

Abstract
As a part of our program to develop OX1-CB1 bivalent ligands, we required a better understanding of the basic structure-activity relationships (SARs) of orexin antagonists. A series of SB-334867 analogues were synthesized and evaluated in calcium mobilization assays. SAR results suggest that the 2-methylbenzoxazole moiety may be replaced with a disubstituted 4-aminophenyl group without loss of activity and an electron-deficient system is generally preferred at the 1,5-naphthyridine moiety for OX1 antagonist activity. In particular, substitution of larger potential linkers such as n-hexyl provided compound 33 with equivalent activity at the OX1 receptor compared to the lead compound SB-334867. These compounds should be of value in the development of ligands targeting the orexin-1 receptor and its potential heterodimers.
Keywords: Orexin, Antagonist, SB-334867, Structure-activity relationships
Orexin-A and -B (also known as hypocretins 1 and 2) are two hypothalamic neuropeptides that were independently discovered by two groups in 1998.1,2 Orexin-A (33 amino acids) and orexin-B (28 amino acids) are highly conserved across mammalian species and are derived by proteolytic cleavage from a common 130-amino acid precursor produced in the hypothalamus named prepro-orexin. Orexin-A and -B are the endogenous ligands for two G protein-coupled receptors (GPCRs), orexin 1 (OX1R) and orexin 2 (OX2R). Orexin-A is equipotent at both receptors, whereas orexin-B displays moderate selectivity for the OX2R.2 Orexin-expressing neurons are located predominantly in a small area in the hypothalamus and locus coeruleus.2–5 However, the nerve fibers of orexin neurons project throughout the central nervous system (CNS), suggesting that orexins have multiple CNS functions.3,6–8 In fact, the orexin system has already been shown to modulate a variety of important biological processes, including sleep/wake cycles,9,10 feeding,2 drug addiction and reward,11–13 as well as energy homeostasis.2
A significant body of evidence indicates that many GPCRs can form heterodimers or oligomers, and these heterodimers/oligomers often display unique binding, distinct phenotypic trafficking, and altered signaling properties than their individual monomers.14–16 In particular, OX1R has an overlapping pattern of distribution with the cannabinoid receptor 1 (CB1) receptor in a number of brain regions including the lateral hippocampus.17 In addition, the OX1R has been shown to form heterodimers in vitro with the CB1 receptor, as demonstrated by co-expression, co-immunoprecipitation and resonance energy transfer studies.18 Biochemical, pharmacological and functional evidence also suggests a crosstalk between OX1R and CB1R in a Chinese Hamster Ovary (CHO) heterologous expression system, where potency of direct activation of OX1R to activate the mitogen-activated protein kinase pathway was affected by the CB1 receptor.19 Evidence supporting the in vivo relevance of OX1-CB1 dimerization includes the observation that in pre-fed rats, pretreatment with subeffective doses of the CB1 antagonist SR141716 attenuates the orexigenic actions of orexin A.20
Considering both the physiological functions of the orexin system and the potential to modulate OX1-CB1 heterodimers with small molecules we have pursued a program to develop bivalent ligands,21 which feature an OX1R antagonist and a CB1 antagonist linked together by a spacer. Although several structural classes of orexin antagonists have been developed,22–26 there is little structure activity data available that can be used to develop bivalent ligands. Therefore, identifying a region of bulk tolerance without altering OXR activity is critical. SB-334867 (1), developed by GlaxoSmithKline (GSK), was the first selective OX1 antagonist described (Fig 1).27 It has ~50-fold higher affinity for OX1R than for OX2R and was selective for OXR over more than 50 GPCRs and ion channels.27 GSK later reported other OX1 selective antagonists, including SB-408124 (2), SB-410220 (3) and SB-674042 (4) (Fig. 1);28 however, SB-334867 remains the most widely studied OX1 antagonist and represents a viable pharmacological tool for evaluating the physiological role of the OX1R specific pathway in vivo. As a part of our program to construct OX1-CB1 bivalent ligands to study GPCR heterodimerization we required a better understanding of the basic structure-activity relationships (SARs) of SB-334867. Here we describe the synthesis and biological evaluation of a series of diaryl urea analogues of SB-334867.
Figure 1.

Structures of SB334867 and other OX1 selective antagonists.
