Google Scholar]
- 14.Rosenfeld SS, Fordyce PM, Jefferson GM, King PH, Block SM. Stepping and stretching. How kinesin uses internal strain to walk processively. J Biol Chem. 2003;278:18550–6. doi: 10.1074/jbc.M300849200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hackney DD, Stock MF, Moore J, Patterson RA. Modulation of kinesin half-site ADP release and kinetic processivity by a spacer between the head groups. Biochemistry. 2003;42:12011–8. doi: 10.1021/bi0349118. [DOI] [PubMed] [Google Scholar]
- 16.Yildiz A, Tomishige M, Gennerich A, Vale RD. Intramolecular strain coordinates kinesin stepping behavior along microtubules. Cell. 2008;134:1030–41. doi: 10.1016/j.cell.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guydosh NR, Block SM. Backsteps induced by nucleotide analogs suggest the front head of kinesin is gated by strain. Proc Natl Acad Sci U S A. 2006;103:8054–9. doi: 10.1073/pnas.0600931103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muthukrishnan G, Zhang Y, Shastry S, Hancock WO. The processivity of kinesin-2 motors suggests diminished front-head gating. Curr Biol. 2009;19:442–7. doi: 10.1016/j.cub.2009.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirose K, Lockhart A, Cross RA, Amos LA. Three-dimensional cryoelectron microscopy of dimeric kinesin and ncd motor domains on microtubules. Proc Natl Acad Sci U S A. 1996;93:9539–44. doi: 10.1073/pnas.93.18.9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nogales E, Whittaker M, Milligan RA, Downing KH. High-resolution model of the microtubule. Cell. 1999;96:79–88. doi: 10.1016/s0092-8674(00)80961-7. [DOI] [PubMed] [Google Scholar]
- 21.Kikkawa M, Sablin EP, Okada Y, Yajima H, Fletterick RJ, Hirokawa N. Switch-based mechanism of kinesin motors. Nature. 2001;411:439–45. doi: 10.1038/35078000. [DOI] [PubMed] [Google Scholar]
- 22.Sindelar CV, Downing KH. The beginning of kinesin’s force-generating cycle visualized at 9-A resolution. J Cell Biol. 2007;177:377–85. doi: 10.1083/jcb.200612090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomishige M, Vale RD. Controlling kinesin by reversible disulfide cross-linking. Identifying the motility-producing conformational change. J Cell Biol. 2000;151:1081–92. doi: 10.1083/jcb.151.5.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romberg L, Pierce DW, Vale RD. Role of the kinesin neck region in processive microtubule-based motility. J Cell Biol. 1998;140:1407–16. doi: 10.1083/jcb.140.6.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hyeon C, Onuchic JN. Internal strain regulates the nucleotide binding site of the kinesin leading head. Proc Natl Acad Sci U S A. 2007;104:2175–80. doi: 10.1073/pnas.0610939104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kellermayer MS, Smith SB, Granzier HL, Bustamante C. Folding-unfolding transitions in single titin molecules characterized with laser tweezers. Science. 1997;276:1112–6. doi: 10.1126/science.276.5315.1112. [DOI] [PubMed] [Google Scholar]
- 27.Oberhauser AF, Hansma PK, Carrion-Vazquez M, Fernandez JM. Stepwise unfolding of titin under force-clamp atomic force microscopy. Proc Natl Acad Sci U S A. 2001;98:468–72. doi: 10.1073/pnas.021321798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rief M, Gautel M, Oesterhelt F, Fernandez JM, Gaub HE. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997;276:1109–12. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 29.Gruber M, Soding J, Lupas AN. Comparative analysis of coiled-coil prediction methods. J Struct Biol. 2006;155:140–5. doi: 10.1016/j.jsb.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 30.Lupas A. Prediction and analysis of coiled-coil structures. Methods Enzymol. 1996;266:513–25. doi: 10.1016/s0076-6879(96)66032-7. [DOI] [PubMed] [Google Scholar]
- 31.Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 32.Parry DA. Coiled-coils in alpha-helix-containing proteins: analysis of the residue types within the heptad repeat and the use of these data in the prediction of coiled-coils in other proteins. Biosci Rep. 1982;2:1017–24. doi: 10.1007/BF01122170. [DOI] [PubMed] [Google Scholar]
- 33.Delorenzi M, Speed T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics. 2002;18:617–25. doi: 10.1093/bioinformatics/18.4.617. [DOI] [PubMed] [Google Scholar]
- 34.Wolf E, Kim PS, Berger B. MultiCoil: a program for predicting two- and three-stranded coiled coils. Protein Sci. 1997;6:1179–89. doi: 10.1002/pro.5560060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McDonnell AV, Jiang T, Keating AE, Berger B. Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics. 2006;22:356–8. doi: 10.1093/bioinformatics/bti797. [DOI] [PubMed] [Google Scholar]
- 36.Bryson K, McGuffin LJ, Marsden RL, Ward JJ, Sodhi JS, Jones DT. Protein structure prediction servers at University College London. Nucleic Acids Res. 2005;33:W36–8. doi: 10.1093/nar/gki410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones DT. GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol. 1999;287:797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- 38.Jones DT. Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics. 2007;23:538–44. doi: 10.1093/bioinformatics/btl677. [DOI] [PubMed] [Google Scholar]
