

Abstract
Computational protein design facilitates the continued development of methods for the design of biomolecular structure, sequence and function. Recent applications include the design of novel protein sequences and structures, proteins incorporating nonbiological components, protein assemblies, soluble variants of membrane proteins, and proteins that modulate membrane function.
Keywords: Computational protein design, Nanostructures, Structural biology, Self-assembly, Protein library, Protein symmetry, Nanoparticle, Optoelectronic, Cofactor, Porphyrin, Membrane protein
1. Overview
Natural proteins possess a wide variety of selective functionalities, including folding, self-assembly, catalysis and molecular recognition. Since the folded state of a protein is in most cases dictated by the sequence of amino acids, structure can potentially be specified through the careful selection of sequence. Nature leverages the physicochemical properties of the amino acids to arrive at highly functional sequences that spontaneously fold, where structural and functional properties are tuned during the course of evolution. Well-structured proteins may also be realized via the careful design of sequences. This can be nontrivial. Proteins are large, comprising tens to thousands of amino acid monomers, and possess many backbone and side-chain degrees of freedom. As a result the configurational state space for proteins is large, even if the backbone tertiary structure is predetermined. The stabilizing interactions that guide the protein to its native state are largely noncovalent, and quantitative estimates of stability with respect to unfolding can be difficult to infer. In addition, the large number of possible sequences leads to a further combinatorial complexity in protein design: for a modestly sized protein of only 100 amino acids, more than 10130 sequences are possible if only the 20 naturally occurring amino acids are used. Nonetheless, theoretical methods have made accessible the design and study of new proteins and protein-based assemblies. Most such methods begin with a target structure, which can be a naturally occurring computationally modeled. A physically motivated objective function that quantifies consistency of the sequences with the target structure is optimized so as to identify individual sequences or the properties of the ensemble of sequences consistent with the target structure and any desired functional properties. Algorithmic techniques for identifying low-energy sequences include dead-end elimination, Monte Carlo simulated annealing, genetic algorithms, and optimization theory approaches [1–4]. In addition, probabilistic methods characterize the ensemble of designed sequences and may use statistical thermodynamic self-consistent field techniques or Monte Carlo sampling of sequences to estimate the site-specific probabilities of the amino acids at monomer sites targeted for variation [5,6]. Computational protein design may be used to better understand protein folding, to facilitate the study of natural proteins, to enhance or redirect the functionality of natural proteins, and to develop novel nonbiological protein-based molecular systems.
Herein we survey recent applications of computational methods for the design and engineering of proteins, focusing largely on cases where the resulting proteins have been experimentally realized. Many recent efforts have involved protein redesign, where a known protein is re-engineered so as to augment stability and functionality. In addition to protein redesign, new protein structures (and sequences) have been computationally designed, which may incorporate non-biological components. The engineering of new functionality into proteins has seen recent advances, particularly with regard to catalytic activity. Here we focus instead on larger length scale properties involving folding and assembly of proteins and protein complexes. Membrane proteins represent an important new frontier with regard to the design and redesign of proteins.
2. Protein re-engineering
The activation domain of human procarboxypeptidase A2 has been redesigned, resulting in a variant with 68% of the wild-type sequence mutated. The redesigned protein is over 10 kcal/mol more stable than the wild-type protein, and the high-resolution crystal structure and solution NMR structures are effectively superimposable with the computational template [7•].
The entire sequence of a 51 amino acid residue protein, an engrailed homeodomain, has been computationally redesigned [8]. Two sequences were selected for experimental characterization that had only 22% and 24% sequence identity with the wild type. Both proteins were considerably more stable than the naturally occurring protein, having thermal denaturation temperatures greater than 99 °C. The solution structure as determined using NMR spectroscopy closely matches that of the original design template.
There are relatively few examples of designed beta sheet proteins [9], and as a result it is of interest to further explore these structures as design targets. A naturally occurring beta sheet protein, tenascin, has been redesigned using an algorithm that does not include explicit negative design, i.e., the explicit consideration of competing low-energy tertiary structures in the design process [10•]. The resulting proteins are more stable than the wild type. The crystallographic structure of one closely matches the structure of the computationally generated model. Though nature may have partially solved the problem in selecting for a structure such that alternate competing structures are less likely to be populated, the results suggest that explicit negative design may not be necessary to arrive at well-folded beta proteins.
