

Abstract
Marijuana exposure during the critical period of adolescent brain maturation may disrupt neuro-modulatory influences of endocannabinoids and increase schizophrenia susceptibility. Cannabinoid receptor 1 (CB1/CNR1) is the principal brain receptor mediating marijuana effects. No study to-date has systematically investigated the impact of CNR1 on quantitative phenotypic features in schizophrenia and inter-relationships with marijuana misuse. We genotyped 235 schizophrenia patients using 12 tag single nucleotide polymorphisms (tSNPs) that account for most of CB1 coding region genetic variability. Patients underwent a high-resolution anatomic brain magnetic resonance scan and cognitive assessment. Almost a quarter of the sample met DSM marijuana abuse (14%) or dependence (8%) criteria. Effects of CNR1 tSNPs and marijuana abuse/dependence on brain volumes and neurocognition were assessed using ANCOVA, including co-morbid alcohol/non-marijuana illicit drug misuse as covariates. Significant main effects of CNR1 tSNPs (rs7766029, rs12720071, and rs9450898) were found in white matter (WM) volumes. Patients with marijuana abuse/dependence had smaller fronto-temporal WM volumes than patients without heavy marijuana use. More interestingly, there were significant rs12720071 genotype-by-marijuana use interaction effects on WM volumes and neurocognitive impairment; suggestive of gene-environment interactions for conferring phenotypic abnormalities in schizophrenia. In this comprehensive evaluation of genetic variants distributed across the CB1 locus, CNR1 genetic polymorphisms were associated with WM brain volume variation among schizophrenia patients. Our findings suggest that heavy cannabis use in the context of specific CNR1 genotypes may contribute to greater WM volume deficits and cognitive impairment, which could in turn increase schizophrenia risk.
Keywords: CNR1, magnetic resonance imaging, cognition, candidate gene association study, schizophrenia, substance abuse dependence
1. Introduction
Marijuana use during early adolescence has been associated with two-fold increased risk for schizophrenia (Andreasson, Allebeck, Engstrom, & Rydberg, 1987; Arseneault et al., 2002; Zammit, Allebeck, Andreasson, Lundberg, & Lewis, 2002). Longitudinal assessments of birth cohorts find that adolescent marijuana users are significantly more likely than non-users to be diagnosed with schizophrenia at follow-up. Other large epidemiologic prospective studies have replicated this association between adolescent marijuana use and persistent psychotic disorders (Henquet et al., 2005a; Stefanis et al., 2004; van Os et al., 2002). Recent meta-analyses further indicate that schizophrenia risk rises with increasingly heavy adolescent marijuana exposure, and estimate that adolescent marijuana use may account for 8–14% of schizophrenia cases (Henquet, Murray, Linszen, & van Os, 2005b; Moore et al., 2007).
Although the association between adolescent marijuana use and subsequent schizophrenia is well-replicated, the nature of this relationship remains widely debated (e.g. (Henquet & Van Os, 2008)). The neurobiological mechanisms underlying how early adolescent cannabis exposure may confer increased schizophrenia susceptibility is poorly understood (D’Souza, Sewell, & Ranganathan, 2009; DeLisi, 2008; Kumra, 2007; Sewell, Ranganathan, & D’Souza, 2009). Most schizophrenia patients have no history of adolescent marijuana use, and the majority of adolescents who abuse marijuana do not go on to develop schizophrenia. Some have argued against a causal link (Degenhardt, Hall, & Lynskey, 2003; Hickman et al., 2009), and suggest that individuals with incipient schizophrenia may be self-medicating with marijuana. Other researchers have suggested that adolescence is a sensitive time period during which the brain may be especially vulnerable to deleterious effects of marijuana, which may disrupt normal brain maturation and lead to increased schizophrenia risk (e.g. (Murray, Morrison, Henquet, & Di Forti, 2007)). A growing body of animal studies supports such a hypothesis. Chronic cannabis administration in adolescent rats, but not adult cannabinoid exposure, leads to enduring cognitive deficits in adulthood, including working memory deficits and prepulse inhibition abnormalities commonly observed in schizophrenia probands (O’Shea, McGregor, & Mallet, 2006; O’Shea, Singh, McGregor, & Mallet, 2004; Schneider & Koch, 2007). These persistent cognitive deficits are further associated with altered FOS protein expression within brain regions critical in schizophrenia (such as the hippocampus, nucleus accumbens, caudate and putamen)(Wegener & Koch, 2009).
The effects of exogenous cannabis and endocannabinoids are mediated by cannabinoid receptor 1 (CNR1; a.k.a. CB1 or CB1R), which is widely expressed in the brain, including prefrontal cortex and medial temporal lobe (Pazos, Nunez, Benito, Tolon, & Romero, 2005). CNR1 is localized to Chr 6q14-q15, a schizophrenia susceptibility locus (Kohn & Lerer, 2005). Previous schizophrenia-CNR1 genetic association studies, restricted mostly to two CNR1 variants (i.e. rs1049353 SNP and an (AAT)n trinucleotide repeat), have found mixed results (Chavarría-Siles et al., 2008; Seifert, Ossege, Emrich, Schneider, & Stuhrmann, 2007). No studies have comprehensively investigated the effects of CNR1 on brain morphometric and neurocognitive phenotypic features of schizophrenia. The goal of this study is to examine the interactions between CNR1 genetic variants and heavy marijuana misuse on brain volumes and cognitive function among schizophrenia patients. Our hypothesis is that patients with specific CNR1 genotypes are more vulnerable to the effects of heavy marijuana misuse, and will show greater brain volume deficits and cognitive impairment than patients without marijuana misuse.
2. Patients and Methods
2.1 Subjects
Two hundred and thirty-five patients with schizophrenia-spectrum disorders were recruited through the University of Iowa Mental Health Clinical Research Center (MHCRC). These subjects participated in various MHCRC research studies approved by the University of Iowa human subjects research review board. All study participants gave written informed consent to undergo research assessments, which included a morphometric magnetic resonance (MR) brain scan, standardized neuropsychological test battery and blood sample for DNA analyses. They were further evaluated using a semi-structured interview instrument, Comprehensive Assessment of Symptoms and History (CASH)(Andreasen, Flaum, & Arndt, 1992), on which schizophrenia (N=221) or schizoaffective disorder (N=14) diagnoses meeting DSM-III-R or DSM-IV criteria were based. All subjects were of Caucasian ancestry. Subjects were relatively young (Mean age=27.9 years (SD=9.44)), and had become psychiatrically ill recently (Mean age of illness onset=24.9 years (SD=8.4); Mean duration of illness=3.2 years (SD=5.7)).
2.2 Substance Use
Alcohol and illicit drug use was assessed using the CASH interview. Information from multiple sources (including the subject, available family informants and medical records) was utilized to determine lifetime substance abuse or dependence diagnoses meeting DSM criteria (Ho, Flaum, Hubbard, Arndt, & Andreasen, 2004). The CASH evaluates 8 drug categories: alcohol, barbiturates/hypnotics, opioids, cocaine, amphetamines/stimulants, phencyclidine, hallucinogens and marijuana. For each category, the patient is asked if he/she has ever used the drug, pattern of use, period of heaviest use, and questions relating to DSM abuse and dependence diagnostic criteria. Inter-rater reliability of alcohol/illicit drug abuse/dependence categorization was high (Mean intraclass r=.75 (SD=.16)).
Almost half the sample (N=106) had no prior marijuana exposure. Approximately one-third (N=77) had lifetime marijuana use not meeting DSM marijuana abuse or dependence criteria. Thirty-three patients (14.0%) had marijuana abuse and 19 (8.1%) marijuana dependence. In this study, we contrasted patients with marijuana abuse or dependence (N=52) against 183 patients who never met DSM criteria for marijuana abuse or dependence. Besides marijuana, 50 patients had lifetime alcohol abuse or dependence, 22 amphetamines abuse/dependence, 15 hallucinogen abuse/dependence, 9 cocaine abuse/dependence, 1 phencyclidine abuse/dependence, 1 opioid abuse/dependence, and 1 barbiturates abuse/dependence. Of the 52 patients with marijuana abuse/dependence, 31 patients had concurrent alcohol or non-cannabis illicit drug abuse/dependence.
2.3 Selection of CNR1 SNPs and Genotyping
In this study, we investigated tag single nucleotide polymorphisms (tSNPs) so as to maximally represent CNR1 common variants in the population. Twelve tSNPs (Figure 1) were selected using Haploview (Barrett, Fry, Maller, & Daly, 2005) (aggressive tagging 2-marker haplotype r2≥0.8) and the HapMap CEU population SNP database (http://www.hapmap.org, Release 22/Phase II). These tSNPs accounted for greater than 90% of the variance of all HapMap SNPs (minor allele frequencies ≥5%) within the genomic region surrounding CNR1 Exons 3 and 4. The 12 tSNPs span approximately 18.7kb at Chromosome 6q14-q15 (Mean distance between tSNPs=1.70kb; Median=1.49kb). To genotype study participants, DNA was prepared by high-salt extraction from whole blood (Lahiri & Nurnberger, 1991) and assayed using Illumina Infininum II array BeadChips which were designed, manufactured and completed by Illumina (San Diego, CA). Genotype call rates were 100% for each of the 12 CNR1 tSNPs. Illumina utilizes their proprietary GenCall data analysis software to ascertain quality and reliability of the genotypes called. A 10% GenCall score (i.e. the 10th percentile rank for all GenCall scores of the study samples at a given locus) greater than 0.7 is considered accurate and high-quality genotype data. The mean 10% GenCall scores for the 12 CNR1 tSNPs is 0.89 (SD=0.05; Range=0.80 – 0.96).