2-Methylquinoline analogue (5) was synthesized following a slightly modified literature procedure as shown in Scheme 1.29 2-Methylbenzoxazole acid (8) was obtained in three steps from commercially available methyl 3-hydroxy-4-nitrobenzoate. Reduction of the nitro group gave 6 followed by condensation with ethyl acetimidate to form benzoxazole ring 7, and subsequent ester hydrolysis provided 8. Curtius rearrangement of 8 using diphenylphosphoryl azide formed an in situ isocyanate, which was treated with 2-methyl-4-aminoquinoline to afford the desired urea 5. Compounds 9–23 and 33 were synthesized by combining the requisite 4-aminoquinoline derivative with the appropriate 4-nitrophenylisocyanate (9–13). The resulting 4-nitro compounds were reduced using 10% palladium on carbon and subjected to reductive alkylation (15–17, 19–23, 33) or BOP-mediated amidation (18) (Scheme 2). Similarly, 24–28, 30–32 and 34–37 were prepared by condensation of the appropriate 4-aminoquinoline with the arylisocyanate. Formation of thiourea 26 required sodium hydride activation of the aminoquinoline followed by condensation with 4-dimethylaminophenylisothiocyanate at −20°C. Amide 29 was prepared by BOP coupling of 8-fluoro-2-methyl-4-aminoquinoline with 4-dimethylaminophenylacetic acid. Compounds 38 and 39 were obtained through formation of the phenyl carbamate of aminoquinoline (40) followed by displacement of the phenoxide with the appropriate amine (Scheme 3). All target compounds were fully characterized by NMR and mass spectroscopy,30 and then tested in a calcium mobilization based functional assay.31
Scheme 1.

Reagents and conditions: (a) H2, Pd/C, EtOH; (b) ethyl acetimidate hydrochloride, EtOH; (c) 2N NaOH, MeOH; (d) 2-methyl-4-aminoquinoline, PO(OPh)2N3, Et3N, toluene, DMF.
Scheme 2.

Reagents and conditions: (a) 4-NO2-Ph-NCO, toluene, 80°C; (b) H2 (40 psi), Pd/C, EtOH; (c) aldehyde, Na(OAc)3BH, 1,2-DCE; (d) acid, BOP, Et3N, THF.
Scheme 3.

Reagents and conditions: (a) arylisocyanate, toluene, 80 °C or arylisothiocyanate, NaH, DMF, −20°C; (b) PhOCOCl, Et3N, CH2Cl2; (c) R2-NH2, Et3N, THF.
The primary goal of this study was to develop SAR surrounding the SB-334867 scaffold that could be used to identify suitable sites to attach linkers for bivalent ligand development. Since little SAR is known regarding this class of ligands at OX receptors we broadened our scope to evaluate multiple positions and substitutions. Initial attempts to develop SAR around SB-334867 included replacing the naphthyridine and the 2-methylbenzoxazole rings, respectively, with other aromatic systems (Table 1). The naphthyridine in SB-334867 was first replaced with 2-methylquinoline (5), which led to a modest reduction in activity (12 fold). Since 2-methyl-4-aminoquinoline 5 had reasonable activity and was commercially available, this group was employed as the core scaffold for evaluating substitutions on the 2-methylbenzoxazole moiety. The 4-nitro derivatives (9–13) were inactive as antagonists at OX1R. Aniline analogue 14 was also inactive under the test conditions in both OX1 and OX2 assays. When 14 was dimethylated (15), activity was restored albeit 7-fold weaker than SB-334867. Introduction of an ortho methyl (16) or methoxy (17) group to 15 significantly decreased potency. Monoalkylation, acylation or dialkylation with long chain alkyl groups (18–22) resulted in markedly reduced activity. Finally, replacing the 2-methylbenzoxazole ring in SB-334867 with a 4-dimethylaminophenyl group (23) restored the activity and selectivity for the OX1R, which is consistent with previous data for SB-408124 and SB-410220.28
Table 1.
Activity of quinoline urea analogues against OX1R and OX2R.
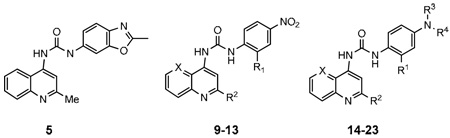 | |||||||
|---|---|---|---|---|---|---|---|
| No. | R1 | R2 | R3 | R4 | X | Ke (nM) | |
| OX1a | OX2b | ||||||
| 1 | 45±12 | 1194 | |||||
| 5 | 682.6±231 | >10,000 | |||||
| 9 | H | H | NO2 | N | >10,000 | nt | |
| 10 | H | Me | NO2 | CH | >10,000 | nt | |
| 11 | Cl | Me | NO2 | CH | >10,000 | nt | |
| 12 | Me | Me | NO2 | CH | >10,000 | nt | |
| 13 | OMe | Me | NO2 | CH | >10,000 | nt | |
| 14 | H | Me | H | H | CH | >10,000 | >10,000 |
| 15 | H | Me | Me | Me | CH | 356.7±89 | >10,000 |
| 16 | Me | Me | Me | Me | CH | 2561±266 | >10,000 |
| 17 | OMe | Me | Me | Me | CH | 3945±2517 | >10,000 |
| 18 | H | Me | H | Hexanoyl | CH | >10,000 | >10,000 |
| 19 | H | Me | H | 3-Ph-propyl | CH | 3278±639 | >10,000 |
| 20 | H | Me | Me | 3-Ph-propyl | CH | >10,000 | >10,000 |
| 21 | H | Me | H | Hexyl | CH | >10,000 | >10,000 |
| 22 | H | Me | Me | Hexyl | CH | 398±16 | >10,000 |
| 23 | H | H | Me | Me | N | 46.3±8 | >10,000 |
values are the mean of at least three independent experiments in duplicate.