Specificity of interactions as well as structure has been studied with the aid of computational protein design. A scheme for computationally designing specificity into protein interactions has been developed and used to identify partners for basic-region leucine zipper (bZIP) transcription factors [11•]. With the aid of experimental studies using protein microarrays, many of the designed peptide ligands, including those that bind important oncoproteins, were found to be selective for their intended targets. The results suggest that bZIPs have sparsely sampled the range of possible interactions. The computational method may yield routes to explore the interplay between stability and specificity in these systems using protein design.
Computational protein design may also be used to design libraries of sequences. This has been illustrated in a study comparing different methods of generating combinatorial libraries of green fluorescent protein variants [12]. The fraction of fluorescent, functional proteins was largest for the library designed using a structure-based computational method. A trend was observed toward greater diversity of color in designed libraries that maintained fluorescence. In contrast to many other methods for generating libraries, with structure-based computational methods function was preserved even as mutation level increased among the libraries.
Lipid transfer proteins bind and transfer lipids and have been re-engineered to provide a novel biosensor. Computational protein design has been used to remove a disulfide bridge and attach a thio-reactive fluorophore. Ligand binding likely causes a conformational change that affects fluorescence. These variants have affinity to palmitate that is consistent with wild type [13].
3. De novo designed proteins
In addition to redesign of existing proteins, new proteins may be realized when structure and/or new functionality is included as part of the design. Four-helix bundle motifs can be geometrically parameterized using a few variables that describe the global structure. Such methods have been used to arrive at model di-iron and di-manganese proteins, which provide useful platforms for engineering and investigating the versatile range of binding and catalytic properties exhibited by this metalloprotein motif [14–17]. A 114-residue protein that presents such a di-nuclear binding site has been computationally designed, and was found to be highly stable, particularly with the metals present, despite having a large number of ionizable groups within its interior [18]. The structure of the Zn-bound version of the protein has been determined using a synthesis of NMR spectroscopy and structural refinement using quantum chemical calculations, where the calculations are used to redress structural incongruities among the model structures, especially near the dimetal site. This resulting structure is fully consistent with the NMR spectra and in excellent agreement with the template structure [19•] (Fig. 1).
Fig. 1.
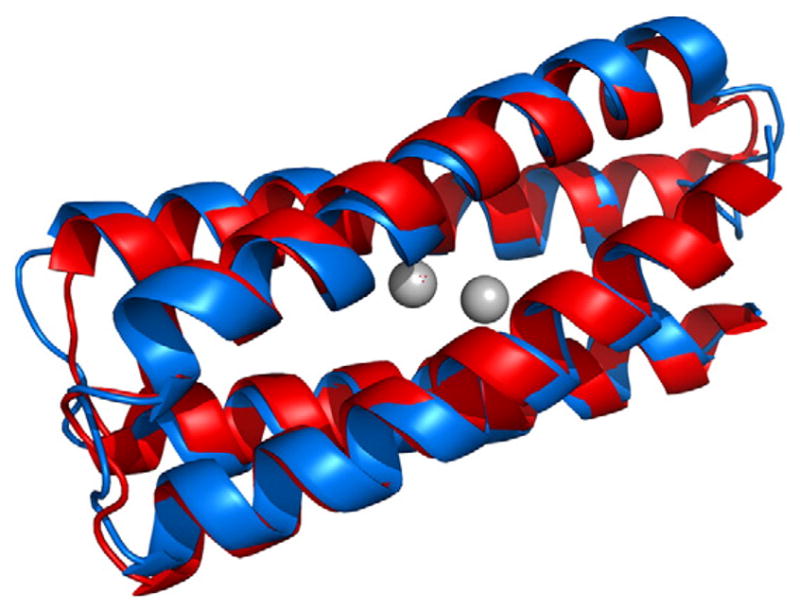
Superposition of the computational model used to design the single-chain di-nuclear metalloprotein DFsc (red) [18] and the experimentally determined structure obtained using NMR spectroscopy with QM/MM refinement (blue) [19•].