Figure 1.
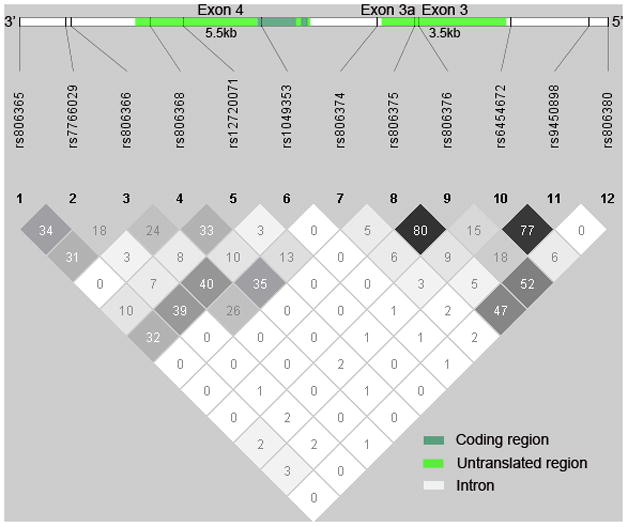
Cannabinoid receptor 1 (CNR1) gene structure in relation to tag single nucleotide polymorphisms (tSNPs) and pairwise linkage disequilibrium (D′) among 12 CNR1 tSNPs
2.4 MRI Acquisition and Image Processing
High-resolution morphometric brain MR data were collected using one of two imaging protocols. For subjects enrolled into the study before calendar year 2000, MRI brain scans were acquired on a 1.5-Tesla GE (General Electric Medical Systems, Milwaukee, Wisconsin) Signa MR scanner. In this earlier imaging protocol (termed ‘MR5’), three-dimensional T1-weighted images were acquired in the coronal plane using a spoiled GRASS sequence (SPGR). The parameters were: echo time (TE)=5ms, repetition time (TR)=24ms, numbers of excitations (NEX)=2, rotation angle=40 degrees, field of view (FOV)=26×19×18.6 cm, and a matrix of 256×192×124. Two-dimensional PD and T2 sequences were acquired as follows: 3.0 or 4.0 mm thick coronal slices, TR=3000ms, TE=36ms (for PD) and 96ms (for T2), NEX=1, FOV=26×26 cm, matrix=256×192. For subjects recruited in 2000 or later, we used a 1.5-Tesla Siemens Avanto scanner. In this more recent imaging protocol (termed ‘MR6’), the T1 sequence was obtained as a 3D volume in the coronal plane using a FLASH sequence with the following parameters: TE=6 ms, TR=20 ms, flip angle=30°, FOV=260×260×192 mm, matrix = 256×256×124, NEX=2, slice thickness 3 mm, ETL=5. The MR6 T2 images were acquired using a 2D fast spin-echo sequence in the coronal plane. The parameters were: TE=85 ms, TR=4800 ms, slice thickness/gap=1.8/0.0 mm, FOV=160×160 mm, matrix=256×256, NEX=3, number of echoes=8, number of slices=124.
MR images were processed using our locally developed BRAINS2 (Brain Research: Analysis of Images, Networks, and Systems, Version 2) software package (Magnotta et al., 2002). Detailed descriptions of image analysis methods have been provided elsewhere (Andreasen et al., 1993; Andreasen et al., 1994; Andreasen et al., 1996; Harris et al., 1999). In brief, the T1-weighted images were spatially normalized and re-sampled so that the anterior-posterior axis of the brain was realigned parallel to the anterior-posterior commissure line, and the interhemispheric fissure was aligned on the other two axes. The T2-weighted images were aligned to the spatially normalized T1-weighted image using an automated image registration program (Woods, Cherry, & Mazziotta, 1992). These images were then subjected to a linear transformation into standardized stereotaxic Talairach atlas space (Talairach & Tournoux, 1988) to generate automated measurements of frontal, temporal, parietal and occipital lobes (Andreasen et al., 1996). To further classify tissue volumes into gray matter (GM), white matter (WM) and cerebrospinal fluid (CSF), we employed a discriminant analysis method of tissue segmentation based on automated training class selection that utilized data from the T1 and T2 sequences (Harris et al., 1999). In this study, we examined total and lobar (Talairach atlas-based frontal, temporal and parietal sub-divisions) GM and WM brain volumes and lateral ventricles.
To enhance MR5 and MR6 data compatibility, MR6 scans were re-sampled into the same resolution and image size as MR5 scans so as to simulate similar amounts of partial volume effects in voxels that border two tissue types. To verify our ability to combine data from the 2 MR protocols, we have acquired both MR5 and MR6 scans on 60 patients (Ho, Andreasen, Ziebell, Pierson, & Magnotta, In Press). Brain volume differences between the two imaging sequences were small (Median percent difference=0.19%). Intra-class correlations were high across regions of interest (Median ICC=0.97). Hence, MR5 and MR6 data are compatible for combined statistical analyses.
2.5 Neurocognitive Assessment
All patients except 13 also underwent neurocognitive assessment administered by psychometrists who have been trained in standardized neuropsychological assessment and scoring procedures. Testing generally took approximately 4 hours to complete and, when necessary, occurred over several sessions. Assessment was performed when the subject was cooperative, clinically stable and psychiatric symptoms would not interfere with testing.
Full scale IQ scores were derived from the Wechsler Adult Intelligence Scale – Revised Edition (WAIS-R). Based on a priori theoretical considerations (Green et al., 2004; Hill, Schuepbach, Herbener, Keshavan, & Sweeney, 2004; Kareken et al., 1995; Milev, Ho, Arndt, & Andreasen, 2005; Saykin et al., 1994), 40 test variables from the standardized neuropsychological battery were grouped into 6 cognitive domains: Verbal Memory, Processing Speed/Attention, Problem Solving, Language, Visuospatial Abilities and Motor Skills. These cognitive domain groupings had good internal consistency (Median Cronbach’s alpha=0.80; Range=0.75–0.85)(Ho et al., 2003; Milev et al., 2005). The component neuropsychological test variables used to compute each cognitive domain score have been previously described (Milev et al., 2005). For Processing Speed/Attention domain score, these were: WAIS-R Digit Span, WAIS-R Digit Symbol, Trail Making A, and Stroop Color and Word Test (trials 1–3). Problem Solving domain score comprised of Wisconsin Card Sorting Test (WCST) number of categories attained, WCST number of perseverative errors, Shipley Institute of Living Scale abstractions subtest, WAIS-R Comprehension, Similarities, Picture Completion and Picture Arrangement subtests. Prior to deriving cognitive domain scores, raw test score from each neuropsychological test variable was converted to a z score (Mean=0, SD=1) based on norms established using 382 healthy volunteers. Scores were reversed where necessary. A larger negative score indicates poorer performance below the mean. Using these z scores, each domain score is the summed average of its component neuropsychological test variables.
2.6 Statistical analyses
Analyses were performed using Haploview (Barrett et al., 2005) and SAS software (version 9.2; SAS Institute, Cary, NC). Inter-correlations between the 12 CNR1 tSNPs were analyzed with pair-wise linkage disequilibrium (LD) statistics within Haploview. Because only a minority of the sample had heavy marijuana misuse, we grouped patients with marijuana abuse and patients with marijuana dependence together (N=52) for statistical analyses. Furthermore, since there were no significant group differences in sociodemographics, illness characteristics, MRI brain volumes or CNR1 tSNP allele frequencies between patients without prior marijuana exposure and patients whose marijuana use had not meet DSM marijuana abuse or dependence (data not shown; available upon request), these patients were grouped together (N=183) for comparison against patients with marijuana abuse or dependence. Group differences on categorical variables were tested using χ2-test and continuous variables independent group T-test or ANCOVA.
To assess brain volume-CNR1 relationships, statistical analyses were conducted in stages to reduce Type I error. We first tested whether there was an overall effect of each CNR1 genotype (minor allele carriers versus major allele homozygotes) on total cerebral GM or on total cerebral WM volumes. We used the adaptive false discovery rate (FDR) (Benjamini & Hochberg, 2000) procedure to control for potential type I error rate inflation due to multiple testing. Compared to familywise error rate controlling methods, FDR-controlling methods generally provide greater statistical power (Sabatti, Service, & Freimer, 2003). In each general linear model, total cerebral brain volume was entered as dependent measure, and genotype the independent variable. As a second step, for CNR1 genotypes in which the total cerebral brain volume test was statistically significant (FDR-adjusted p≤.05), follow-up analyses were performed to determine differential brain volume-CNR1 relationships across patients with versus patients without marijuana abuse/dependence. In each follow-up ANCOVA, the dependent variable was frontal, temporal or parietal lobar brain volume. Genotype, marijuana misuse (presence versus absence of lifetime marijuana abuse or dependence) and genotype-by-marijuana misuse interaction terms were the independent measures (p≤.05). Covariates in the total cerebral and follow-up lobar brain volume analyses were intracranial volume, age, gender, imaging protocol, antipsychotic treatment (lifetime antipsychotic exposure) and alcohol/non-cannabis illicit substance abuse/dependence. Intracranial volume adjusted for individual differences in overall cranial size. Gender, age and antipsychotic treatment are known to influence brain volumes. To further ensure that scan sequence variation was not producing erroneous results, we included imaging protocol (i.e. ‘MR5’ versus ‘MR6’ scanning protocol) as a covariate in the analyses. Alcohol/other illicit substance use (presence versus absence of lifetime alcohol abuse/dependence or non-marijuana illicit substance abuse/dependence) may affect brain volumes and potentially confound brain volume-CNR1 relationships.
