

Abstract
The bicuspid aortic valve (BAV) is a common congenital cardiac anomaly, having a prevalence of 0.9% to 1.37% in the general population and a male preponderance ratio of 2:1. The recognition of a BAV is clinically relevant because of its association with aortic stenosis or regurgitation, aortic aneurysm or dissection, and infective endocarditis. Although some patients with a BAV may go undetected without clinical complications for a lifetime, the vast majority will require intervention, most often surgery, at some point. In fact, the natural history of BAV is that of valve calcification and progressive stenosis that typically occurs at a faster rate than in tricuspid aortic valves (AVs). This pattern of presentation supports the hypothesis that shear stress in patients with congenitally abnormal AV may contribute to an earlier leaflet calcification. However, there is emerging research data showing that the valve calcification process might have a similar pathophysiologic process as that of vascular atherosclerosis. This review focuses on the current knowledge of the cellular mechanisms of BAV.
Keywords: Valvular Heart Disease, Lipids, Bicuspid Aortic Valve
Bicuspid Aortic Valve Disease
With the decline incidence of rheumatic carditis, bicuspid aortic valve disease(BAVD) has become the most common indication for surgical valve replacement in the US. Bicuspid aortic valve disease (BAVD) covers a spectrum of disease from early atherosclerotic changes in the valve leaflets, aortic sclerosis which is characterized by early calcification and thickening, and finally, to outflow obstruction and severe aortic stenosis. The later stages are characterized by thickening of the valve leaflets secondary to extracellular matrix synthesis1, angiogenesis2 and new bone formation3 present throughout the valve leaflets, but concentrated near the aortic valve surface. Severe aortic stenosis is characterized by symptoms of chest pain, shortness of breath and syncope. Surgery is indicated at the time of symptom onset in this patient population.
The bicuspid aortic valve (BAV) is a common congenital cardiac anomaly, having a prevalence of 2% to 3% in the general population and a male preponderance ratio of 2:14. Patients with bicuspid aortic valve severe stenosis present at an earlier age (approximately 45–55 years of age), than patients with tricuspid aortic valve stenosis (approximately 65–75 years of age)5. Therefore, these patients with severe stenosis and symptoms need to have valve surgery at an earlier age (45–55 years of age)6. The current recommendation for valve prosthesis is a mechanical aortic valve for patients before the age of 65, because the average life time of a bioprosthetic heart valve is 10–15 years6. 50% of patients also present with an aortopathy which ranges from dilatation of the sinus of Valsalva and or the ascending aorta, or coarctation which is probably an underlying biochemical abnormality of the aortic wall. Figure 1, demonstrates a bicuspid calcified valve removed a patient at the time of surgical valve replacement. For years, valvular heart disease has been thought to be a degenerative process. However, epidemiologic studies have demonstrated that risk factors for atherosclerotic vascular disease are similar to calcific aortic valve disease. These risk factors include elevated low-density lipoproteins, hypertension, male gender and smoking. In the past five years emerging evidence towards understanding this disease process have been published. These studies have been published in the genetic literature and also ex vivo tissue analysis of bicuspid aortic valves. This review will discuss the evolving understanding of the genetics and also the signaling pathways important in this disease process.
Figure 1.
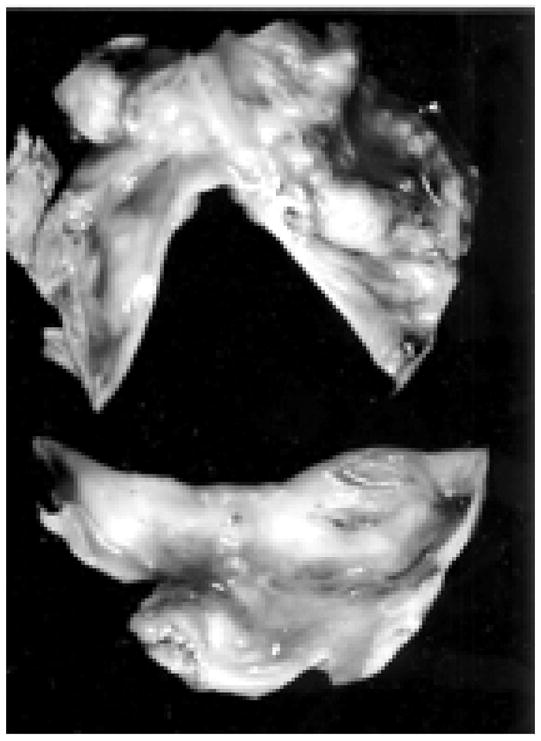
Bicuspid Aortic Valve Removed from Surgical Valve Replacement demonstrating extensive calcification and bone present in the bicuspid valve removed at the time of surgical valve replacement.5 (Used with Permission from Wolters Kluwer Health, Publisher for Circulation.)
Normal Valve Development
Bischoff7, has provided important studies in the understanding of the developmental paradigms important in normal heart valve development. The heart tube is composed of myocardium and an inner lining endocardial cells, separated by an extensive extracellular matrix (ECM) referred to as the cardiac jelly. After rightward looping of the heart, the cardiac jelly overlying the future atrioventricular canal and outflow track expands into swellings known as cardiac cushions. The formation of the cardiac cushions is a complex event characterized by endothelial cells that are specified in the cushion-forming regions to delaminate and invade the cardiac jelly, where they subsequently proliferate and complete their differentiation into mesenchymal cells. The locally expanded swellings of cardiac jelly and mesenchymal cells are referred to as cardiac cushions. These cardiac cushions undergo extensive remodeling from bulbous swellings to eventual thinly tapered heart valves secondary to a complex series of sequentially timed events under the direction of specific signaling pathways.