4. Nonbiological assemblies of proteins
Computational de novo design accelerates the creation of novel protein assemblies, which may not have analogs seen in nature and have applications as biomaterials and electrochemical devices. The association of nonbiological cofactors and other macromolecules with proteins opens the possibility of realizing biomolecular systems with properties not accessible with naturally occurring amino acids or with biological cofactors. For sufficiently complex cofactors it may not be possible to find appropriate scaffolds among natural protein structures and as a result, novel structures must also be created. Computational design has been applied to arrive at a protein framework that encapsulates a synthetic cofactor [20], yielding a computationally designed protein that selectively binds a nonbiological cofactor, a diphenyl iron porphyrin (DPP-Fe). This design strategy has been elaborated to realize modular metalloporphyrin peptide arrays of varying lengths [21•]. The two-metalloporphyrin array has been extended into to a four-metalloporphyrin array consistent with a coiled-coil repeat unit. Experimental studies of these elongated proteins reveal the targeted cofactor-binding stoichiometry and cofactor specificity. This work illustrates how the computational design may be used to create extended linear assemblies of organized electronically and optically active cofactors. Many nonbiological cofactors of interest may be asymmetric, and as a result moving to protein scaffolds that are not symmetric is desirable. One route to the realization of protein structures that present interiors complementary to asymmetric scaffolds is through the computational design of single-chain four-helix bundles. Such proteins may be genetically over-expressed and noncovalently self-assembled with fully synthetic non-natural porphyrin cofactors. Experimental characterization of such a computationally designed protein is consistent with high specificity of binding to the desired cofactors and a well-structured protein [22•]. This designed protein presents an example of a scaffold that may be used to explore asymmetric mutations to enable a variety of properties, including surface-immobilization and selective binding of different cofactors and chromophores. Designed amphipathic helical bundle proteins binding similar cofactors are capable of self-ordering at aqueous interfaces, conferring vectorial orientation to the bundles and contained cofactors [23,24] (Fig. 3).
Fig. 3.

Proteins designed to accommodate nonbiological cofactors (diphenyl iron porphyrins). (a) Single-chain protein binding two equivalents of the cofactor [22•]. (b) Homotetrameric alpha-helical bundle that presents a linear array of the nonbiological cofactors [21•].
5. Multimeric protein assemblies
In addition to individual proteins and helix bundles, there has been much recent interest in multimeric assemblies of proteins. Computational studies of protein structure and iterative design have revealed that a bias toward low-energy complexes can result in symmetric assemblies of proteins from random ensembles in which symmetric complexes are essentially absent initially [25].
Protein–protein interactions have also recently been subjects of computational protein design. Structure-based computational design has yielded tetrapeptides that efficiently depolymerize serine-protease inhibitor (serpin) fibrils, which have been associated with maladies such as cirrhosis and emphysema [26]. The study reveals how small, designed peptides may be used to modulate the polymerization/depolymerization of fibrillar aggregates and provide evidence that peptide-induced depolymerization takes place via a heterogeneous, multi-step process that begins with the internal fragmentation of the fibrils. One designed tetrapeptide is the most potent antitrypsin depolymerizer yet identified.
Protein dimers have been generated from monomeric proteins using computational protein docking and sequence design. A docking algorithm was used to generate a dimeric model of the a1 domain of streptococcal protein G. Computational design of 24/56 residues yielded a heterodimer comparing subunits with eight and twelve mutations relative to wild type. The dimer is weakly bound (0.3 mM dissociation constant), but for one of the designed proteins, NMR spectral changes observed in the presence of its binding partner suggest specific dimer formation [27].
Proteins having complex quaternary structures have also been subjects of design. The symmetry of some large protein assemblies may be leveraged to address large numbers of simultaneous mutations. The DNA protection protein Dps is a dodecameric structure having a 4.5 nm diameter interior cavity. This cavity has been computationally redesigned by varying up to 120 sites (10 per protein subunit) to confer a large hydrophobic interior surface, illustrating the versatility of ferritin scaffolds for engineering proteins containing large cavities and having potential applications as nanoscale containers [28].