Neurocognition-CNR1 relationships focused on SNPs where there were significant genotype-by-marijuana misuse interaction effects on brain volumes (p≤.05). In these ANCOVAs, the dependent measure was cognitive domain score (Verbal Memory, Processing Speed/Attention, Problem Solving, Language, Visuospatial Abilities or Motor Skills) with covariates being WAIS-R Full Scale IQ, gender, age and alcohol/non-cannabis illicit substance abuse/dependence.
3. Results
Allele frequency and pair-wise LD between the 12 CNR1 tSNPs are summarized in Table 1. Several of the tSNPs had comparatively lower allele frequencies than other tSNPs. Such differences led to inflated D′ estimates of LD with corresponding low r2 LD values (e.g. rs12720071 and rs1049353: D′=1.00 versus r2=.03). Therefore, r2 yielded a better measure of LD in our study sample. There were high inter-correlations between 2 pairs of tSNPs: rs806375/rs806376 (r2=.80) and rs6454672/rs9450898 (r2=.77). To reduce redundancy, rs806375 and rs6454672 were excluded from subsequent analyses. Genotype distributions in all 12 tSNPs were in Hardy-Weinberg equilibrium (p≥.06).
Table 1.
Pairwise linkage disequilibrium (r2 below and D′ above diagonal) between 12 cannabinoid receptor 1 (CNR1) tag single nucleotide polymorphisms (tSNP), SNP location on Chromosome 6q14-q15, and SNP allele frequencies in 235 schizophrenia patients
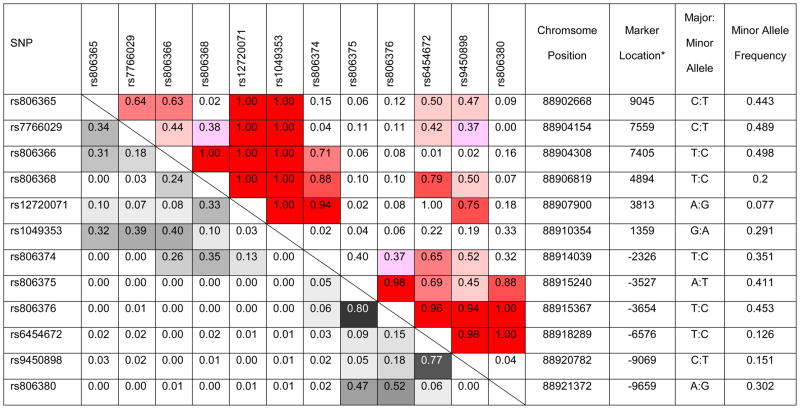 |
indicated as distances from translation start site
3.1 Differences between patients with and without lifetime history of marijuana abuse/dependence
Compared to patients without prior heavy marijuana use, patients with lifetime history of marijuana abuse/dependence were younger, more likely to be male and to have comorbid alcohol and/or non-marijuana illicit substance misuse (Table 2, p≤.001). Otherwise, the 2 comparison groups were comparable with respect to other sociodemographic measures, illness characteristics and antipsychotic treatment (p≥.10).
Table 2.
Comparison of sociodemographics, clinical characteristics, antipsychotic treatment and cannabinoid receptor 1 tag single nucleotide polymorphism allele frequencies between schizophrenia patients grouped by absence or presence of lifetime diagnosis of marijuana abuse/dependence
| No Marijuana Abuse/Dependence | With Marijuana Abuse/Dependence | T/χ2 (p) | |
|---|---|---|---|
| N | 183 | 52 | |
| Mean Age (years) | 29.0 (9.9) | 24.0 (6.5) | 4.29 (<.0001) |
| Males (N (%)) | 127 (69.4) | 48 (92.3) | 11.2 (.001) |
| Mean Parental SES | 2.9 (0.7) | 2.8 (0.6) | 1.32 (.10) |
| Mean Premorbid Adjustment1 | 5.4 (3.0) | 4.7 (2.4) | 1.57 (.12) |
| Mean Illness Duration (years) | 3.3 (6.0) | 2.5 (4.5) | 1.04 (.30) |
| Other Substance Use2 (N (%)) | 32 (17.5) | 31 (59.6) | 36.6 (<.0001) |
| Mean Symptom Severity3 | |||
| Psychotic symptoms | 6.5 (2.7) | 6.8 (2.4) | 0.77 (.44) |
| Disorganized symptoms | 4.8 (3.0) | 5.1 (3.0) | 0.63 (.53) |
| Negative symptoms | 11.4 (3.8) | 11.2 (3.4) | 0.36 (.72) |
| Antipsychotic naïve (N (%)) | 25 (13.7) | 8 (15.4) | 0.10 (.75) |
| Ever needed clozapine (N (%)) | 20 (10.9) | 4 (7.7) | 0.46 (.50) |
| Mean lifetime antipsychotic exposure4 | 16.8 (49.0) | 7.0 (16.2) | 0.94 (.35)5 |
| Mean daily antipsychotic dose6 | 257.9 (341.8) | 215.3 (235.2) | 0.11 (.91)5 |
| Typical antipsychotics7 | 47.2 (45.0) | 36.6 (40.9) | 1.40 (.16) |
| Non-clozapine atypical antipsychotics8 | 48.4 (45.7) | 60.2 (43.1) | 1.61 (.11) |
| Minor Allele Frequency (%) | |||
| rs806365 (T) | 44.0 | 45.2 | 0.04 (.82) |
| rs7766029 (T) | 48.1 | 51.9 | 0.47 (.48) |
| rs806366 (C) | 48.6 | 53.9 | 0.88 (.34) |
| rs806368 (C) | 19.4 | 22.1 | 0.37 (.54) |
| rs12720071 (G) | 7.7 | 7.7 | 0.00 (.98) |
| rs1049353 (A) | 31.2 | 22.1 | 3.19 (.07) |
| rs806374 (C) | 35.0 | 35.6 | 0.01 (.90) |
| rs806376 (C) | 46.5 | 41.4 | 0.85 (.35) |
| rs9450898 (T) | 15.0 | 15.4 | 0.00 (.92) |
| rs806380 (G) | 30.9 | 27.9 | 0.34 (.55) |
Modified Premorbid Adjustment Scale(Hollingshead & Redlich, 1958)
Lifetime alcohol and/or non-marijuana illicit drug abuse/dependence
Scale for the Assessment of Positive Symptoms and Scale for the Assessment of Negative Symptoms; Psychotic symptom dimension: Sum of delusions and hallucination global ratings; Disorganized symptom dimension: Sum of bizarre behavior, formal thought disorder and inappropriate affect global ratings; Negative symptom dimension: Sum of alogia, affective flattening, avolition/apathy and anhedonia/asociality global ratings
Dose years (1 dose year is equal to 100 mg chlorpromazine-equivalent per day for 1 year)
Wilcoxon two-sample rank sums test
Chlorpromazine-milligram equivalents per day
Mean proportion of lifetime antipsychotic exposure that comprised treatment using typical antipsychotics (%)
Mean proportion of lifetime antipsychotic exposure that comprised treatment using second generation antipsychotics (%; excluding clozapine)
Patients with marijuana abuse/dependence had smaller frontal WM (Adjusted Mean=175.6 versus 180.6cc in patients without marijuana abuse/dependence; F=3.10, p=.07; Figure 2a) and smaller temporal WM volumes (Adjusted Mean=68.6 and 70.0cc respectively; F=5.49, p=.02; Figure 2b). Gray matter, parietal WM and lateral ventricle volumes did not differ significantly between patients with and without marijuana abuse/dependence (F≤2.73, p≥.09).
Figure 2.

Comparison of (a) frontal and (b) temporal lobe white matter volumes between patients with and without marijuana abuse/dependence
Allele frequency distributions for CNR1 tSNPs also did not differ significantly between patients with and without marijuana abuse/dependence (Table 2; χ2≤3.19, p≥.07). For rs1049353, G-allele frequency was non-significantly more prevalent among patients with marijuana misuse (77.9% versus 68.8% in patients without marijuana abuse/dependence; p=.07). rs1049353 genotype frequencies across the two comparison groups did not differ significantly either (χ2=3.25, p=.20).
3.2 Relationships between CNR1 tSNPs and brain volumes across marijuana misuse groupings
After adjusting for multiple comparisons, the main effects of 3 CNR1 tSNPs on total cerebral WM brain volumes remained statistically significant (Table 3): rs7766029 (F=6.00, FDR-adjusted p=.05), rs12720071 (F=5.82, FDR-adjusted p=.05), and rs9450898 (F=8.24, FDR-adjusted p=.04). Pair-wise LD analysis of these 3 tSNPs found low inter-correlations (Table 1 and Figure 1; r2≤.07; Median r2=.02). For the other 7 tSNPs, there were no significant associations between CNR1 genotype and total cerebral WM or total cerebral GM brain volumes (Table 3; F≤2.34, FDR-adjusted p≥.13).
Table 3.