The cushions protrude from the underlying myocardium, forming thin, tapered leaflets with a single endothelial cell layer and a central matrix comprised of collagen, elastin, and glycosaminoglycans. These delamination and remodeling events depend on further cell differentiation, apoptosis and ECM remodeling. The final atrioventricular canal (mitral and tricuspid) valves are derived entirely from endocardial tissue. The end stages in development of the outflow tract (aortic and pulmonic) valves are more controversial. A population of neural crest cells derived form the branchial arches migrates to the distal outflow tract and is required for aortopulmonary septation7. These series of events are critical for the normal development of the cardiac valves in the heart. Bischoff et al, have also described the events that play a role in abnormal endothelial- mesenchymal transition that are important in the abnormal development of valvular heart disease. Many of these events are related to an abnormal regulation of these signaling pathways which may be initiated by oxidative stress which may predispose the valves to abnormal development7.
The Role of Endothelial Nitric Oxide Synthase in Bicuspid Aortic Valve Disease: Nos3tm1Unc mouse Expression of the Bicuspid Aortic Valve Phenotype
Mice homozygous for the Nos3tm1Unc targeted mutation are viable and fertile, and develop the bicuspid aortic valve phenotype in approximately 25% of the mice8. These mice have unique phenotypic features which include mild elevations of systolic blood pressure, lower heart rates, insulin resistance with hyperglycemic-euglycemic clamp, abnormal vascular development8 and respiratory development. These mice also paradoxically do not develop atherosclerosis when treated with a western diet. These investigators propose a dual role for eNOS in the vasculature: 1) the beneficial effects of the presence of eNOS on attenuation of vascular disease and 2) the lack of the eNOS-driven oxidation of accumulated LDL in the vessel wall may contribute to the reduction of eNOS-deficient mice in atherosclerotic lesion formation9. These mice also have bicuspid aortic valves8 and other congenital heart defects10 in a subset of eNOS null mice. While studying these mice using histologic techniques, they found these abnormalities in these mice. The incidence of the bicuspid aortic valve phenotype is 5/12 as reported in the initial study. However, how these mice develop the bicuspid phenotype has not been explored to date nor has the mechanism of stenosis in bicuspid aortic valves been discovered. Figure 2 demonstrates the bicuspid valve phenotype present in the subset of eNOS null mice. A recent study in human diseased valve demonstrates the eNOS protein expression was decreased in the BAV versus the tricuspid aortic valves11. This data provides further evidence of the potential functional importance of eNOS enzymatic activity in the bicuspid versus tricuspid aortic valves. This role may be at the developmental level for the formation of the congenital heart abnormality in addition to the actual role in the calcification process.
Figure 2.
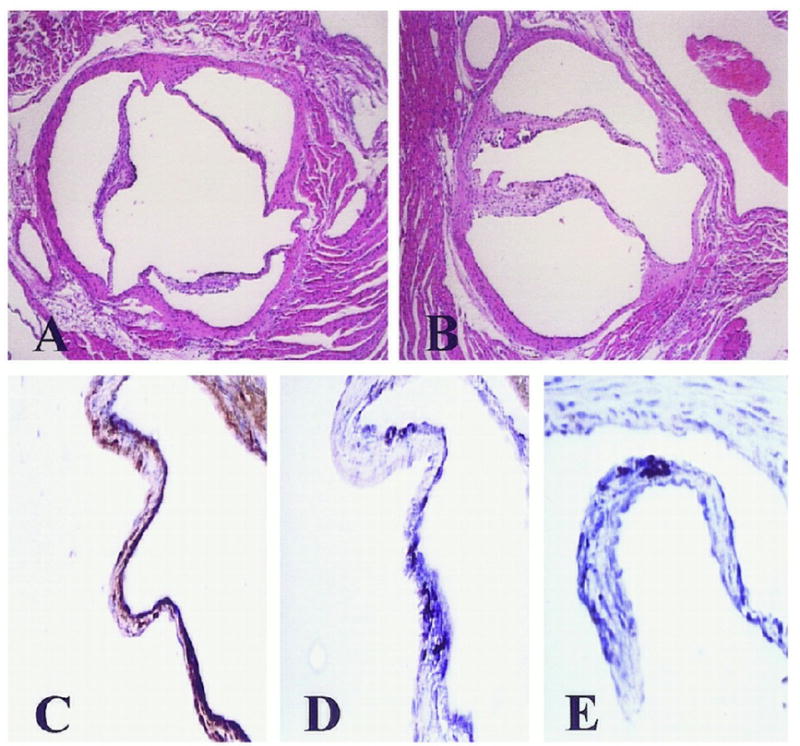
Photo demonstrates the bicuspid valve phenotype present in the subset of eNOS null mice found randomly in the mice by evaluating the valves in the eNOS null mice8. (Used with Permission from Wolters Kluwer Health, Publisher for Circulation.)
Evidence for the Role of Nitric Oxide in Bicuspid Aortic Valve Disease
Nitric oxide (NO) is a key regulator of normal endothelial function in the vasculature. Nitric oxide synthase (NOS) is responsible for the production of NO. There are multiple mechanisms of regulation of this enzyme through transcriptional, post-transcriptional, and post-translational levels12, 13. Endothelial nitric oxide synthase (eNOS) is constitutively expressed in endothelium. In cultured cells, eNOS expression decreases with exposure to high concentrations of oxidized low density lipoprotein14. Under these conditions, post-transcriptional changes in mRNA half-life play an important role in the down-regulation of eNOS expression 15–17. This inactivation of eNOS leads to a decrease in the availability of nitric oxide. Our group has demonstrated an association of abnormalities in eNOS enzymatic activity can induce calcification in the aortic valve18 in the presence of hypercholesterolemia. It is known that this decrease in available nitric oxide causes the stimulation of valvular myofibroblast proliferation and extracellular matrix production which leads to the development of atherosclerosis in the aortic valve18, 19.