values are the mean of at least two independent experiments in duplicate.
nt = not tested.
Since the 4-dimethylaminophenyl group retained comparable activity as the 2-methylbenzoxazole (5 vs. 15) this moiety was used to further probe the SAR of the naphthyridine ring (Table 2). Considering electron rich 2-methylquinoline had reduced activity (5), electron deficient aromatic rings were explored. Introduction of an 8-fluoro group to quinoline 24 or 2-methylquinoline 25 provided analogues with excellent potency, as well as selectivity for the OX1R. The corresponding thiourea 26 was significantly less active suggesting a steric limitation exists at this region. In addition, the position of the trifluoromethyl substituent was critical, as shown by the loss of activity in 27 compared to 28. Finally, removal of the urea as shown in the corresponding amide provided analogue 29 with no antagonist activity, confirming the importance of the urea group for optimal potency.
Table 2.
Activity of 4-dimethylaminophenyl quinoline ureas against OX1R and OX2R.
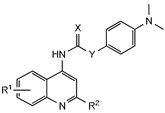 | ||||||
|---|---|---|---|---|---|---|
| No. | R1 | R2 | X | Y | Ke (nM) | |
| OX1a | OX2b | |||||
| 24 | 8-F | H | O | NH | 2.4±1.7 | 171±25 |
| 25 | 8-F | Me | O | NH | 65±33 | >10,000 |
| 26 | 8-F | H | S | NH | 1115.1 | >10,000 |
| 27 | 6-CF3 | H | O | NH | >10,000 | >10,000 |
| 28 | 8-CF3 | H | O | NH | 131±33 | >10,000 |
| 29 | 8-F | H | O | CH2 | >10,000 | >10,000 |
values are the mean of at least three independent experiments in duplicate.
values are the mean of at least two independent experiments in duplicate
Having identified an 8-fluoroquinoline core with improved activity, the effects of substitution on the 2-methylbenzoxazole ring was re-examined (table. 3). While the 4-dimethylaminophenyl derivative (24) showed the best activity, its conformationally constrained analog indoline 32 had almost identical potency. The piperidine derivative (30) was 100-fold less potent than 24 and activity was completely abolished when N-methylpiperazine (31) was substituted for the dimethylamino group, indicating that cyclic hydrophobic structures at this position are not well tolerated. When a longer n-hexyl chain was substituted for methyl on the aniline nitrogen (33), a reduction in potency compared to 24 was seen but it retained comparable activity with SB-334867, suggesting flexibility in the hydrophobic chain on the aniline nitrogen is critical for potent activity. This finding was significant since compound 33 contained a potential site for linker attachment for bivalent ligands. Favorable antagonist activity was also observed for oxygen-containing substituents. Although the dimethoxy analogue (36) was an weak antagonist and the 3,4,5-trimethoxy derivative (37) was inactive, constraining the two oxygens as in the 5 or 6 membered rings of 34 and 35 gave good activity. The simple 4-ethyl analogue (38) showed surprisingly good activity. Finally, cyclohexyl derivative (39) was completely inactive.
Table 3.
Activity of 8-fluoroquinoline urea analogues against OX1R and OX2R.