Relationships* between 10 CNR1 tag SNPs and total cerebral brain volumes
| Genotype | Total Cerebral GM (p) | Total Cerebral WM (p) | ||
|---|---|---|---|---|
| Uncorrected | FDR-Adjusted | Uncorrected | FDR-Adjusted | |
| rs806365 | .43 | .97 | .61 | .69 |
| rs7766029 | .01 | .13 | .01 | .05 |
| rs806366 | .74 | .99 | .13 | .29 |
| rs806368 | .99 | .99 | .82 | .75 |
| rs12720071 | .80 | .99 | .02 | .05 |
| rs1049353 | .58 | .97 | .36 | .54 |
| rs806374 | .58 | .97 | .83 | .75 |
| rs806376 | .23 | .97 | .23 | .42 |
| rs9450898 | .56 | .97 | .005 | .04 |
| rs806380 | .94 | .99 | .55 | .69 |
Analysis of covariance (uncorrected and false discovery rate (FDR)-adjusted p-values) assessing the main effects of CNR1 genotype on total cerebral GM or WM volumes (Covariates: Intracranial volume, age, sex, imaging protocol, antipsychotic treatment and alcohol/non-cannabis drug abuse/dependence); GM: gray matter; WM: white matter
Follow-up analyses investigating the inter-relationships between these 3 CNR1 tSNPs on lobar WM volumes across patients with versus without marijuana misuse are summarized in Table 4. Compared to rs12720071-A homozygotes, G-allele carriers had significantly smaller frontal and temporal WM volumes (p≤.05; Table 4 and Figures 3a and 3b). Most interestingly, there were significant rs12720071-genotype-by-marijuana misuse interaction effects on parietal WM volumes (p=.05; Table 4), which are suggestive of gene×environment interactions for conferring brain volume deficits in schizophrenia. rs12720071-G-allele carriers with marijuana abuse/dependence had the smallest mean parietal WM volumes than the other 3 subgroups (i.e. patients without marijuana abuse/dependence and rs12720071-A homozygotes with marijuana abuse/dependence; Figure 3c). Temporal and parietal WM volumes in rs7766029-C homozygotes were significantly smaller than WM volumes in rs7766029-T-allele carriers (p≤.05; Table 4 and Figure 4). rs9450898-C homozygotes had significantly smaller frontal and parietal WM volumes than T-allele carriers (p≤.05; Table 4 and Figure 4).
Table 4.
Follow-up analyses of relationships* between MRI lobar white matter brain volumes with CNR1 SNP genotype, marijuana abuse/dependence and interaction term
| Brain Volumes | Independent measure | rs12720071 | rs7766029 | rs9450898 |
|---|---|---|---|---|
| Frontal WM | Genotype | .01 | .08 | .03 |
| Marijuana Use | .01 | .06 | .09 | |
| G × MJ | .11 | .52 | .80 | |
| Temporal WM | Genotype | .05 | .02 | .05 |
| Marijuana Use | .01 | .03 | .25 | |
| G × MJ | .27 | .88 | .08 | |
| Parietal WM | Genotype | .09 | .05 | .0005 |
| Marijuana Use | .16 | .87 | .77 | |
| G × MJ | .05 | .86 | .42 | |
Analysis of covariance (p-value): Data are shown only for the 3 CNR1 tag SNPs (rs7766029, rs12720071, and rs9450898) which had statistically significant main effects on total cerebral WM volumes (Covariates: Intracranial volume, age, sex, imaging protocol, antipsychotic treatment and alcohol/non-cannabis drug abuse/dependence);
WM: White matter; Marijuana use: Patients with marijuana abuse/dependence versus patients without marijuana abuse/dependence; G × MJ: Genotype by marijuana use grouping interaction term
Figure 3.

Lobar white matter volume deficits (a, b and c), and cognitive impairment (Processing Speed/Attention (d) and Problem Solving (e)) among rs12720071-G-allele carriers with marijuana abuse/dependence (Abbreviations: cc, cubic centimeters; MJ Abuse/Dep, Marijuana Abuse/Dependence; AA, rs12720071-A-allele homozygotes (N=156 and 44 without and with MJ Abuse/Dep respectively for brain volumes; N=144 and 43 without and with MJ Abuse/Dep respectively for neurocognition); G-Carriers, rs12720071-G-allele carriers (N=27 and 8 without and with MJ Abuse/Dep respectively for brain volumes and for neurocognition)). Error bar represents standard deviation of the mean.
Figure 4.

Lobar white matter volume deficits among rs7766029-C-allele homozygotes (a and b; CC, rs7766029-C-allele homozygotes (N=46 and 15 without and with MJ Abuse/Dep respectively); T-Carriers, rs7766029-T-allele carriers (N=137 and 37 without and with MJ Abuse/Dep respectively)) and rs9450898-C-allele homozygotes (c and d; CC, rs9450898-C-allele homozygotes (N=134 and 40 without and with MJ Abuse/Dep respectively); T-Carriers, rs9450898-T-allele carriers (N=49 and 12 without and with MJ Abuse/Dep respectively)) (Abbreviations: cc, cubic centimeters; MJ Abuse/Dep, Marijuana Abuse/Dependence;). Error bar represents standard deviation of the mean.
3.3 Relationships between rs12720071 and neurocognition
There were significant rs12720071 genotype effects on Processing Speed/Attention and Problem Solving cognitive domain scores (F≥5.56, p≤.03; Figures 3d and 3e). G-allele carriers (who had smaller frontal and temporal WM volumes) had poorer performance on tests assessing Processing Speed/Attention and Problem Solving skills. Genotype-by-marijuana misuse interaction main effects were significant for Problem Solving (F=5.02, p=.04) but not for Processing Speed/Attention (F=1.24, p=.27). Again, rs12720071-G-allele carriers with marijuana abuse/dependence (who had the smallest parietal WM volumes) also had the worst Problem Solving test performance (Mean z=−1.78 versus −1.12, −1.19 and −1.23 in A homozygotes with and without marijuana misuse and G-allele carriers without marijuana misuse respectively; Figure 3e). There was no statistically significant rs12720071 genotype or genotype by-marijuana misuse interaction main effects on Verbal Memory, Language, Visuospatial Abilities and Motor Skills cognitive domain scores (F≤2.41, p≥.12).
Because there was significantly more males among patients with marijuana misuse (92.3% versus 69.4% in patients without marijuana misuse), we repeated the statistical analyses using only male subjects. Genotype effects on WM brain volumes and on neurocognition among males were similar to those using the entire study sample (data available upon request). As an alternative to single-marker analyses, we conducted exploratory haplotype analyses (see Supplementary Information). Haplotype-based findings were consistent with those from single-marker tests.
4. Discussion
In this study, we investigated 12 tSNPs representing much of the genetic variability surrounding the genomic region encoding CB1, the principal receptor for endogenous and exogenous cannabinoids. Of these, three independent CNR1 tSNPs had significant effects on brain volumes in schizophrenia patients. CNR1 rs12720071-G-allele carriers, rs7766029-C homozygotes, and rs9450898-C homozygotes were associated with smaller WM brain volumes than their respective counterparts. CNR1 also interacts with heavy marijuana use to influence white matter volume deficits and cognitive dysfunction; these results are suggestive of combined gene×environment influences in mediating phenotypic features of schizophrenia. Specifically, rs12720071 SNP G-allele carriers may be especially vulnerable to the impact of marijuana misuse on influencing parietal lobe WM volumes and on impairing problem solving skills. Lastly, we found schizophrenia patients with marijuana abuse/dependence had smaller frontotemporal WM volumes than patients without heavy marijuana use.
CB1 is the primary brain receptor activated by marijuana. It is down-regulated in the brain in response to delta-9-tetrahydrocannabinol (Breivogel et al., 1999; Romero, Berrendero, Garcia-Gil, Ramos, & Fernandez-Ruiz, 1998), the psychoactive component within marijuana. CB1 is a member of the Gi/Go-protein-coupled receptor superfamily (Pertwee, 1997). Its activation triggers a diverse range of cellular responses, including multiple second messenger transduction mechanisms important in regulating dopaminergic and GABAergic neurons (Eggan & Lewis, 2007; Gardner, 2005; Laviolette & Grace, 2006; Lewis & Hashimoto, 2007; Price et al., 2007). CB1 is widely expressed in the dorsolateral prefrontal cortex, hippocampus, thalamus and cerebellum (Pazos et al., 2005); which are also brain regions implicated in the pathophysiology of schizophrenia (Lewis & Gonzalez-Burgos, 2008) and in addiction/reward circuitry (Haber & Knutson, 2009). CB1 receptors are localized predominantly in neurons with the highest concentrations in afferent axon terminals, neuronal cell bodies, and dendrites (Ong & Mackie, 1999). However, recent studies have found CB1 in oligodendrocytes (Moldrich & Wenger, 2000; Rodriguez, Mackie, & Pickel, 2001) as well as in oligodendrocyte progenitor cells within the subventricular zones where post-natal glial proliferation occurs (Berrendero et al., 1998). Cannabinoid-mediated CB1 signaling has been shown to control post-natal subventricular zone oligodendrogenesis (Arevalo-Martin et al., 2007), and enhance oligodendrocyte (Molina-Holgado et al., 2002) and neuronal (Galve-Roperh, Palazuelos, Aguado, & Guzman, 2009; Harkany, Keimpema, Barabas, & Mulder, 2008) lineage cell survival during neurodevelopment. Thus, our findings of associations between CNR1 genetic variations and white brain volumes are consistent with the role of CB1 signaling in maintaining neural integrity and function.