Computational design has also been applied to other ferritin proteins with applications to nanotechnology. Human H ferritin forms an assembly with 24 four-helix bundle subunits having an 8 nm diameter cavity. This system has been redesigned to confer noble metal ion (Au3+, Ag+) binding, reduction, and nanoparticle formation within the cavity [29•]. Binding to these metals is mediated largely by exposed thiol groups introduced (via cysteine mutations) to the interior surface of the cavity. A variant with histidines removed from the exterior surface and 96 non-native cysteines added to the interior surface retained wild-type stability and structure, as confirmed by X-ray crystallography and spectroscopic studies. This designed variant promoted the formation of silver and gold metal nanoparticles within the protein cavity (Fig. 2).
Fig. 2.
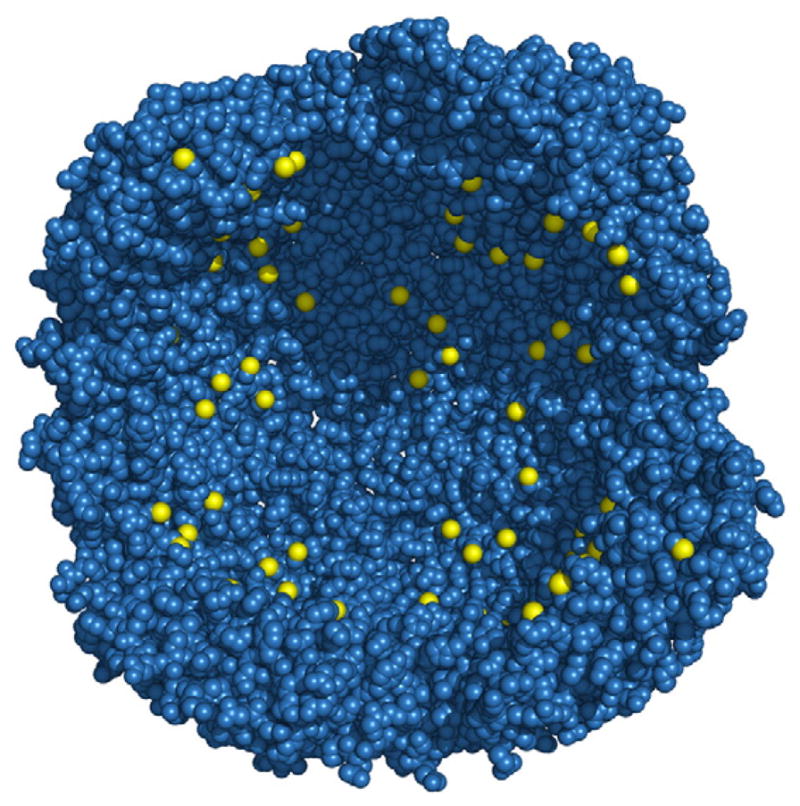
Redesigned interior of human ferritin [29•]. Sulfur atoms of cysteines are rendered in yellow. The parent wild type has no cysteines on the interior surface. Eight of the twenty-four subunits have been deleted to expose the interior.
6. Membrane proteins
Membrane-associated proteins control the structure and function of biological membranes. The design of novel membrane proteins has applications toward better understanding natural membranes and membrane proteins, controlling the activity of membrane-associated biomolecular processes, and developing molecular agents for controlling the structure and function of nonbiological membrane structures, e.g., vesicles and bilayers. Membrane proteins are essential to a variety of cellular processes and as a result are targets of a large number of drugs and therapeutics. Many of these proteins span the membrane and have large numbers of exterior hydrophobic residues that maintain structure and registry in the membrane bilayer; as a result, these proteins are typically aggregation prone and are difficult to obtain and purify using standard solution phase methods. Most efforts involving biophysical studies of membrane proteins involve the aqueous dispersion of membrane proteins, usually using detergents, lipids, auxiliary proteins, and reconstitution, e.g., so as to obtain diffraction-quality crystals or solubilized proteins suitable for NMR studies. Obtaining such solubilizing conditions for biophysical and structural studies remains subtle and time intensive.