- 39.Jones DT, Taylor WR, Thornton JM. A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry. 1994;33:3038–49. doi: 10.1021/bi00176a037. [DOI] [PubMed] [Google Scholar]
- 40.McGuffin LJ, Jones DT. Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics. 2003;19:874–81. doi: 10.1093/bioinformatics/btg097. [DOI] [PubMed] [Google Scholar]
- 41.Kull FJ, Sablin EP, Lau R, Fletterick RJ, Vale RD. Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature. 1996;380:550–5. doi: 10.1038/380550a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sack S, Muller J, Marx A, Thormahlen M, Mandelkow EM, Brady ST, Mandelkow E. X-ray structure of motor and neck domains from rat brain kinesin. Biochemistry. 1997;36:16155–65. doi: 10.1021/bi9722498. [DOI] [PubMed] [Google Scholar]
- 43.Song YH, Marx A, Muller J, Woehlke G, Schliwa M, Krebs A, Hoenger A, Mandelkow E. Structure of a fast kinesin: implications for ATPase mechanism and interactions with microtubules. EMBO J. 2001;20:6213–25. doi: 10.1093/emboj/20.22.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kozielski F, Sack S, Marx A, Thormahlen M, Schonbrunn E, Biou V, Thompson A, Mandelkow EM, Mandelkow E. The crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell. 1997;91:985–94. doi: 10.1016/s0092-8674(00)80489-4. [DOI] [PubMed] [Google Scholar]
- 45.Hoeng JC, Dawson SC, House SA, Sagolla MS, Pham JK, Mancuso JJ, Lowe J, Cande WZ. High Resolution Crystal Structure and in vivo Function of a Kinesin-2 Homolog in Giardia intestinalis. Mol Biol Cell. 2008 doi: 10.1091/mbc.E07-11-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turner J, Anderson R, Guo J, Beraud C, Fletterick R, Sakowicz R. Crystal structure of the mitotic spindle kinesin Eg5 reveals a novel conformation of the neck-linker. J Biol Chem. 2001;276:25496–502. doi: 10.1074/jbc.M100395200. [DOI] [PubMed] [Google Scholar]
- 47.Cole DG. Kinesin-II, coming and going. J Cell Biol. 1999;147:463–6. doi: 10.1083/jcb.147.3.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okada Y, Hirokawa N. A processive single-headed motor: kinesin superfamily protein KIF1A. Science. 1999;283:1152–7. doi: 10.1126/science.283.5405.1152. [DOI] [PubMed] [Google Scholar]
- 49.Tomishige M, Klopfenstein DR, Vale RD. Conversion of Unc104/KIF1A Kinesin into a Processive Motor After Dimerization. Science. 2002;297:2263–7. doi: 10.1126/science.1073386. [DOI] [PubMed] [Google Scholar]
- 50.Kashina AS, Baskin RJ, Cole DG, Wedaman KP, Saxton WM, Scholey JM. A bipolar kinesin. Nature. 1996;379:270–2. doi: 10.1038/379270a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Valentine MT, Fordyce PM, Krzysiak TC, Gilbert SP, Block SM. Individual dimers of the mitotic kinesin motor Eg5 step processively and support substantial loads in vitro. Nat Cell Biol. 2006;8:470–6. doi: 10.1038/ncb1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kwok BH, Kapitein LC, Kim JH, Peterman EJ, Schmidt CF, Kapoor TM. Allosteric inhibition of kinesin-5 modulates its processive directional motility. Nat Chem Biol. 2006;2:480–5. doi: 10.1038/nchembio812. [DOI] [PubMed] [Google Scholar]
- 53.Kim Y, Heuser JE, Waterman CM, Cleveland DW. CENP-E combines a slow, processive motor and a flexible coiled coil to produce an essential motile kinetochore tether. J Cell Biol. 2008;181:411–9. doi: 10.1083/jcb.200802189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yardimci H, van Duffelen M, Mao Y, Rosenfeld SS, Selvin PR. The mitotic kinesin CENP-E is a processive transport motor. Proc Natl Acad Sci U S A. 2008;105:6016–21. doi: 10.1073/pnas.0711314105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thorn KS, Ubersax JA, Vale RD. Engineering the processive run length of the kinesin motor. J Cell Biol. 2000;151:1093–100. doi: 10.1083/jcb.151.5.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fersht A. Structure and Mechanism in Protein Science. New York: W.H. Freeman and Company; 1998. p. 631. [Google Scholar]
- 57.Howard J. Mechanics of Motor Proteins and the Cytoskeleton. 1. Sunderland, MA: Sinauer Associates, Inc; 2001. p. 367. [Google Scholar]
- 58.Pauling L, Corey RB, Branson HR. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci U S A. 1951;37:205–11. doi: 10.1073/pnas.37.4.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nagy A, Grama L, Huber T, Bianco P, Trombitas K, Granzier HL, Kellermayer MS. Hierarchical extensibility in the PEVK domain of skeletal-muscle titin. Biophys J. 2005;89:329–36. doi: 10.1529/biophysj.104.057737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu KP, Finn G, Lee TH, Nicholson LK. Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol. 2007;3:619–29. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 61.Valiaev A, Lim DW, Oas TG, Chilkoti A, Zauscher S. Force-induced prolyl cis-trans isomerization in elastin-like polypeptides. J Am Chem Soc. 2007;129:6491–7. doi: 10.1021/ja070147r. [DOI] [PubMed] [Google Scholar]
- 62.Richardson JS, Richardson DC. Amino acid preferences for specific locations at the ends of alpha helices. Science. 1988;240:1648–52. doi: 10.1126/science.3381086. [DOI] [PubMed] [Google Scholar]