Previous schizophrenia-CNR1 genetic association studies, restricted mostly to two CNR1 variants, have found mixed results. Several studies report no significant associations between schizophrenia and a synonymous SNP (rs1049353) within the CNR1 coding region on Exon 4 (Leroy et al., 2001; Seifert et al., 2007; Ujike et al., 2002). The (AAT)n trinucleotide repeat located 18kb downstream from the Exon 4 translational start site (Zhang et al., 2004) has been associated with schizophrenia and with hebephrenic schizophrenia subtype in several (Chavarría-Siles et al., 2008; Martinez-Gras et al., 2006; Ujike et al., 2002) but not all studies (Tsai, Wang, & Hong, 2000). Few prior schizophrenia-CNR1 genetic association studies have included schizophrenia patients with comorbid substance misuse. Martinez-Gras and colleagues did not find significant differences in (AAT)n trinucleotide variant allele frequencies between patients with and without substance abuse (Martinez-Gras et al., 2006). Leroy et al (Leroy et al., 2001) reported preponderance of rs1049353-G-allele among substance abusing schizophrenia patients. Similarly, patients with marijuana abuse/dependence in our study were also more likely to have the rs1049353-G-allele although this difference did not achieve statistical significance.
Associations between CNR1 and substance misuse have also not been consistently replicated. The (AAT)n variant has been linked with cocaine, heroin, alcohol and polysubstance abuse (Ballon et al., 2006; Comings et al., 1997; Johnson et al., 1997). However, these reports differed regarding the number of (AAT)n repeats that is associated with drug misuse risk. Although the majority of studies examining CNR1 SNPs reported significant associations with substance misuse, no uniform patterns of associations with specific SNPs have emerged (Agrawal et al., 2009; Chen et al., 2008; Hopfer et al., 2006; Hutchison et al., 2008; Schmidt et al., 2002; Zhang et al., 2004; Zuo, Kranzler, Luo, Covault, & Gelernter, 2007). Other investigators have failed to find significant relationships between substance use and (AAT)n tandem repeat (Covault, Gelernter, & Kranzler, 2001; Heller, Schneider, Seifert, Cimander, & Stuhrmann, 2001; Li et al., 2000; Zhang et al., 2004) or with CNR1 SNPs (Heller et al., 2001; Herman, Kranzler, Cubells, Gelernter, & Covault, 2006). This conflicting literature may in part be related to differences in study samples (e.g. participants differed in the type and/or severity of illicit drug use, ancestry/ethnicity, etc.), high comorbidity of different illicit drugs within a given patient, as well as overlapping and specific genetic influences mediating each type of illicit drug misuse (Kendler, Jacobson, Prescott, & Neale, 2003; Tsuang et al., 1998; Young, Rhee, Stallings, Corley, & Hewitt, 2006).
SNPs may be functional in exerting their effects on disease phenotype if the variant either 1) substitutes an amino acid and changes protein structure (Cargill et al., 1999; Sunyaev, Ramensky, & Bork, 2000), 2) alters mRNA expression, stability or localization through regulating transcription (e.g. disrupt transcription factor binding sites at intronic enhancer or promoter regions (Prokunina et al., 2002)) or via alternate splicing (e.g. affecting exonic splicing enhancers or silencers (Cartegni, Chew, & Krainer, 2002)), or 3) if the variant is in linkage disequilibrium with another functional SNP. There are two known SNPs within the CNR1 coding region, i.e. rs1049353 (which we examined in this study) and rs3505747. However, both SNPs are synonymous variants that do not result in amino acid substitution or protein structure alterations. In our study, we did not find significant rs1049353 genotype effects on MRI brain volumes or on marijuana misuse among schizophrenia patients.
The three CNR1 SNPs implicated in our study (i.e. rs7766029, rs12720071, and rs9450898) are localized to introns or within the untranslated region of Exon 4. No studies to-date has directly examined how these 3 SNPs may mediate CNR1 gene expression and function. There have not been genetic association studies linking these 3 SNPs to schizophrenia or to marijuana misuse either. Thus, the impact of these 3 SNPs on the pathophysiology of schizophrenia remains unknown. Nevertheless, previous studies have identified regulatory regions within cnr1/CNR1 (Borner, Bedini, Hollt, & Kraus, 2008; Zhang et al., 2004) that may provide useful indicators regarding the potential functional roles of rs9450898 and rs12720071 in mediating the neurobiology of schizophrenia. Based on the HapMap CEU population SNP database (http://www.hapmap.org), rs9450898 is in perfect LD with another CNR1 SNP rs2023239 (D′=1.0; r2=1.0). The latter has been associated with altered CNR1 Exon 3 RNA levels in the human brain (Zhang et al., 2004) as well as with prefrontal CB1 receptor expression differences (Hutchison et al., 2008). Thus, phenotypic abnormalities associated with rs9450898 observed in the current study may be related to LD with known functional variants, or to yet unknown direct effects of rs9450898 on altering transcriptional regulation. The genomic sequence surrounding rs12720071 (i.e. GATTC) may be a potential transcription factor binding site (Heinemeyer et al., 1998). Presence of the A-allele on rs12720071 predicts a transcription factor binding site for CCAAT/enhancer-binding protein beta (C/EBPbeta)(Akira et al., 1990). C/EBPbeta is a member of the transcription factor family comprising of basic leucine-zipper DNA-binding proteins that recognizes a common DNA-binding sequence (Vinson, Sigler, & McKnight, 1989). These C/EBP transcription factors can promote or repress gene expression to regulate embryonic neurogenesis, adult neuroplasticity, learning/memory and neuronal regeneration following injury (Alberini, Ghirardi, Metz, & Kandel, 1994; Cortes-Canteli, Pignatelli, Santos, & Perez-Castillo, 2002; Menard et al., 2002; Sterneck & Johnson, 1998; Taubenfeld, Milekic, Monti, & Alberini, 2001). Thus, the A>G substitution on rs12720071 may potentially disrupt C/EBPbeta transcription factor binding, and leads to WM volume deficits and cognitive dysfunction among rs12720071-G-alleles carriers with heavy marijuana use.
The limitations of this study include our relatively small sample of patients with marijuana misuse and absence of comparison groups. Future studies will need to include healthy controls and marijuana abusers without schizophrenia in order to determine the specificity of the effects of CNR1 gene polymorphisms on brain structure and function. Furthermore, our statistical analyses used FDR to adjust for multiple comparisons. Although FDR-controlling methods yield greater statistical power, these are less stringent than Bonferroni-correction or familywise error rate controlling methods. We chose a two-step analytic approach to limit Type I errors in order to explore the influence of CNR1 on carefully selected phenotypic measures. No prior studies have genotyped CNR1 to this level of genomic coverage in relation to phenotypic features of schizophrenia. Nonetheless, our findings of genotype-phenotype associations are preliminary and will require future replication. There is often substantial co-occurrence of alcohol and other illicit drug use among individuals with marijuana abuse/dependence. Even though our study sample is representative of this population, comorbid alcohol and non-marijuana substance misuse remains as a potential confounding factor in genotype-phenotype associations despite inclusion of the former as a covariate in our statistical analyses. Further studies will need to investigate marijuana abuse/dependent patients without other comorbid substance misuse in order to more definitively assess gene-marijuana interactive effects on brain morphology and cognition.
In conclusion, our study indicates that marijuana misuse in combination with specific CNR1 genotypes may contribute to WM volume deficits and cognitive impairment for a subgroup of schizophrenia patients. These findings suggest that for a proportion of patients within the heterogeneous grouping of schizophrenia, heavy marijuana misuse may be a necessary and/or a sufficient factor in mediating phenotypic features of the disorder.