Using computational design, water-soluble variants of integral membrane proteins have been designed, potentially facilitating studies of their structures and functions. Recently the solution structure of a computationally designed water-soluble variant of the bacterial potassium ion channel KcsA has been determined [30,31•]. The structure is in striking agreement with the tertiary and quaternary structure of the membrane-soluble wild-type structure. These findings highlight the promise of developing water-soluble variants of membrane proteins—for biophysical studies and for therapeutic development—using computational redesign of sequence (Fig. 4).
Fig. 4.
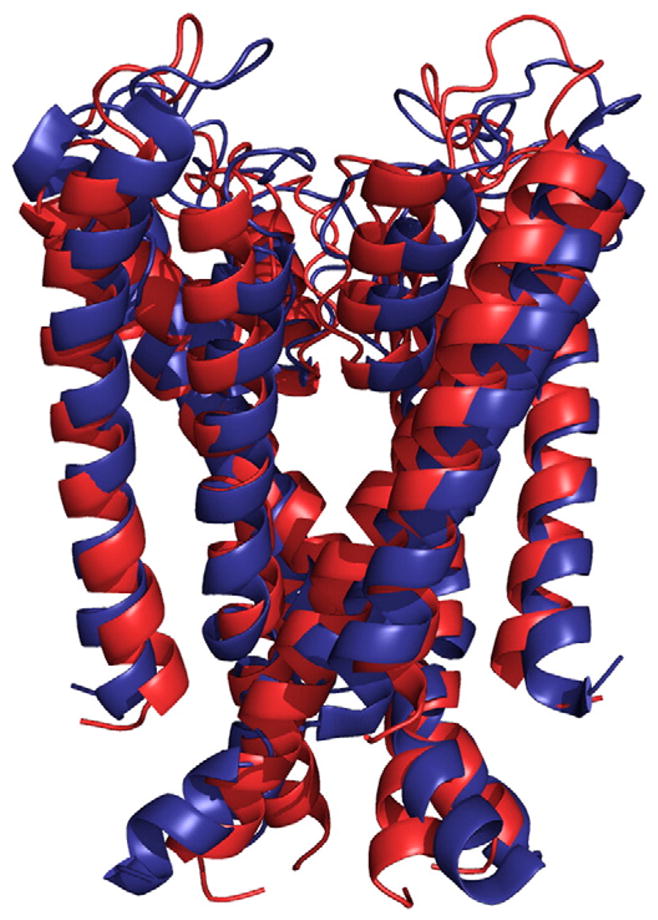
Superposition of the model structure of water-soluble variant of the transmembrane portion of the bacterial potassium ion channel KcsA (red) [30] and its experimentally determined structure (blue) (pdb accession code: 1K4E) [31•].
Computational protein design has been used to test and refine models of G-protein coupled receptors, where the effects of mutations on oligomerization state are compared with mutagenesis studies and are used to confirm atomistic models of these important receptors [32].
Proteins have been created that modulate the activity of membrane proteins. Membrane spanning peptides have been computationally designed that target transmembrane helices of integrins in a sequence specific manner. The designed peptides are able to discriminate transmembrane helices of two closely related integrins (alpha(IIb)beta(3) and alpha(v)beta(3)), where the specificity is realized via complementary peptide–helix steric interactions [33•]. This approach has been extended to address direct interactions of a designed anti-RIIb peptide with isolated full-length integrin RIIb in detergent micelles. The properties of the designed peptides in phospholipid bilayers are essentially identical with those in detergent micelles. The peptides take on a transmembrane alpha-helical conformation that does not disrupt the bilayer [34]. Proteins may also be designed to provide controllable integrity of a bilayer. A natural alpha-helical cell-lytic peptide, mastoparan X, has been re-engineered to bind metal cations. Binding of Zn(II) or Ni(II) stabilizes the peptide’s amphiphilic structure that leads to the lysis of cells and vesicles [35].
7. Outlook
Recent achievements in computational protein design provide examples of how designed proteins can be crafted to exhibit targeted structures, properties, and new functionalities. Mutagenesis is a central tool for facilitating the study of proteins and for investigating protein structure and function; protein design stands to make such mutation-based studies more informative and effective. Natural proteins can be redesigned so as to confer new functionalities, including how to modulate and control the properties of vesicles, membranes, and interfaces. The design of proteins that mimic the efficiency and specificity of natural proteins is likely to remain challenging, but based on recent successes, a synthesis of computational design and experimental studies is likely to yield important advances in the identification of novel proteins and in furthering our understanding of nature’s proteins. Lastly, incorporating nonbiological components, e.g., nonbiological amino acids and cofactors, will yield protein systems poised to present new functions that have not previously been accessible.