The Genetic Evidence for the role of Notch1 receptor in Human Bicuspid Aortic Valve Calcification
In 2005, Garg et al, discovered mutations in the signaling and transcriptional regulator Notch1 to cause a spectrum of developmental aortic valve anomalies and severe valve calcification. Notch1 encodes a transmembrane receptor (2,556 amino acids) that function in a highly conserved intracellular signaling pathway involved in cellular differentiation, cell fate and lateral inhibition20. Notch1 is a single pass transmembrane receptor that is activated by direct contact with the membrane-bound ligands Delta 1–4 and Serrate/Jagged 1 and 2. Notch-ligand interaction results in the proteolytic cleavage at the juxtamembrane portion of the Notch receptor and release of the intracellular domain (NICD), which translocates to the nucleus. Therefore, NICD is the constitutively active domain of the Notch receptor after ligand activation. The Notch signaling pathway is among the most commonly used pathway in animal cells. Recent studies have demonstrated that this pathway is indispensable for cells in various stages of maturation, including ontogenesis20, 21. Upon ligand binding, Notch receptors undergo proteolytic cleavage leading to the release of their intracellular domain (NICD). Overexpression of Notch1 impairs osteoblastogenesis, but the mechanisms are not understood. In the Garg study22, they show that Notch1 transcripts were most abundant in the developing aortic valve of mice. Notch1 repressed the activity of Runx2 or Cbfa1, a central transcriptional regulator of the osteoblast cell. They tested the hairy-related family of transcriptional repressors (Hrt) mechanism in COS cells and found that they physically interacted with Cbfa1 and repressed Cbfa1 transcriptional activity independent of histone deacytylase activity. The investigators identified patients with aortic valve calcification and BAV in a population based study called the Texas Heart Study. They tested these patients and found that these patients have the R1108X mutation. The investigators demonstrate an autosomal–dominant inheritance of the disease phenotype with complete penetrance as shown in Figure 3 22. These studies in humans and the verification of the mechanism in the COS cells provides the foundation for future understanding of accelerated BAV calcification from genetic lessons and also the implications in Cbfa1 transcriptional factor regulation as tested in the COS cells.
Figure 3.
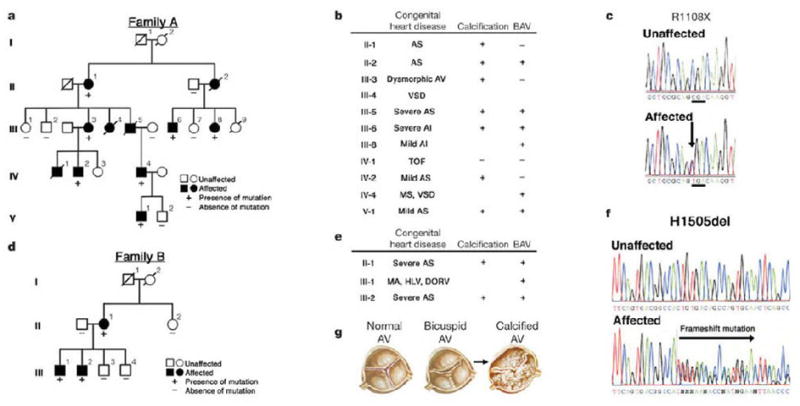
The autosomal–dominant inheritance of the disease phenotype with complete penetrance of the Notch1 loss of function mutation22. The Notch1 mutations segregate with the family members with aortic valve disease. There are also family members who expressed congenital heart abnormalities. (Used with Permission from Nature Publishing Group, Publisher for Nature.)
Evidence for the role of Lrp5/beta-catenin Activation and Notch1 in Cardiovascular Calcification and Osteoblast Bone Formation
The Lrp5 co-receptor has been shown to be an important development pathway in abnormal skeletal bone formation23, 24. Bone and cartilage are major tissues in the vertebrate skeletal system, which is primarily composed of three cell types: osteoblasts, chrondrocytes, and osteoclasts. In the developing embryo, osteoblast and chrondrocytes, both differentiate from common mesenchymal progenitors in situ, where as osteoclasts are of hematopoietic origin and brought in later by invading blood vessels. Osteoblast differentiation and maturation lead to bone formation controlled by two distinct mechanisms: intramembranous and endochondral ossification, both starting from mesenchymal condensations.
To date only two osteoblast-specific transcripts have been identified: 1) Cbfa1 and 2) osteocalcin (OC). The transcription factor Cbfa125 has all the attributes of a ‘master gene’ differentiation factor for the osteoblast lineage and bone matrix gene expression. During embryonic development, Cbfa1 expression precedes osteoblast differentiation and is restricted to mesenchymal cells destined to become osteoblast. In addition to its critical role in osteoblast commitment and differentiation, Cbfa1 appears to control osteoblast activity, i.e., the rate of bone formation by differentiated osteoblasts 25. We have shown previously that experimental cholesterol increases Cbfa1 gene expression in the aortic valve and atorvastatin decreases the gene expression1 in an animal model. We have also demonstrated that Sox9 and Cbfa1 are expressed in human degenerative valves removed at the time of surgical valve replacement26. This osteogenic regulatory mechanism for bone formation begins in osteoblast progenitor cells which differentiate into terminally differentiated cells. This process is a well orchestrated and well studied pathway which involves cellular proliferation events, bone matrix protein synthesis, which requires the actions of specific paracrine/hormonal factors and the activation of a number of different signaling pathways including Wnt, Notch1, BMP and TGFbeta 21, 23, 27–30.