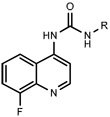 | |||
|---|---|---|---|
| Ke (nM) | |||
| No. | R | OX1a | OX2b |
| 24 |  |
2.4±1.7 | 171±25 |
| 30 | 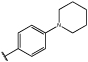 |
395.6±111 | >10,000 |
| 31 | 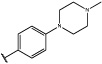 |
>10,000 | >10,000 |
| 32 | 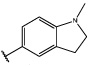 |
4.9±2 | 491±90 |
| 33 | 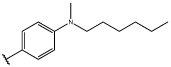 |
64.2±12 | 2410±250 |
| 34 | 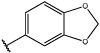 |
13.4±9 | 1617±50 |
| 35 | 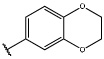 |
10.3±5 | 306±30 |
| 36 | 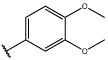 |
965±419 | >10,000 |
| 37 | 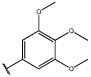 |
>10,000 | >10,000 |
| 38 | 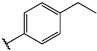 |
50.5±18 | 3199±207 |
| 39 | 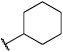 |
4563±1797 | >10,000 |
values are the mean of at least three independent experiments in duplicate.
values are the mean of at least two independent experiments in duplicate
All target compounds showed a strong selectivity for OX1R over OX2R, with the majority showing no activity at OX2R and those that did show activity on OX2 possessed at least a 30-fold selectivity for OX1R. All compounds were also evaluated for agonist activity and none of the analogues demonstrated any appreciable agonist stimulation at 10 µM.
In conclusion, we have synthesized a series of analogues of SB-334867 in order to define critical receptor tolerances to substitution for potential use as bivalent ligands. We have characterized our analogues using a calcium mobilization functional assay and identified several important structural features. SAR results suggest that the 2-methylbenzoxazole moiety may be replaced with a disubstituted 4-aminophenyl group without loss of activity and an electron-deficient system is generally preferred at the 1,5-naphthyridine moiety for OX1 antagonist activity. In particular, substitution of a larger n-hexyl chain for methyl provided compound 33 with roughly equal activity at the OX1 receptor compared to the lead compound SB-334867 suggesting a region of bulk tolerance that can be exploited at this position. Further modification of this scaffold is underway to optimize OX potency and develop bivalent ligands containing OX1 receptor ligands. Future compounds based on these scaffolds should be of value in the development of ligands targeting the orexin-1 receptor and its potential heterodimers.
Acknowledgments
This work was supported by National Institute on Drug Abuse, National Institute of Health, USA (Grant No. DA026582). We thank Ms. Tiffany Langston, Mr. Keith Warner and Ms. Allyson Smith for their valuable technical assistance. We also thank Dr. Hernan A. Navarro and Dr. James B. Thomas for their help in the calcium mobilization assay development.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, 2nd, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. Proc Natl Acad Sci U S A. 1998;95:322. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Cell. 1998;92:573. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 3.Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. J Neurosci. 1998;18:9996. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Date Y, Ueta Y, Yamashita H, Yamaguchi H, Matsukura S, Kangawa K, Sakurai T, Yanagisawa M, Nakazato M. Proc Natl Acad Sci U S A. 1999;96:748. doi: 10.1073/pnas.96.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nambu T, Sakurai T, Mizukami K, Hosoya Y, Yanagisawa M, Goto K. Brain Res. 1999;827:243. doi: 10.1016/s0006-8993(99)01336-0. [DOI] [PubMed] [Google Scholar]
- 6.Sutcliffe JG, de Lecea L. Nat Rev Neurosci. 2002;3:339. doi: 10.1038/nrn808. [DOI] [PubMed] [Google Scholar]
- 7.Sakurai T. Sleep Med Rev. 2005;9:231. doi: 10.1016/j.smrv.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 8.de Lecea L, Sutcliffe JG. FEBS J. 2005;272:5675. doi: 10.1111/j.1742-4658.2005.04981.x. [DOI] [PubMed] [Google Scholar]
- 9.Kilduff TS, Peyron C. Trends Neurosci. 2000;23:359. doi: 10.1016/s0166-2236(00)01594-0. [DOI] [PubMed] [Google Scholar]
- 10.Ohno K, Sakurai T. Front Neuroendocrinol. 2008;29:70. doi: 10.1016/j.yfrne.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 11.Harris GC, Wimmer M, Aston-Jones G. Nature. 2005;437:556. doi: 10.1038/nature04071. [DOI] [PubMed] [Google Scholar]
- 12.Tsujino N, Sakurai T. Pharmacol Rev. 2009;61:162. doi: 10.1124/pr.109.001321. [DOI] [PubMed] [Google Scholar]