Acknowledgments
The author gratefully acknowledges support from the US Department of Energy (DE-FG02-04ER46156), the National Institutes of Health (P01 GM55876), the University of Pennsylvania’s Nano/Bio Interface Center through the National Science Foundation (NSF) NSEC DMR-0425780 and the Laboratory for Research on the Structure of Matter through NSF MRSEC DMR-0520020. The figures were rendered using PyMol (DeLano Scientific LLC).
References
• Each is work of interest.
- 1.Park S, Fu X, Wang W, Yang X, Saven JG. Computational protein design and discovery. Annu Rep Prog Chem Sect C. 2004;100:195–236. [Google Scholar]
- 2.Floudas CA, Fung HK, McAllister SR, Monnigmann M, Rajgaria R. Advances in protein structure prediction and de novo protein design: a review. Chem Eng Sci. 2006;61:966–88. [Google Scholar]
- 3.Saven JG. Computational protein design. In: Schwede T, Peitsch MC, editors. Computational structural biology: methods and applications. World Scientific Publishing Co; 2008. pp. 401–24. [Google Scholar]
- 4.Lehmann A, Lanci CL, Petty TJ, II, Kang SG, Saven JG. Protein design: tailoring sequence, structure and folding properties. In: Muñoz V, editor. Protein folding, misfolding and aggregation: classical themes and novel approaches. Royal Society of Chemistry Biomolecular Sciences; 2008. pp. 188–213. [Google Scholar]
- 5.Park S, Kono H, Wang W, Boder ET, Saven JG. Progress in the development and application of computational methods for probabilistic protein design. Comput Chem Eng. 2005;29:407–21. [Google Scholar]
- 6.Yang X, Saven JG. Computational methods for protein design and protein sequence variability: biased Monte Carlo and replica exchange. Chem Phys Lett. 2005;401:205–10. [Google Scholar]
- 7•.Dantas G, Corrent C, Reichow SL, Havranek JJ, Eletr ZM, Isern NG, et al. High-resolution structural and thermodynamic analysis of extreme stabilization of human procarboxypeptidase by computational protein design. J Mol Biol. 2007;366:1209–21. doi: 10.1016/j.jmb.2006.11.080. The authors show that computational design is capable of essentially redesigning the sequence of an enzyme fragment. Excellent agreement between the computational and experimentally determined structures is obtained. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah PS, Hom GK, Ross SA, Lassila JK, Crowhurst KA, Mayo SL. Full-sequence computational design and solution structure of a thermostable protein variant. J Mol Biol. 2007;372:1–6. doi: 10.1016/j.jmb.2007.06.032. [DOI] [PubMed] [Google Scholar]
- 9.Nanda V, Rosenblatt MM, Osyczka A, Kono H, Getahun Z, Dutton PL, et al. De novo design of a redox-active minimal rubredoxin mimic. J Am Chem Soc. 2005;127:5804–5. doi: 10.1021/ja050553f. [DOI] [PubMed] [Google Scholar]
- 10•.Hu XZ, Wang HC, Ke HM, Kuhlman B. Computer-based redesign of a beta sandwich protein suggests that extensive negative design is not required for de novo beta sheet design. Structure. 2008;16:1799–805. doi: 10.1016/j.str.2008.09.013. Many current methods for computational design of sequence succeed despite the fact that they do not explicitly consider tertiary structures that may compete with the targeted structure. The authors speculate on why such methods are effective and provide an example of the application of computational design to a beta structured protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11•.Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458:859–64. doi: 10.1038/nature07885. The authors probe specificity of protein interactions via protein design. An elegant use of computational and microarray data explores the effectiveness of design and the range of possible interactions between bZIP domain partners. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Treynor TP, Vizcarra CL, Nedelcu D, Mayo SL. Computationally designed libraries of fluorescent proteins evaluated by preservation and diversity of function. Proc Natl Acad Sci U S A. 2007;104:48–53. doi: 10.1073/pnas.0609647103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi EJ, Mao J, Mayo SL. Computational design and biochemical characterization of maize nonspecific lipid transfer protein variants for biosensor applications. Protein Sci. 2007;16:582–8. doi: 10.1110/ps.062607007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maglio O, Nastri F, Calhoun JR, Lahr S, Wade H, Pavone V, et al. Artificial di-iron proteins: solution characterization of four helix bundles containing two distinct types of inter-helical loops. J Biol Inorg Chem. 2005;10:539–49. doi: 10.1007/s00775-005-0002-8. [DOI] [PubMed] [Google Scholar]
- 15.Calhoun JR, Nastri F, Maglio O, Pavone V, Lombardi A, DeGrado WF. Artificial diiron proteins: from structure to function. Biopolymers. 2005;80:264–78. doi: 10.1002/bip.20230. [DOI] [PubMed] [Google Scholar]
- 16.Wei PP, Skulan AJ, Wade H, DeGrado WF, Solomon EI. Spectroscopic and computational studies of the de novo designed protein DF2t: correlation to the biferrous active site of ribonucleotide reductase and factors that affect O-2 reactivity. J Am Chem Soc. 2005;127:16098–106. doi: 10.1021/ja053661a. [DOI] [PubMed] [Google Scholar]
- 17.Wade H, Stayrook SE, DeGrado WF. The structure of a designed diiron(III) protein: implications for cofactor stabilization and catalysis. Angew Chem Int Ed. 2006;45:4951–4. doi: 10.1002/anie.200600042. [DOI] [PubMed] [Google Scholar]
- 18.Calhoun JR, Kono H, Lahr S, Wang W, DeGrado WF, Saven JG. Computational design and characterization of a monomeric helical dinuclear metalloprotein. J Mol Biol. 2003;334:1101–15. doi: 10.1016/j.jmb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 19•.Calhoun JR, Liu W, Spiegel K, Dal Peraro M, Klein ML, Valentine KG, et al. Solution NMR structure of a designed metalloprotein and complementary molecular dynamics refinement. Structure. 2008;16:210–5. doi: 10.1016/j.str.2007.11.011. This work results in an experimentally determined structure of a di-nuclear metalloprotein that is in excellent agreement with that resulting from the computational design. In addition, the authors show how quantum-mechanics/molecular-mechanics (QM/MM) modeling can be used to obtained high quality models consistent with NMR spectra, particularly for cases where the quality of the molecular force field used in refinement may be poor or incomplete. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cochran FV, Wu SP, Wang W, Nanda V, Saven JG, Therien MJ, et al. Computational de novo design and characterization of a four-helix bundle protein that selectively binds a nonbiological cofactor. J Am Chem Soc. 2005;127:1346–7. doi: 10.1021/ja044129a. [DOI] [PubMed] [Google Scholar]
- 21•.McAllister KA, Zou HL, Cochran FV, Bender GM, Senes A, Fry HC, Nanda V, Keenan PA, Lear JD, Saven JG, et al. Using alpha-helical coiled-coils to design nanostructured metalloporphyrin arrays. J Am Chem Soc. 2008;130:11921–7. doi: 10.1021/ja800697g. The authors expand upon the computational design of helix bundle proteins to obtain extended systems will well positioned redox-active nonbiological chromophores. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22•.Bender GM, Lehmann A, Zou H, Cheng H, Fry HC, Engel D, Therien MJ, Blasie JK, Roder H, Saven JG, et al. De novo design of a single-chain diphenylporphyrin metalloprotein. J Am Chem Soc. 2007;129:10732–40. doi: 10.1021/ja071199j. By designing helical structures, loop conformation, and sequence, the authors arrive at an asymmetric, single-chain structure and sequence that binds two equivalents of a nonbiological porphyrin cofactor. The resulting protein is more stable than similar oligomeric proteins and exhibits a dramatic increase in folded state stability upon cofactor binding. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zou HL, Therien MJ, Blasie JK. Structure and dynamics of an extended conjugated NLO chromophore within an amphiphilic 4-helix bundle peptide by molecular dynamics simulation. J Phys Chem B. 2008;112:1350–7. doi: 10.1021/jp076643j. [DOI] [PubMed] [Google Scholar]