Genes which code for the bone extracellular matrix proteins in osteoblast cells include alkaline phosphatase (AP), osteopontin (OP), osteocalcin (OC), and bone sialoprotein (BSP). This data supports a potential regulatory mechanism that these matrix proteins play a critical role in the development of biomineralization. To date, many of these markers have been shown to be critical in the extracellular mineralization and bone formation that develops in normal osteoblast differentiation. A link between lipids and osteoporosis has been studied extensively31–36. These groups have shown in in vitro and in vivo studies that lipids decrease bone formation and increase vascular calcification. Hurska’s group from the University of Washington have studied this important hypothesis in the LDLR−/ − mice with renal disease31. This study correlated the important understanding of chronic kidney disease with decreased bone formation rates and increase in vascular calcification. This study demonstrates that accelerated vascular calcification found in patients with end stage renal disease may be related to multifactorial mechanisms including traditional atherosclerotic risk factors and elevated serum phosphate levels. Giachelli has also studied extensively the hypothesis of a sodium phosphate abnormality in the vascular smooth muscle cell37. Her group has also shown that osteopontin expression by vascular smooth muscle cells may have an inhibitory effect in the development of calcification38 which further defines the complexity of the matrix synthesis phase of bone formation. Demer’s laboratory has also studied extensively the correlation of lipids with vascular calcification and osteoporosis via inhibition of Cbfa1 in osteoblast cells36, 39. This paradoxical finding between the calcifying vascular aorta and osteoporosis is an important link in the hypercholesterolemia hypothesis. The development of cardiovascular calcification is a multifactorial process which includes a number of mechanisms. Studies in the different laboratories provide important evidence towards the development of therapies depending on the patient population i.e. end stage renal disease versus treatment of the traditional risk factors for vascular disease.
Recently, our lab (43) and Towler’s laboratory (44) have shown that the Lrp5/Wnt/beta-catenin pathway plays an important role in the development of vascular and valvular calcification. Studies have shown that different mutations in Lrp5, an LDL receptor related protein; develop a high bone mass phenotype and an osteoporotic phenotype (45, 46). In the presence of the palmitoylation of Wnt an active beta-catenin accumulates in the cytoplasm, presumably in a signaling capacity, and eventually translocates to the nucleus via binding to nucleoporins40, where it can interacts with LEF-1/TCFs in an inactive transcription complex 41, 42, The Wnt/Lrp5/frizzled complex turns on downstream components such as Dishevelled (Dvl/Dsh) which leads to repression of the glycogen synthase kinase-3 (GSK3) 43. Inhibition of GSK3 allows beta-catenin to accumulate in the nucleus, interacting with members of the LEF/TCF class of architectural HMG box of transcription factors including Cbfa1 involved in cell differentiation and osteoblast activation 44, 45, 46–48 and Sox 9, a HMG box transcription factor, is required for chondrocyte cell fate determination and marks early chondrocytic differentiation of mesenchymal progenitors49.
The presence of an osteogenic phenotype in tricuspid and bicuspid aortic valve disease was delineated in a study evaluating the phenotypic spectrum of bone formation in left sided valve lesions including tricuspid aortic valve disease, bicuspid aortic valve disease and myxomatous mitral valve disease. Figure 4, demonstrates the immunohistochemistry of the bicuspid aortic valves from the study26. The specific stains measure the different regulatory pathways critical in the development of bone formation within the valve leaflet. PCNA or proliferating cell nuclear antigen is a marker of DNA proliferation. Alcian blue is a marker of chondrogenesis and alizarin red a marker of mineralization. In the study there was more alizarin red than the Alcian blue stain indicating calcification. Lrp5/Wnt3a are the extracellular membrane components important in the activation of the canonical Wnt pathway of bone formation in mesenchymal cells. Bone sialoprotein(BSP), osteocalcin(OC), osteopontin(OPN) are increased and are markers of extracellular matrix synthesis in the valve. Notch1 is decreased in the valves as shown in Figure 4 by immunohistochemistry(Data not shown for the PCR and western blot analysis)26. The protein and RNA expression also demonstrated a decrease in overall Notch1 in these disease tissues implicating the loss of normal Notch1 is necessary for valve calcification similar to the implications of the loss of function mutation in the genetic study. Overall the loss of Notch1 function and the increase in Lrp5 signaling demonstrates the role of these important regulators of bone metabolism in these diseases tissues. Figure 5, demonstrates a diagram for the mechanism of bicuspid aortic valve disease includes the early event of abnormal oxidative stress which results in a decrease in Notch1 function, and an increase in Lrp5 expression which activates bone formation within the valve myofibroblast.
Figure 4.