- 13.Boutrel B, de Lecea L. Physiol Behav. 2008;93:947. doi: 10.1016/j.physbeh.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minneman KP. Biochem Pharmacol. 2007;73:1043. doi: 10.1016/j.bcp.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuxe K, Marcellino D, Rivera A, Diaz-Cabiale Z, Filip M, Gago B, Roberts DC, Langel U, Genedani S, Ferraro L, de la Calle A, Narvaez J, Tanganelli S, Woods A, Agnati LF. Brain Res Rev. 2008;58:415. doi: 10.1016/j.brainresrev.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 16.Szidonya L, Cserzo M, Hunyady L. J Endocrinol. 2008;196:435. doi: 10.1677/JOE-07-0573. [DOI] [PubMed] [Google Scholar]
- 17.Hervieu GJ, Cluderay JE, Harrison DC, Roberts JC, Leslie RA. Neuroscience. 2001;103:777. doi: 10.1016/s0306-4522(01)00033-1. [DOI] [PubMed] [Google Scholar]
- 18.Ellis J, Pediani JD, Canals M, Milasta S, Milligan G. J Biol Chem. 2006;281:38812. doi: 10.1074/jbc.M602494200. [DOI] [PubMed] [Google Scholar]
- 19.Hilairet S, Bouaboula M, Carriere D, Le Fur G, Casellas P. J Biol Chem. 2003;278:23731. doi: 10.1074/jbc.M212369200. [DOI] [PubMed] [Google Scholar]
- 20.Crespo I, Gomez de Heras R, Rodriguez de Fonseca F, Navarro M. Neuropharmacology. 2008;54:219. doi: 10.1016/j.neuropharm.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Gilliam A, Maitra R, Damaj MI, Tajuba JM, Seltzman HH, Thomas BF. J Med Chem. 2010;53:7048. doi: 10.1021/jm1006676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roecker AJ, Coleman PJ. Curr Top Med Chem. 2008;8:977. doi: 10.2174/156802608784936746. [DOI] [PubMed] [Google Scholar]
- 23.Boss C, Brisbare-Roch C, Jenck F. J Med Chem. 2009;52:891. doi: 10.1021/jm801296d. [DOI] [PubMed] [Google Scholar]
- 24.Kodadek T, Cai D. Mol Biosyst. 2010;6:1366. doi: 10.1039/c003468a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gatfield J, Brisbare-Roch C, Jenck F, Boss C. ChemMedChem. 2010;5:1197. doi: 10.1002/cmdc.201000132. [DOI] [PubMed] [Google Scholar]
- 26.Scammell TE, Winrow CJ. Annu Rev Pharmacol Toxicol. 2011;51:243. doi: 10.1146/annurev-pharmtox-010510-100528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smart D, Sabido-David C, Brough SJ, Jewitt F, Johns A, Porter RA, Jerman JC. Br J Pharmacol. 2001;132:1179. doi: 10.1038/sj.bjp.0703953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langmead CJ, Jerman JC, Brough SJ, Scott C, Porter RA, Herdon HJ. Br J Pharmacol. 2004;141:340. doi: 10.1038/sj.bjp.0705610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porter RA, Chan WN, Coulton S, Johns A, Hadley MS, Widdowson K, Jerman JC, Brough SJ, Coldwell M, Smart D, Jewitt F, Jeffrey P, Austin N. Bioorg Med Chem Lett. 2001;11:1907. doi: 10.1016/s0960-894x(01)00343-2. [DOI] [PubMed] [Google Scholar]
- 30.Analytical data for compound 33: 1H NMR (CDCl3) δ 0.91 (t, 3H), 1.33 (m, 8H), 3.02 (s, 3H), 3.39 (t, 2H), 6.77 (m, 3H), 7.31 (m, 4H), 8.31 (d, 1H), 8.85 (d, 1H); MS (EI) 395 (M+1).
- 31.Functional determinations: Activity of the target compounds at the OX1 and OX2 receptors utilized RD-HGA16 cells (Molecular Devices), a CHO cell line stably over-expressing the promiscuous Gq-protein Ga16. Two individual cell lines were used that stably express either OX1 or OX2 receptors. Cells are loaded with the calcium sensitive dye for 1 h and compounds are assayed in separate experiments for intrinsic activity and for the ability to inhibit orexin-A activity as measured by increased fluorescence intensity, a measure of increased internal calcium concentrations, in the FlexStation assay. Test compound Ke values were determined by running 8-point half-log orexin-A concentration response curves in the presence or absence of a single concentration of test compound. EC50 values were calculated for orexin-A (A) and orexin-A + test compound (A’), and these were used to calculate the test compound Ke. The concentration/response data were fit to a three-parameter logistic equation using GraphPad Prism (v5 for Windows, GraphPad Software; San Diego, CA) to calculate the EC50 values. At least two different concentrations of test compound were used for these experiments, and these were chosen such that they caused a 4-fold or greater rightward shift in the Orexin-A EC50. The Ke was calculated from the formula: Ke = [L]/(DR-1), where [L] equals the concentration of test compound in the assay and DR equals the dose ratio (A’/A). The data represent the mean from at least three independent experiments.