- 24.Zou HL, Strzalka J, Xu T, Tronin A, Blasie JK. Three-dimensional structure and dynamics of a de novo designed, amphiphilic, metalloporphyrin-binding protein maquette at soft interfaces by molecular dynamics simulations. J Phys Chem B. 2007;111:1823–33. doi: 10.1021/jp0666378. [DOI] [PubMed] [Google Scholar]
- 25.Andre I, Strauss CEM, Kaplan DB, Bradley P, Baker D. Emergence of symmetry in homooligomeric biological assemblies. Proc Natl Acad Sci U S A. 2008;105:16148–52. doi: 10.1073/pnas.0807576105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chowdhury P, Wang W, Lavender S, Bunagan MR, Klemke JW, Tang J, et al. Fluorescence correlation spectroscopic study of serpin depolymerization by computationally designed peptides. J Mol Biol. 2007;369:462–73. doi: 10.1016/j.jmb.2007.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang PS, Love JJ, Mayo SL. A de novo designed protein–protein interface. Protein Sci. 2007;16:2770–4. doi: 10.1110/ps.073125207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swift J, Wehbi WA, Kelly BD, Stowell XF, Saven JG, Dmochowski IJ. Design of functional ferritin-like proteins with hydrophobic cavities. J Am Chem Soc. 2006;128:6611–9. doi: 10.1021/ja057069x. [DOI] [PubMed] [Google Scholar]
- 29•.Butts CA, Swift J, Kang SG, Di Costanzo L, Christianson DW, Saven JG, et al. Directing noble metal ion chemistry within a designed ferritin protein. Biochemistry. 2008;47:12729–39. doi: 10.1021/bi8016735. The authors use computational design to arrive at human H ferritin variants that promote the formation of Ag and Au nanoparticles on their interiors, yielding bio-nano hybrid systems. The high-resolution crystallographic structures of the 24 subunit assemblies are in good agreement with the targeted protein structural modifications. [DOI] [PubMed] [Google Scholar]
- 30.Slovic AM, Kono H, Lear JD, Saven JG, DeGrado WF. Computational design of water-soluble analogues of the potassium channel KcsA. Proc Natl Acad Sci U S A. 2004;101:1828–33. doi: 10.1073/pnas.0306417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31•.Ma DJ, Tillman TS, Tang P, Meirovitch E, Eckenhoff R, Carnini A, et al. NMR studies of a channel protein without membranes: structure and dynamics of water-solubilized KcsA. Proc Natl Acad Sci U S A. 2008;105:16537–42. doi: 10.1073/pnas.0805501105. NMR based studies of a computationally designed water soluble variant of a bacterial potassium ion channel is in excellent agreement with the structure of the parent membrane bound form. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor MS, Fung HK, Rajgaria R, Filizola M, Weinstein H, Floudas CA. Mutations affecting the oligomerization interface of G-protein-coupled receptors revealed by a novel de novo protein design framework. Biophys J. 2008;94:2470–81. doi: 10.1529/biophysj.107.117622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33•.Yin H, Slusky JS, Berger BW, Walters RS, Vilaire G, Litvinov RI, et al. Computational design of peptides that target transmembrane helices. Science. 2007;315:1817–22. doi: 10.1126/science.1136782. Computational protein design is used to obtain membrane embedded proteins that selectively bind to integral membrane proteins, in this case integrins, and modulate their functions. [DOI] [PubMed] [Google Scholar]
- 34.Caputo GA, Litvinov RI, Li W, Bennett JS, DeGrado WF, Yin H. Computationally designed peptide inhibitors of protein–protein interactions in membranes. Biochemistry. 2008;47:8600–6. doi: 10.1021/bi800687h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Signarvic RS, DeGrado WF. Metal-binding dependent disruption of membranes by designed helices. J Am Chem Soc. 2009;131:3377–84. doi: 10.1021/ja809580b. [DOI] [PMC free article] [PubMed] [Google Scholar]