Supplementary Material
Acknowledgments
This research was supported in part by NIMH Grants MH68380, MH31593, MH40856, MH80128 and MH43271, and Ortho-McNeil Janssen Scientific Affairs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Parts of this research were presented at the 48th Annual Meeting of the American College of Neuropsychopharmacology December 7, 2009; Hollywood, Florida
References
- Agrawal A, Wetherill L, Dick DM, Xuei X, Hinrichs A, Hesselbrock V, Kramer J, Nurnberger JI, Jr, Schuckit M, Bierut LJ, Edenberg HJ, Foroud T. Evidence for association between polymorphisms in the cannabinoid receptor 1 (CNR1) gene and cannabis dependence. Am J Med Genet B Neuropsychiatr Genet. 2009;150B (5):736–40. doi: 10.1002/ajmg.b.30881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Isshiki H, Sugita T, Tanabe O, Kinoshita S, Nishio Y, Nakajima T, Hirano T, Kishimoto T. A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. Embo J. 1990;9 (6):1897–906. doi: 10.1002/j.1460-2075.1990.tb08316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberini CM, Ghirardi M, Metz R, Kandel ER. C/EBP is an immediate-early gene required for the consolidation of long-term facilitation in Aplysia. Cell. 1994;76 (6):1099–114. doi: 10.1016/0092-8674(94)90386-7. [DOI] [PubMed] [Google Scholar]
- Andreasen NC, Cizadlo T, Harris G, Swayze V, O’Leary DS, Cohen G, Ehrhardt J, Yuh WT. Voxel processing techniques for the antemortem study of neuroanatomy and neuropathology using magnetic resonance imaging. J Neuropsychiatry Clin Neurosci. 1993;5 (2):121–30. doi: 10.1176/jnp.5.2.121. [DOI] [PubMed] [Google Scholar]
- Andreasen NC, Flashman L, Flaum M, Arndt S, Swayze V, 2nd, O’Leary DS, Ehrhardt JC, Yuh WT. Regional brain abnormalities in schizophrenia measured with magnetic resonance imaging. JAMA. 1994;272 (22):1763–9. [PubMed] [Google Scholar]
- Andreasen NC, Flaum M, Arndt S. The Comprehensive Assessment of Symptoms and History (CASH). An instrument for assessing diagnosis and psychopathology. Arch Gen Psychiatry. 1992;49 (8):615–23. doi: 10.1001/archpsyc.1992.01820080023004. [DOI] [PubMed] [Google Scholar]
- Andreasen NC, Rajarethinam R, Cizadlo T, Arndt S, Swayze VW, 2nd, Flashman LA, O’Leary DS, Ehrhardt JC, Yuh WT. Automatic atlas-based volume estimation of human brain regions from MR images. J Comput Assist Tomogr. 1996;20 (1):98–106. doi: 10.1097/00004728-199601000-00018. [DOI] [PubMed] [Google Scholar]
- Andreasson S, Allebeck P, Engstrom A, Rydberg U. Cannabis and schizophrenia. A longitudinal study of Swedish conscripts. Lancet. 1987;2 (8574):1483–6. doi: 10.1016/s0140-6736(87)92620-1. [DOI] [PubMed] [Google Scholar]
- Arevalo-Martin A, Garcia-Ovejero D, Rubio-Araiz A, Gomez O, Molina-Holgado F, Molina-Holgado E. Cannabinoids modulate Olig2 and polysialylated neural cell adhesion molecule expression in the subventricular zone of post-natal rats through cannabinoid receptor 1 and cannabinoid receptor 2. Eur J Neurosci. 2007;26 (6):1548–59. doi: 10.1111/j.1460-9568.2007.05782.x. [DOI] [PubMed] [Google Scholar]
- Arseneault L, Cannon M, Poulton R, Murray R, Caspi A, Moffitt TE. Cannabis use in adolescence and risk for adult psychosis: longitudinal prospective study. BMJ. 2002;325 (7374):1212–3. doi: 10.1136/bmj.325.7374.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballon N, Leroy S, Roy C, Bourdel MC, Charles-Nicolas A, Krebs MO, Poirier MF. (AAT)n repeat in the cannabinoid receptor gene (CNR1): association with cocaine addiction in an African-Caribbean population. Pharmacogenomics J. 2006;6 (2):126–30. doi: 10.1038/sj.tpj.6500352. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21 (2):263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. On the Adaptive Control of the False Discovery Rate in Multiple Testing With Independent Statistics. J Educ Behav Stat. 2000;25 (1):60–83. [Google Scholar]
- Berrendero F, Garcia-Gil L, Hernandez ML, Romero J, Cebeira M, de Miguel R, Ramos JA, Fernandez-Ruiz JJ. Localization of mRNA expression and activation of signal transduction mechanisms for cannabinoid receptor in rat brain during fetal development. Development. 1998;125 (16):3179–88. doi: 10.1242/dev.125.16.3179. [DOI] [PubMed] [Google Scholar]
- Borner C, Bedini A, Hollt V, Kraus J. Analysis of promoter regions regulating basal and interleukin-4-inducible expression of the human CB1 receptor gene in T lymphocytes. Mol Pharmacol. 2008;73 (3):1013–9. doi: 10.1124/mol.107.042945. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Vogt LJ, Sim-Selley LJ. Chronic delta9-tetrahydrocannabinol treatment produces a time-dependent loss of cannabinoid receptors and cannabinoid receptor-activated G proteins in rat brain. J Neurochem. 1999;73 (6):2447–59. doi: 10.1046/j.1471-4159.1999.0732447.x. [DOI] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Shaw N, Lane CR, Lim EP, Kalyanaraman N, Nemesh J, Ziaugra L, Friedland L, Rolfe A, Warrington J, Lipshutz R, Daley GQ, Lander ES. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22 (3):231–8. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3 (4):285–98. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- Chavarría-Siles I, Contreras-Rojas J, Hare E, Walss-Bass C, Quezada P, Dassori A, Contreras S, Medina R, Ramírez M, Salazar R, Raventos H, Escamilla MA. Cannabinoid receptor 1 gene (CNR1) and susceptibility to a quantitative phenotype for hebephrenic schizophrenia. Am J Med Genet Part B: Neuropsychiatric Genet. 2008;147B (3):279–284. doi: 10.1002/ajmg.b.30592. [DOI] [PubMed] [Google Scholar]
- Chen X, Williamson VS, An SS, Hettema JM, Aggen SH, Neale MC, Kendler KS. Cannabinoid receptor 1 gene association with nicotine dependence. Arch Gen Psychiatry. 2008;65 (7):816–24. doi: 10.1001/archpsyc.65.7.816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comings DE, Muhleman D, Gade R, Johnson P, Verde R, Saucier G, MacMurray J. Cannabinoid receptor gene (CNR1): association with i.v. drug use. Mol Psychiatry. 1997;2 (2):161–8. doi: 10.1038/sj.mp.4000247. [DOI] [PubMed] [Google Scholar]
- Cortes-Canteli M, Pignatelli M, Santos A, Perez-Castillo A. CCAAT/enhancer-binding protein beta plays a regulatory role in differentiation and apoptosis of neuroblastoma cells. J Biol Chem. 2002;277 (7):5460–7. doi: 10.1074/jbc.M108761200. [DOI] [PubMed] [Google Scholar]
- Covault J, Gelernter J, Kranzler H. Association study of cannabinoid receptor gene (CNR1) alleles and drug dependence. Mol Psychiatry. 2001;6 (5):501–2. doi: 10.1038/sj.mp.4000925. [DOI] [PubMed] [Google Scholar]
- D’Souza DC, Sewell RA, Ranganathan M. Cannabis and psychosis/schizophrenia: human studies. Eur Arch Psychiatry Clin Neurosci. 2009;259 (7):413–31. doi: 10.1007/s00406-009-0024-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt L, Hall W, Lynskey M. Testing hypotheses about the relationship between cannabis use and psychosis. Drug and Alcohol Dependence. 2003;71 (1):37–48. doi: 10.1016/s0376-8716(03)00064-4. [DOI] [PubMed] [Google Scholar]
- DeLisi LE. The effect of cannabis on the brain: can it cause brain anomalies that lead to increased risk for schizophrenia? Curr Opin Psychiatry. 2008;21 (2):140–50. doi: 10.1097/YCO.0b013e3282f51266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggan SM, Lewis DA. Immunocytochemical distribution of the cannabinoid CB1 receptor in the primate neocortex: a regional and laminar analysis. Cereb Cortex. 2007;17 (1):175–91. doi: 10.1093/cercor/bhj136. [DOI] [PubMed] [Google Scholar]
- Galve-Roperh I, Palazuelos J, Aguado T, Guzman M. The endocannabinoid system and the regulation of neural development: potential implications in psychiatric disorders. Eur Arch Psychiatry Clin Neurosci. 2009;259 (7):371–82. doi: 10.1007/s00406-009-0028-y. [DOI] [PubMed] [Google Scholar]
- Gardner EL. Endocannabinoid signaling system and brain reward: emphasis on dopamine. Pharmacol Biochem Behav. 2005;81 (2):263–84. doi: 10.1016/j.pbb.2005.01.032. [DOI] [PubMed] [Google Scholar]
- Green MF, Nuechterlein KH, Gold JM, Barch DM, Cohen J, Essock S, Fenton WS, Frese F, Goldberg TE, Heaton RK, Keefe RS, Kern RS, Kraemer H, Stover E, Weinberger DR, Zalcman S, Marder SR. Approaching a consensus cognitive battery for clinical trials in schizophrenia: the NIMH-MATRICS conference to select cognitive domains and test criteria. Biol Psychiatry. 2004;56 (5):301–7. doi: 10.1016/j.biopsych.2004.06.023. [DOI] [PubMed] [Google Scholar]