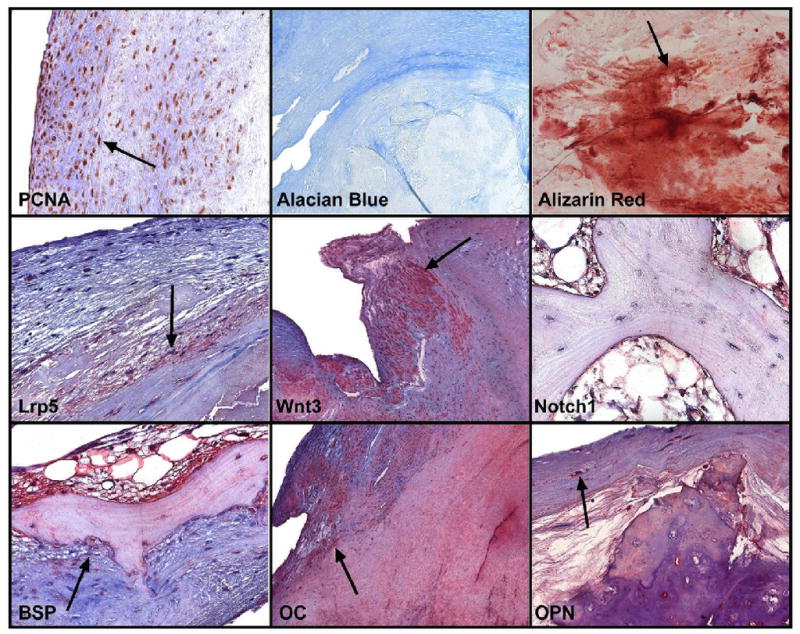
Bone immunohistochemistry for extracellular matrix and Lrp5/Notch1 signaling markers in Bicuspid Aortic Valves26. PCNA- Proliferating nuclear cell antigen indicates upregulation of DNA polymerase during active proliferation. Alcian blue stains for cartilage which is minimal. The stain binds to proteoglycans. The Alizarin red is a stain for calcium and hydroxyapatite in the valves which was increased markedly in the valves. The Lrp5 and Wnt3a stain is localized in the myofibroblast cells which are differentiating to an osteoblast like phenotype. The Notch1 stain is much less than the canonical Wnt markers in the areas of complex bone formation. The Bone sialoprotein, Osteocalcin and Osteopontin stains are all upregulated in bicuspid tissues indicating osteogenic differentiation and later stage mineralization. (Used with Permission from Elsevier, Publisher for Journal of the American College of Cardiology.)
Figure 5.
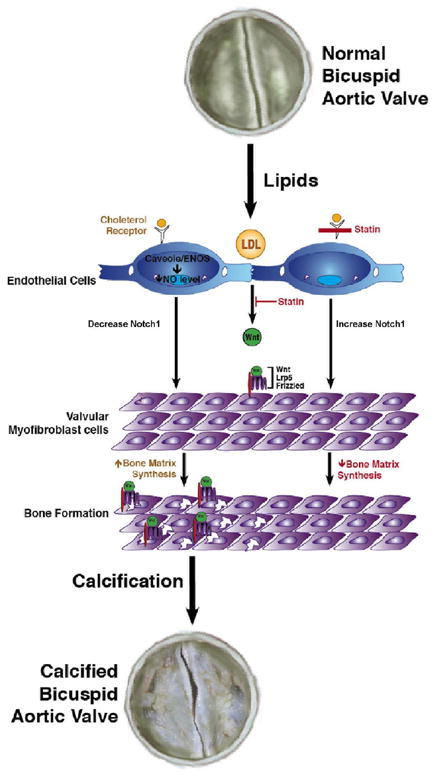
Proposed Cellular mechanisms of Bicuspid Aortic Valve Disease.
Development of Future Medical Therapies for Calcific Aortic Stenosis
The natural history studies of valvular aortic stenosis as defined by clinical and histopathologic parameters have provided landmark developments towards the understanding of this disease. HMG CoA reductase inhibitors may provide an innovative therapeutic approach by employing both lipid lowering and possibly non-lipid lowering effects to forestall critical stenosis in the aortic valve. Studies have shown that atorvastatin has a number of effects in the aortic valve including: 1) inhibition of foam cell accumulation 1, 2) inhibition of Cbfa1 activation1, 3) eNOS enzymatic activation18 and 4) attenuation of Lrp5 receptor activation50. Statins have potent LDL lowering effects via inhibition of the rate-limiting step in cholesterol synthesis. There are a number of experimental models which demonstrate the potential for treating the vasculature with statins to inhibit matrix formation51–53, cellular proliferation1 and vascular aneurysm formation54. Other laboratories have shown that statins have pleiotropic effects via purine nucleotides55 and ROCK56. Although valve replacement is the current treatment of choice for severe critical aortic stenosis, future insights into the mechanisms of calcification and its progression may indicate a role for lipid lowering therapy in modifying the rate of progression of stenosis.
Summary
Bicuspid aortic valve disease is the most common indication for surgical valve replacement for aortic stenosis. The cellular mechanisms are evolving and in the last decade, scientific progress has demonstrated the active biology of this disease process. In the future, as further understanding of the design for clinical trials in valvular heart disease evolves, the cellular biology of this disease may be targeted and slowing of the progression of the valve lesion and the aortopathy may occur to help this patient population in the future.
Acknowledgments
This work was completed with the support of an American Heart Association Grant-in-Aid (0555714Z) and a grant from the National Institute of Health (5K08HL073927-04, 1R01HL085591-01A1). Nalini M. Rajamannan is an inventor on a patent for the use of statins in degeneration of aortic valve disease. This patent is owned by the Mayo Clinic and Dr. Rajamannan does not receive any royalties from this patent.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rajamannan NM, Subramaniam M, Springett M, Sebo TC, Niekrasz M, McConnell JP, Singh RJ, Stone NJ, Bonow RO, Spelsberg TC. Atorvastatin inhibits hypercholesterolemia-induced cellular proliferation and bone matrix production in the rabbit aortic valve. Circulation. 2002;105(22):2260–2265. doi: 10.1161/01.cir.0000017435.87463.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rajamannan NM, Nealis TB, Subramaniam M, Pandya S, Stock SR, Ignatiev CI, Sebo TJ, Rosengart TK, Edwards WD, McCarthy PM, Bonow RO, Spelsberg TC. Calcified rheumatic valve neoangiogenesis is associated with vascular endothelial growth factor expression and osteoblast-like bone formation. Circulation. 2005;111(24):3296–3301. doi: 10.1161/CIRCULATIONAHA.104.473165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, Orszulak T, Fullerton DA, Tajik AJ, Bonow RO, Spelsberg T. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation. 2003;107(17):2181–2184. doi: 10.1161/01.CIR.0000070591.21548.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts WC. The congenitally bicuspid aortic valve. A study of 85 autopsy cases. The American Journal of Cardiology. 1970;26:72–83. doi: 10.1016/0002-9149(70)90761-7. [DOI] [PubMed] [Google Scholar]
- 5.Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation. 2005;111(7):920–925. doi: 10.1161/01.CIR.0000155623.48408.C5. [DOI] [PubMed] [Google Scholar]
- 6.Bonow RO, Carabello BA, Kanu C, de Leon AC, Jr, Faxon DP, Freed MD, Gaasch WH, Lytle BW, Nishimura RA, O'Gara PT, O'Rourke RA, Otto CM, Shah PM, Shanewise JS, Smith SC, Jr, Jacobs AK, Adams CD, Anderson JL, Antman EM, Fuster V, Halperin JL, Hiratzka LF, Hunt SA, Nishimura R, Page RL, Riegel B. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): developed in collaboration with the Society of Cardiovascular Anesthesiologists: endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. Circulation. 2006;114(5):e84–231. doi: 10.1161/CIRCULATIONAHA.106.176857. [DOI] [PubMed] [Google Scholar]
- 7.Armstrong EJ, Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circulation Research. 2004;95(5):459–470. doi: 10.1161/01.RES.0000141146.95728.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee TC, Zhao YD, Courtman DW, Stewart DJ. Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation. 2000;101(20):2345–2348. doi: 10.1161/01.cir.101.20.2345. [DOI] [PubMed] [Google Scholar]
- 9.Shi W, Wang X, Shih DM, Laubach VE, Navab M, Lusis AJ. Paradoxical reduction of fatty streak formation in mice lacking endothelial nitric oxide synthase. Circulation. 2002;105(17):2078–2082. doi: 10.1161/01.cir.0000015853.59427.32. [DOI] [PubMed] [Google Scholar]
- 10.Feng Q, Song W, Lu X, Hamilton JA, Lei M, Peng T, Yee SP. Development of heart failure and congenital septal defects in mice lacking endothelial nitric oxide synthase. Circulation. 2002;106(7):873–879. doi: 10.1161/01.cir.0000024114.82981.ea. [DOI] [PubMed] [Google Scholar]
- 11.Aicher D, Urbich C, Zeiher A, Dimmeler S, Schafers HJ. Endothelial nitric oxide synthase in bicuspid aortic valve disease. The Annals of Thoracic Surgery. 2007;83(4):1290–1294. doi: 10.1016/j.athoracsur.2006.11.086. [DOI] [PubMed] [Google Scholar]
- 12.Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97(12):1129–1135. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 13.Venema RC, Sayegh HS, Kent JD, Harrison DG. Identification, characterization, and comparison of the calmodulin-binding domains of the endothelial and inducible nitric oxide synthases. Journal of Biological Chemistry. 1996;271(11):6435–6440. doi: 10.1074/jbc.271.11.6435. [DOI] [PubMed] [Google Scholar]
- 14.Blair A, Shaul PW, Yuhanna IS, Conrad PA, Smart EJ. Oxidized low density lipoprotein displaces endothelial nitric-oxide synthase (eNOS) from plasmalemmal caveolae and impairs eNOS activation. The Journal of Biological Chemistry. 1999;274(45):32512–32519. doi: 10.1074/jbc.274.45.32512. [DOI] [PubMed] [Google Scholar]
- 15.Mital S, Magneson A, Loke KE, Liao J, Forfia PR, Hintze TH. Simvastatin acts synergistically with ACE inhibitors or amlodipine to decrease oxygen consumption in rat hearts. Journal of Cardiovascular Pharmacology. 2000;36(2):248–254. doi: 10.1097/00005344-200008000-00016. [DOI] [PubMed] [Google Scholar]