- Haber SN, Knutson B. The Reward Circuit: Linking Primate Anatomy and Human Imaging. Neuropsychopharmacology. 2009;35 (1):4–26. doi: 10.1038/npp.2009.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkany T, Keimpema E, Barabas K, Mulder J. Endocannabinoid functions controlling neuronal specification during brain development. Mol Cell Endocrinol. 2008;286 (1–2 Suppl 1):S84–90. doi: 10.1016/j.mce.2008.02.011. [DOI] [PubMed] [Google Scholar]
- Harris G, Andreasen NC, Cizadlo T, Bailey JM, Bockholt HJ, Magnotta VA, Arndt S. Improving tissue classification in MRI: a three-dimensional multispectral discriminant analysis method with automated training class selection. J Comput Assist Tomogr. 1999;23 (1):144–54. doi: 10.1097/00004728-199901000-00030. [DOI] [PubMed] [Google Scholar]
- Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Ignatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, Podkolodny NL, Kolchanov NA. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998;26 (1):362–7. doi: 10.1093/nar/26.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller D, Schneider U, Seifert J, Cimander KF, Stuhrmann M. The cannabinoid receptor gene (CNR1) is not affected in German i.v. drug users. Addict Biol. 2001;6 (2):183–187. doi: 10.1080/13556210020040271. [DOI] [PubMed] [Google Scholar]
- Henquet C, Krabbendam L, Spauwen J, Kaplan C, Lieb R, Wittchen HU, van Os J. Prospective cohort study of cannabis use, predisposition for psychosis, and psychotic symptoms in young people. BMJ. 2005a;330 (7481):11. doi: 10.1136/bmj.38267.664086.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquet C, Murray R, Linszen D, van Os J. The environment and schizophrenia: the role of cannabis use. Schizophr Bull. 2005b;31 (3):608–12. doi: 10.1093/schbul/sbi027. [DOI] [PubMed] [Google Scholar]
- Henquet C, Van Os J. Letter to the Editor: The coherence of the evidence linking cannabis with psychosis. Psychological Medicine. 2008;38 (03):461–464. doi: 10.1017/S0033291707002279. [DOI] [PubMed] [Google Scholar]
- Herman AI, Kranzler HR, Cubells JF, Gelernter J, Covault J. Association study of the CNR1 gene exon 3 alternative promoter region polymorphisms and substance dependence. Am J Med Genet B Neuropsychiatr Genet. 2006;141B (5):499–503. doi: 10.1002/ajmg.b.30325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman M, Vickerman P, Macleod J, Lewis G, Zammit S, Kirkbride J, Jones P. If cannabis caused schizophrenia - how many cannabis users may need to be prevented in order to prevent one case of schizophrenia? England and Wales calculations. Addiction. 2009;104 (11):1856–1861. doi: 10.1111/j.1360-0443.2009.02736.x. [DOI] [PubMed] [Google Scholar]
- Hill SK, Schuepbach D, Herbener ES, Keshavan MS, Sweeney JA. Pretreatment and longitudinal studies of neuropsychological deficits in antipsychotic-naive patients with schizophrenia. Schizophr Res. 2004;68 (1):49–63. doi: 10.1016/S0920-9964(03)00213-5. [DOI] [PubMed] [Google Scholar]
- Ho BC, Alicata D, Ward J, Moser DJ, O’Leary DS, Arndt S, Andreasen NC. Untreated initial psychosis: relation to cognitive deficits and brain morphology in first-episode schizophrenia. Am J Psychiatry. 2003;160 (1):142–8. doi: 10.1176/appi.ajp.160.1.142. [DOI] [PubMed] [Google Scholar]
- Ho BC, Andreasen NC, Ziebell S, Pierson R, Magnotta V. Long-term Antipsychotic Treatment and Brain Volume: A Longitudinal Study of First-Episode Schizophrenia. Arch Gen Psychiatry. doi: 10.1001/archgenpsychiatry.2010.199. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho BC, Flaum M, Hubbard W, Arndt S, Andreasen NC. Validity of symptom assessment in psychotic disorders: information variance across different sources of history. Schizophr Res. 2004;68 (2–3):299–307. doi: 10.1016/j.schres.2003.07.006. [DOI] [PubMed] [Google Scholar]
- Hollingshead AB, Redlich FC. Social Class and Mental Illness: A Community Study. John Wiley & Sons, Inc; New York: 1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfer CJ, Young SE, Purcell S, Crowley TJ, Stallings MC, Corley RP, Rhee SH, Smolen A, Krauter K, Hewitt JK, Ehringer MA. Cannabis receptor haplotype associated with fewer cannabis dependence symptoms in adolescents. Am J Med Genet B Neuropsychiatr Genet. 2006;141B (8):895–901. doi: 10.1002/ajmg.b.30378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison KE, Haughey H, Niculescu M, Schacht J, Kaiser A, Stitzel J, Horton WJ, Filbey F. The incentive salience of alcohol: translating the effects of genetic variant in CNR1. Arch Gen Psychiatry. 2008;65 (7):841–50. doi: 10.1001/archpsyc.65.7.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JP, Muhleman D, MacMurray J, Gade R, Verde R, Ask M, Kelley J, Comings DE. Association between the cannabinoid receptor gene (CNR1) and the P300 event-related potential. Mol Psychiatry. 1997;2 (2):169–71. doi: 10.1038/sj.mp.4000246. [DOI] [PubMed] [Google Scholar]
- Kareken DA, Gur RC, Mozley D, Mozley LH, Saykin AJ, Shtasel DL, Gur RE. Cognitive functioning and neuroanatomic volume measures in schizophrenia. Neuropsychology. 1995;9 (2):211–219. [Google Scholar]
- Kendler KS, Jacobson KC, Prescott CA, Neale MC. Specificity of genetic and environmental risk factors for use and abuse/dependence of cannabis, cocaine, hallucinogens, sedatives, stimulants, and opiates in male twins. Am J Psychiatry. 2003;160 (4):687–95. doi: 10.1176/appi.ajp.160.4.687. [DOI] [PubMed] [Google Scholar]
- Kohn Y, Lerer B. Excitement and confusion on chromosome 6q: the challenges of neuropsychiatric genetics in microcosm. Mol Psychiatry. 2005;10 (12):1062–73. doi: 10.1038/sj.mp.4001738. [DOI] [PubMed] [Google Scholar]
- Kumra S. Schizophrenia and cannabis use. Minn Med. 2007;90 (1):36–8. [PubMed] [Google Scholar]
- Lahiri DK, Nurnberger JI., Jr A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res. 1991;19 (19):5444. doi: 10.1093/nar/19.19.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviolette SR, Grace AA. The roles of cannabinoid and dopamine receptor systems in neural emotional learning circuits: implications for schizophrenia and addiction. Cell Mol Life Sci. 2006;63 (14):1597–613. doi: 10.1007/s00018-006-6027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy S, Griffon N, Bourdel MC, Olie JP, Poirier MF, Krebs MO. Schizophrenia and the cannabinoid receptor type 1 (CB1): association study using a single-base polymorphism in coding exon 1. Am J Med Genet. 2001;105 (8):749–52. doi: 10.1002/ajmg.10038. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Gonzalez-Burgos G. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology. 2008;33 (1):141–65. doi: 10.1038/sj.npp.1301563. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T. Deciphering the disease process of schizophrenia: the contribution of cortical GABA neurons. Int Rev Neurobiol. 2007;78:109–31. doi: 10.1016/S0074-7742(06)78004-7. [DOI] [PubMed] [Google Scholar]
- Li T, Liu X, Zhu ZH, Zhao J, Hu X, Ball DM, Sham PC, Collier DA. No association between (AAT)n repeats in the cannabinoid receptor gene (CNR1) and heroin abuse in a Chinese population. Mol Psychiatry. 2000;5 (2):128–30. doi: 10.1038/sj.mp.4000670. [DOI] [PubMed] [Google Scholar]
- Magnotta VA, Harris G, Andreasen NC, O’Leary DS, Yuh WTC, Heckel D. Structural MR image processing using the BRAINS2 toolbox. Comput Med Imaging Graph. 2002;26 (4):251. doi: 10.1016/s0895-6111(02)00011-3. [DOI] [PubMed] [Google Scholar]
- Martinez-Gras I, Hoenicka J, Ponce G, Rodriguez-Jimenez R, Jimenez-Arriero MA, Perez-Hernandez E, Ampuero I, Ramos-Atance JA, Palomo T, Rubio G. (AAT)n repeat in the cannabinoid receptor gene, CNR1: association with schizophrenia in a Spanish population. Eur Arch Psychiatry Clin Neurosci. 2006;256 (7):437–41. doi: 10.1007/s00406-006-0665-3. [DOI] [PubMed] [Google Scholar]
- Menard C, Hein P, Paquin A, Savelson A, Yang XM, Lederfein D, Barnabe-Heider F, Mir AA, Sterneck E, Peterson AC, Johnson PF, Vinson C, Miller FD. An essential role for a MEK-C/EBP pathway during growth factor-regulated cortical neurogenesis. Neuron. 2002;36 (4):597–610. doi: 10.1016/s0896-6273(02)01026-7. [DOI] [PubMed] [Google Scholar]
- Milev P, Ho BC, Arndt S, Andreasen NC. Predictive values of neurocognition and negative symptoms on functional outcome in schizophrenia: a longitudinal first-episode study with 7-year follow-up. Am J Psychiatry. 2005;162 (3):495–506. doi: 10.1176/appi.ajp.162.3.495. [DOI] [PubMed] [Google Scholar]
- Moldrich G, Wenger T. Localization of the CB1 cannabinoid receptor in the rat brain. An immunohistochemical study. Peptides. 2000;21 (11):1735–42. doi: 10.1016/s0196-9781(00)00324-7. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado E, Vela JM, Arevalo-Martin A, Almazan G, Molina-Holgado F, Borrell J, Guaza C. Cannabinoids promote oligodendrocyte progenitor survival: involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J Neurosci. 2002;22 (22):9742–53. doi: 10.1523/JNEUROSCI.22-22-09742.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore TH, Zammit S, Lingford-Hughes A, Barnes TR, Jones PB, Burke M, Lewis G. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet. 2007;370 (9584):319–28. doi: 10.1016/S0140-6736(07)61162-3. [DOI] [PubMed] [Google Scholar]
- Murray RM, Morrison PD, Henquet C, Di Forti M. Cannabis, the mind and society: the hash realities. Nat Rev Neurosci. 2007;8 (11):885–95. doi: 10.1038/nrn2253. [DOI] [PubMed] [Google Scholar]
- O’Shea M, McGregor IS, Mallet PE. Repeated cannabinoid exposure during perinatal, adolescent or early adult ages produces similar longlasting deficits in object recognition and reduced social interaction in rats. J Psychopharmacol. 2006;20 (5):611–21. doi: 10.1177/0269881106065188. [DOI] [PubMed] [Google Scholar]
- O’Shea M, Singh ME, McGregor IS, Mallet PE. Chronic cannabinoid exposure produces lasting memory impairment and increased anxiety in adolescent but not adult rats. J Psychopharmacol. 2004;18 (4):502–8. doi: 10.1177/026988110401800407. [DOI] [PubMed] [Google Scholar]
- Ong WY, Mackie K. A light and electron microscopic study of the CB1 cannabinoid receptor in primate brain. Neuroscience. 1999;92 (4):1177–91. doi: 10.1016/s0306-4522(99)00025-1. [DOI] [PubMed] [Google Scholar]
- Pazos MR, Nunez E, Benito C, Tolon RM, Romero J. Functional neuroanatomy of the endocannabinoid system. Pharmacol Biochem Behav. 2005;81 (2):239–47. doi: 10.1016/j.pbb.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther. 1997;74 (2):129–80. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- Price DA, Owens WA, Gould GG, Frazer A, Roberts JL, Daws LC, Giuffrida A. CB1-independent inhibition of dopamine transporter activity by cannabinoids in mouse dorsal striatum. J Neurochem. 2007;101 (2):389–96. doi: 10.1111/j.1471-4159.2006.04383.x. [DOI] [PubMed] [Google Scholar]
- Prokunina L, Castillejo-Lopez C, Oberg F, Gunnarsson I, Berg L, Magnusson V, Brookes AJ, Tentler D, Kristjansdottir H, Grondal G, Bolstad AI, Svenungsson E, Lundberg I, Sturfelt G, Jonssen A, Truedsson L, Lima G, Alcocer-Varela J, Jonsson R, Gyllensten UB, Harley JB, Alarcon-Segovia D, Steinsson K, Alarcon-Riquelme ME. A regulatory polymorphism in PDCD1 is associated with susceptibility to systemic lupus erythematosus in humans. Nat Genet. 2002;32 (4):666–9. doi: 10.1038/ng1020. [DOI] [PubMed] [Google Scholar]
- Rodriguez JJ, Mackie K, Pickel VM. Ultrastructural localization of the CB1 cannabinoid receptor in mu-opioid receptor patches of the rat Caudate putamen nucleus. J Neurosci. 2001;21 (3):823–33. doi: 10.1523/JNEUROSCI.21-03-00823.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero J, Berrendero F, Garcia-Gil L, Ramos JA, Fernandez-Ruiz JJ. Cannabinoid receptor and WIN-55,212-2-stimulated [35S]GTP gamma S binding and cannabinoid receptor mRNA levels in the basal ganglia and the cerebellum of adult male rats chronically exposed to delta 9-tetrahydrocannabinol. J Mol Neurosci. 1998;11 (2):109–19. doi: 10.1385/JMN:11:2:109. [DOI] [PubMed] [Google Scholar]
- Sabatti C, Service S, Freimer N. False Discovery Rate in Linkage and Association Genome Screens for Complex Disorders. Genetics. 2003;164 (2):829–833. doi: 10.1093/genetics/164.2.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saykin AJ, Shtasel DL, Gur RE, Kester DB, Mozley LH, Stafiniak P, Gur RC. Neuropsychological deficits in neuroleptic naive patients with first-episode schizophrenia. Arch Gen Psychiatry. 1994;51 (2):124–31. doi: 10.1001/archpsyc.1994.03950020048005. [DOI] [PubMed] [Google Scholar]
- Schmidt LG, Samochowiec J, Finckh U, Fiszer-Piosik E, Horodnicki J, Wendel B, Rommelspacher H, Hoehe MR. Association of a CB1 cannabinoid receptor gene (CNR1) polymorphism with severe alcohol dependence. Drug Alcohol Depend. 2002;65 (3):221–4. doi: 10.1016/s0376-8716(01)00164-8. [DOI] [PubMed] [Google Scholar]
- Schneider M, Koch M. The effect of chronic peripubertal cannabinoid treatment on deficient object recognition memory in rats after neonatal mPFC lesion. Eur Neuropsychopharmacol. 2007;17 (3):180–6. doi: 10.1016/j.euroneuro.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Seifert J, Ossege S, Emrich HM, Schneider U, Stuhrmann M. No association of CNR1 gene variations with susceptibility to schizophrenia. Neurosci Lett. 2007;426 (1):29–33. doi: 10.1016/j.neulet.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Sewell RA, Ranganathan M, D’Souza DC. Cannabinoids and psychosis. Int Rev Psychiatry. 2009;21 (2):152–62. doi: 10.1080/09540260902782802. [DOI] [PubMed] [Google Scholar]
- Stefanis NC, Delespaul P, Henquet C, Bakoula C, Stefanis CN, Van Os J. Early adolescent cannabis exposure and positive and negative dimensions of psychosis. Addiction. 2004;99 (10):1333–41. doi: 10.1111/j.1360-0443.2004.00806.x. [DOI] [PubMed] [Google Scholar]
- Sterneck E, Johnson PF. CCAAT/enhancer binding protein beta is a neuronal transcriptional regulator activated by nerve growth factor receptor signaling. J Neurochem. 1998;70 (6):2424–33. doi: 10.1046/j.1471-4159.1998.70062424.x. [DOI] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Bork P. Towards a structural basis of human non-synonymous single nucleotide polymorphisms. Trends in Genetics. 2000;16 (5):198–200. doi: 10.1016/s0168-9525(00)01988-0. [DOI] [PubMed] [Google Scholar]
- Talairach J, Tournoux P. Co-Planar Stereotaxic Atlas of the Human Brain. Thieme Medical Publishers; New York: 1988. [Google Scholar]
- Taubenfeld SM, Milekic MH, Monti B, Alberini CM. The consolidation of new but not reactivated memory requires hippocampal C/EBPbeta. Nat Neurosci. 2001;4 (8):813–8. doi: 10.1038/90520. [DOI] [PubMed] [Google Scholar]
- Tsai SJ, Wang YC, Hong CJ. Association study of a cannabinoid receptor gene (CNR1) polymorphism and schizophrenia. Psychiatric Genetics. 2000;10 (3):149–51. doi: 10.1097/00041444-200010030-00008. [DOI] [PubMed] [Google Scholar]
- Tsuang MT, Lyons MJ, Meyer JM, Doyle T, Eisen SA, Goldberg J, True W, Lin N, Toomey R, Eaves L. Co-occurrence of abuse of different drugs in men: the role of drug-specific and shared vulnerabilities. Arch Gen Psychiatry. 1998;55 (11):967–72. doi: 10.1001/archpsyc.55.11.967. [DOI] [PubMed] [Google Scholar]
- Ujike H, Takaki M, Nakata K, Tanaka Y, Takeda T, Kodama M, Fujiwara Y, Sakai A, Kuroda S. CNR1, central cannabinoid receptor gene, associated with susceptibility to hebephrenic schizophrenia. Mol Psychiatry. 2002;7 (5):515–8. doi: 10.1038/sj.mp.4001029. [DOI] [PubMed] [Google Scholar]
- van Os J, Bak M, Hanssen M, Bijl RV, de Graaf R, Verdoux H. Cannabis use and psychosis: a longitudinal population-based study. Am J Epidemiol. 2002;156 (4):319–27. doi: 10.1093/aje/kwf043. [DOI] [PubMed] [Google Scholar]
- Vinson CR, Sigler PB, McKnight SL. Scissors-grip model for DNA recognition by a family of leucine zipper proteins. Science. 1989;246 (4932):911–6. doi: 10.1126/science.2683088. [DOI] [PubMed] [Google Scholar]
- Wegener N, Koch M. Behavioural disturbances and altered Fos protein expression in adult rats after chronic pubertal cannabinoid treatment. Brain Res. 2009;1253:81–91. doi: 10.1016/j.brainres.2008.11.081. [DOI] [PubMed] [Google Scholar]
- Woods RP, Cherry SR, Mazziotta JC. Rapid automated algorithm for aligning and reslicing PET images. J Comput Assist Tomogr. 1992;16 (4):620–33. doi: 10.1097/00004728-199207000-00024. [DOI] [PubMed] [Google Scholar]
- Young SE, Rhee SH, Stallings MC, Corley RP, Hewitt JK. Genetic and environmental vulnerabilities underlying adolescent substance use and problem use: general or specific? Behavior Genetics. 2006;36 (4):603–15. doi: 10.1007/s10519-006-9066-7. [DOI] [PubMed] [Google Scholar]
- Zammit S, Allebeck P, Andreasson S, Lundberg I, Lewis G. Self reported cannabis use as a risk factor for schizophrenia in Swedish conscripts of 1969: historical cohort study. BMJ. 2002;325 (7374):1199. doi: 10.1136/bmj.325.7374.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang PW, Ishiguro H, Ohtsuki T, Hess J, Carillo F, Walther D, Onaivi ES, Arinami T, Uhl GR. Human cannabinoid receptor 1: 5′ exons, candidate regulatory regions, polymorphisms, haplotypes and association with polysubstance abuse. Mol Psychiatry. 2004;9 (10):916–31. doi: 10.1038/sj.mp.4001560. [DOI] [PubMed] [Google Scholar]
- Zuo L, Kranzler HR, Luo X, Covault J, Gelernter J. CNR1 variation modulates risk for drug and alcohol dependence. Biol Psychiatry. 2007;62 (6):616–26. doi: 10.1016/j.biopsych.2006.12.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.