- 16.Laufs U, Endres M, Stagliano N, Amin-Hanjani S, Chui DS, Yang SX, Simoncini T, Yamada M, Rabkin E, Allen PG, Huang PL, Bohm M, Schoen FJ, Moskowitz MA, Liao JK. Neuroprotection mediated by changes in the endothelial actin cytoskeleton. The Journal of Clinical Investigation. 2000;106(1):15–24. doi: 10.1172/JCI9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. The Journal of Biological Chemistry. 1998;273(37):24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 18.Rajamannan NM, Subramaniam M, Stock SR, Stone NJ, Springett M, Ignatiev KI, McConnell JP, Singh RJ, Bonow RO, Spelsberg TC. Atorvastatin inhibits calcification and enhances nitric oxide synthase production in the hypercholesterolaemic aortic valve. Heart. 2005;91(6):806–810. doi: 10.1136/hrt.2003.029785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller JD, Chu Y, Brooks RM, Richenbacher WE, Pena-Silva R, Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. Journal of the American College of Cardiology. 2008;52(10):843–850. doi: 10.1016/j.jacc.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science (New York, NY) 1999;284(5415):770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 21.Sciaudone M, Gazzerro E, Priest L, Delany AM, Canalis E. Notch 1 impairs osteoblastic cell differentiation. Endocrinology. 2003;144(12):5631–5639. doi: 10.1210/en.2003-0463. [DOI] [PubMed] [Google Scholar]
- 22.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056):270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 23.Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, Manning SP, Swain PM, Zhao SC, Eustace B, Lappe MM, Spitzer L, Zweier S, Braunschweiger K, Benchekroun Y, Hu X, Adair R, Chee L, FitzGerald MG, Tulig C, Caruso A, Tzellas N, Bawa A, Franklin B, McGuire S, Nogues X, Gong G, Allen KM, Anisowicz A, Morales AJ, Lomedico PT, Recker SM, Van Eerdewegh P, Recker RR, Johnson ML. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. American Journal of Human Genetics. 2002;70(1):11–19. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, Zacharin M, Oexle K, Marcelino J, Suwairi W, Heeger S, Sabatakos G, Apte S, Adkins WN, Allgrove J, Arslan-Kirchner M, Batch JA, Beighton P, Black GC, Boles RG, Boon LM, Borrone C, Brunner HG, Carle GF, Dallapiccola B, De Paepe A, Floege B, Halfhide ML, Hall B, Hennekam RC, Hirose T, Jans A, Juppner H, Kim CA, Keppler-Noreuil K, Kohlschuetter A, LaCombe D, Lambert M, Lemyre E, Letteboer T, Peltonen L, Ramesar RS, Romanengo M, Somer H, Steichen-Gersdorf E, Steinmann B, Sullivan B, Superti-Furga A, Swoboda W, van den Boogaard MJ, Van Hul W, Vikkula M, Votruba M, Zabel B, Garcia T, Baron R, Olsen BR, Warman ML Osteoporosis-Pseudoglioma Syndrome Collaborative G. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107(4):513–523. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 25.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation.[see comment] Cell. 1997;89(5):747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 26.Caira FC, Stock SR, Gleason TG, McGee EC, Huang J, Bonow RO, Spelsberg TC, McCarthy PM, Rahimtoola SH, Rajamannan NM. Human degenerative valve disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. Journal of the American College of Cardiology. 2006;47(8):1707–1712. doi: 10.1016/j.jacc.2006.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aubin JE, Liu F, Malaval L, Gupta AK. Osteoblast and chondroblast differentiation. Bone. 1995;17(2 Suppl):77S–83S. doi: 10.1016/8756-3282(95)00183-e. [DOI] [PubMed] [Google Scholar]
- 28.Abe E, Yamamoto M, Taguchi Y, Lecka-Czernik B, O'Brien CA, Economides AN, Stahl N, Jilka RL, Manolagas SC. Essential requirement of BMPs-2/4 for both osteoblast and osteoclast formation in murine bone marrow cultures from adult mice: antagonism by noggin. J Bone Miner Res. 2000;15(4):663–673. doi: 10.1359/jbmr.2000.15.4.663. [DOI] [PubMed] [Google Scholar]
- 29.Deregowski V, Gazzerro E, Priest L, Rydziel S, Canalis E. Notch 1 overexpression inhibits osteoblastogenesis by suppressing Wnt/beta-catenin but not bone morphogenetic protein signaling. The Journal of Biological Chemistry. 2006;281(10):6203–6210. doi: 10.1074/jbc.M508370200. [DOI] [PubMed] [Google Scholar]
- 30.Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna K, Lifton RP. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346(20):1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 31.Davies MR, Lund RJ, Mathew S, Hruska KA. Low turnover osteodystrophy and vascular calcification are amenable to skeletal anabolism in an animal model of chronic kidney disease and the metabolic syndrome. J Am Soc Nephrol. 2005;16(4):917–928. doi: 10.1681/ASN.2004100835. [DOI] [PubMed] [Google Scholar]
- 32.Davies MR, Lund RJ, Hruska KA. BMP-7 is an efficacious treatment of vascular calcification in a murine model of atherosclerosis and chronic renal failure. J Am Soc Nephrol. 2003;14(6):1559–1567. doi: 10.1097/01.asn.0000068404.57780.dd. [DOI] [PubMed] [Google Scholar]
- 33.Parhami F, Garfinkel A, Demer LL. Role of lipids in osteoporosis. Arteriosclerosis, Thrombosis & Vascular Biology. 2000;20(11):2346–2348. doi: 10.1161/01.atv.20.11.2346. [DOI] [PubMed] [Google Scholar]
- 34.Parhami F, Mody N, Gharavi N, Ballard AJ, Tintut Y, Demer LL. Role of the cholesterol biosynthetic pathway in osteoblastic differentiation of marrow stromal cells. Journal of Bone & Mineral Research. 2002;17(11):1997–2003. doi: 10.1359/jbmr.2002.17.11.1997. [DOI] [PubMed] [Google Scholar]
- 35.Parhami F, Morrow AD, Balucan J, Leitinger N, Watson AD, Tintut Y, Berliner JA, Demer LL. Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation. A possible explanation for the paradox of arterial calcification in osteoporotic patients. Arteriosclerosis, Thrombosis, and Vascular biology. 1997;17(4):680–687. doi: 10.1161/01.atv.17.4.680. [DOI] [PubMed] [Google Scholar]
- 36.Parhami F, Tintut Y, Beamer WG, Gharavi N, Goodman W, Demer LL. Atherogenic high-fat diet reduces bone mineralization in mice. J Bone Miner Res. 2001;16(1):182–188. doi: 10.1359/jbmr.2001.16.1.182. [DOI] [PubMed] [Google Scholar]
- 37.Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, Morii H, Giachelli CM. Phosphate regulation of vascular smooth muscle cell calcification. Circulation Research. 2000;87(7):E10–17. doi: 10.1161/01.res.87.7.e10. [DOI] [PubMed] [Google Scholar]
- 38.Wada T, McKee MD, Steitz S, Giachelli CM. Calcification of vascular smooth muscle cell cultures: inhibition by osteopontin. Circulation Research. 1999;84(2):166–178. doi: 10.1161/01.res.84.2.166. [DOI] [PubMed] [Google Scholar]
- 39.Parhami F, Mody N, Gharavi N, Ballard AJ, Tintut Y, Demer LL. Role of the cholesterol biosynthetic pathway in osteoblastic differentiation of marrow stromal cells. J Bone Miner Res. 2002;17(11):1997–2003. doi: 10.1359/jbmr.2002.17.11.1997. [DOI] [PubMed] [Google Scholar]
- 40.Willert K, Nusse R. Beta-catenin: a key mediator of Wnt signaling. Current Opinion in Genetics & Development. 1998;8(1):95–102. doi: 10.1016/s0959-437x(98)80068-3. [DOI] [PubMed] [Google Scholar]
- 41.Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382(6592):638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 42.Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann BG, Kemler R. Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech Dev. 1996;59(1):3–10. doi: 10.1016/0925-4773(96)00597-7. [DOI] [PubMed] [Google Scholar]
- 43.Holmen SL, Salic A, Zylstra CR, Kirschner MW, Williams BO. A novel set of Wnt-Frizzled fusion proteins identifies receptor components that activate beta -catenin-dependent signaling. Journal of Biological Chemistry. 2002;277(38):34727–34735. doi: 10.1074/jbc.M204989200. [DOI] [PubMed] [Google Scholar]
- 44.Caverzasio J. Wnt/LRP5, a new regulation osteoblastic pathway involved in reaching peak bone masses. Revue Medicale de la Suisse Romande. 2004;124(2):81–82. [PubMed] [Google Scholar]
- 45.Kahler RA, Westendorf JJ. Lymphoid enhancer factor-1 and beta-catenin inhibit Runx2-dependent transcriptional activation of the osteocalcin promoter. Journal of Biological Chemistry. 2003;278(14):11937–11944. doi: 10.1074/jbc.M211443200. [DOI] [PubMed] [Google Scholar]
- 46.Smith E, Frenkel B. Glucocorticoids Inhibit the Transcriptional Activity of LEF/TCF in Differentiating Osteoblasts in a Glycogen Synthase Kinase-3{beta}-dependent and -independent Manner. J Biol Chem. 2005;280(3):2388–2394. doi: 10.1074/jbc.M406294200. [DOI] [PubMed] [Google Scholar]
- 47.Wang HY, Malbon CC. Wnt signaling, Ca2+, and cyclic GMP: visualizing Frizzled functions. Science. 2003;300(5625):1529–1530. doi: 10.1126/science.1085259. [DOI] [PubMed] [Google Scholar]
- 48.Gregory CA, Perry AS, Reyes E, Conley A, Gunn WG, Prockop DJ. Dkk-1-derived Synthetic Peptides and Lithium Chloride for the Control and Recovery of Adult Stem Cells from Bone Marrow. J Biol Chem. 2005;280(3):2309–2323. doi: 10.1074/jbc.M406275200. [DOI] [PubMed] [Google Scholar]
- 49.Yano F, Kugimiya F, Ohba S, Ikeda T, Chikuda H, Ogasawara T, Ogata N, Takato T, Nakamura K, Kawaguchi H, Chung UI. The canonical Wnt signaling pathway promotes chondrocyte differentiation in a Sox9-dependent manner. Biochem Biophys Res Commun. 2005;333(4):1300–1308. doi: 10.1016/j.bbrc.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 50.Rajamannan NM, Subramaniam M, Caira F, Stock SR, Spelsberg TC. Atorvastatin inhibits hypercholesterolemia-induced calcification in the aortic valves via the Lrp5 receptor pathway. Circulation. 2005;112(9 Suppl):I229–234. doi: 10.1161/01.CIRCULATIONAHA.104.524306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aikawa M, Rabkin E, Sugiyama S, Voglic SJ, Fukumoto Y, Furukawa Y, Shiomi M, Schoen FJ, Libby P. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation. 2001;103(2):276–283. doi: 10.1161/01.cir.103.2.276. [DOI] [PubMed] [Google Scholar]
- 52.Williams JK, Sukhova GK, Herrington DM, Libby P. Pravastatin has cholesterol-lowering independent effects on the artery wall of atherosclerotic monkeys. Journal of the American College of Cardiology. 1998;31(3):684–691. doi: 10.1016/s0735-1097(97)00537-8. [DOI] [PubMed] [Google Scholar]
- 53.Wu B, Elmariah S, Kaplan FS, Cheng G, Mohler ER., 3rd Paradoxical effects of statins on aortic valve myofibroblasts and osteoblasts: implications for end-stage valvular heart disease. Arteriosclerosis, Thrombosis, and Vascular biology. 2005;25(3):592–597. doi: 10.1161/01.ATV.0000154278.01871.64. [DOI] [PubMed] [Google Scholar]
- 54.Steinmetz EF, Buckley C, Shames ML, Ennis TL, Vanvickle-Chavez SJ, Mao D, Goeddel LA, Hawkins CJ, Thompson RW. Treatment with simvastatin suppresses the development of experimental abdominal aortic aneurysms in normal and hypercholesterolemic mice. Ann Surg. 2005;241(1):92–101. doi: 10.1097/01.sla.0000150258.36236.e0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Osman L, Amrani M, Isley C, Yacoub MH, Smolenski RT. Stimulatory effects of atorvastatin on extracellular nucleotide degradation in human endothelial cells. Nucleosides Nucleotides Nucleic Acids. 2006;25(9–11):1125–1128. doi: 10.1080/15257770600894196. [DOI] [PubMed] [Google Scholar]
- 56.Monzack EL, Gu X, Masters KS. Efficacy of simvastatin treatment of valvular interstitial cells varies with the extracellular environment. Arteriosclerosis, Thrombosis, and Vascular biology. 2009;29(2):246–253. doi: 10.1161/ATVBAHA.108.179218. [DOI] [PMC free article] [PubMed] [Google Scholar]