

Abstract
Continued examination of substituted 6-arylquinazolin-4-amines as Clk4 inhibitors resulted in selective inhibitors of Clk1, Clk4, Dyrk1A and Dyrk1B. Several of the most potent inhibitors were validated as being highly selective within a comprehensive kinome scan.
Keywords: Clk1, Clk2, Clk3, Clk4, Dyrk1A, Dyrk1B, Pre-mRNA splicing, kinase inhibition, quinazoline
The cdc2-like kinase (Clk) family contains four isoforms (Clk1-4) and is proposed to alter the function of the spliceosome by phosphorylating serine-arginine-rich (SR) proteins within the spliceosome assembly.1 The spliceosome regulates the processing, or splicing, of pre-mRNAs, yielding mature protein-encoding mRNAs.2,3 Many human genes express more than one mRNA via alternative splicing, leading to protein diversity,4 however, misregulation of alternative splicing is involved in the pathogenesis of cancer and other diseases.5,6 Studies have revealed that Clk isoforms are associated with alternative splicing of PkcβII7, TF8, β-globin9 and E1A pre-mRNA.10 The Clks also regulate the alternative splicing of microtubule-associated protein tau and are implicated in frontotemporal dementia and Parkinson’s disease through the phosphorylation of splicing factors (SF).11 Inhibitors of Clk isoforms may alter these events and could prove to be useful agents in disease phenotypes characterized by abnormal splicing. Hagiwara reported TG003 (1), a small molecule benzothiazole, as having low-nanomolar IC50 values versus Clk1 and Clk4. The patent literature revealed structurally similar benzothiazole 1a from Sirtris Pharmaceuticals12 and a quinoline 3 from Chronogen, Inc13 reported to have activity versus Clk1 (Figure 1). Indole 2 was recently revealed as a potent (20 nM) ATP competitive Clk1 inhibitor with good selectivity over Clk3 via a unique binding mode.14 We have previously described a series of substituted 6-arylquinazolin-4-amines including NCGC00010037 (4, ML106) as potent inhibitors of Clk1 and Clk4.15
Figure 1.

Structures of known Clk inhibitors including TG003 (1), KH-CB19 (2), NCGC00010037 (4, ML106) and Harmine (5).
In our previous study we examined 1 and 4 within a screen of 402 kinases and found that both agents had impressive activity versus the dual-specificity tyrosine-phosphorylation regulated kinase 1A (Dyrk1A) (IC50 values of 12 and 27 nM for 1 and 4, respectively). Dyrk1A16 has been shown to accumulate in nuclear speckles where it interacts and activates splicing factors.17,18 Although the role of Dyrk1A in the development and function of the brain is not well understood, it is proposed to play a critical role in the development of Down Syndrome (DS) due to the location of the Dyrk1A gene on Chromosome 21 in the Down Syndrome critical region.19–25 Overexpression of Dyrk1A has been associated with DS phenotypes20,26,27 while loss-of-function results in potentially fatal conditions, such as decreased body and brain size.27,28 Arron and coworkers recently found that Dyrk1A regulates calcineurin/NFAT (nuclear factor of activated T cells) signaling, which has critical roles in human development, suggesting that increased levels of Dyrk1A can lead to many of the developmental phenotypes typically associated with DS.29 Given these studies, selective inhibitors of Dyrk1A would be of great use to further elucidate the role this kinase plays in several disease phenotypes.
The Dyrk family contains several additional isoforms (Dyrk1B, Dyrk2, Dyrk3, and Dyrk4).20 The Dyrk1B homolog (also referred to as Mirk) was first isolated and described by Friedman and coworkers.30,31 Several reports suggest that Dyrk1B plays a significant role in cancer biology and muscle differentiation. Extensive studies on the Dyrk1B gene found that overexpression is associated with pancreatic and other cancers potentially due to its downstream effect on oncogenic K-ras and the hedgehog pathway.32–36 It has been hypothesized that inhibition of Dyrk1B would diminish the ability of cancer cells to mitigate the effects of reactive oxygen species resulting in cell damage and induction of apoptosis.32 Accordingly, selective inhibitors of Dyrk1B could be used to understand the role of this kinase in cancer progression. A prior art search of Dyrk1A and Dyrk1B inhibitors revealed the naturally occurring chemotherapeutic harmine (5) as a nonspecific inhibitor with nanomolar potency (Figure 1).22,37–39 Harmine also represents the only reported inhibitor of Dyrk1B, although potency is mild and selectivity is poor.38 Given the potential associated with these targets we aimed to further define small molecules with alternative selectivity profiles. Here we report the results of continued evaluations of substituted 6-arylquinazolin-4-amines as selective and highly potent small molecule inhibitors of Clk1, Clk4, Dyrk1A and Dyrk1B.
Previously, we utilized a synthetic strategy that relied upon an amine displacement step starting with 6-bromo-4-chloroquinazoline followed by an end-stage Suzuki-Miyaura coupling with various aryl boronic acids. In this study, we utilized a Suzuki-Miyaura coupling to introduce a variety of aryl boronic acids (e.g. 3,4-(Methylenedioxy)-phenylboronic acid) to the commercially available 6-bromoquinazolin-4-one (6) scaffold. The resulting substituted quinazolinone 7 was chlorinated using phosphorous oxychloride to yield chloro-pyrimidine 8, which upon reaction with a variety of commercially available amines under mild heating gave the desired analogues.
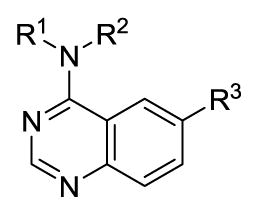
Substituted quinazolines are a common pharmacophore for ATP-competitive kinase inhibitors and our previous work confirmed that these agents are inhibiting Clk isoforms through an ATP-competitive mechanism. Agents such as Tarceva (a clinically used cancer therapeutic that inhibits epidermal growth factor receptor 1) utilize substitutions on both the 6- and 7-position of the core quinazoline structure to boost target affinity and to endow the molecule with improved aqueous solublity.40 We had previously shown that an aryl substitution on the 6-position was a critical element of the pharmacophore of these compounds; however, we were also interested to see if substitution at the 7-position would be tolerated. To examine this we set out to synthesize a variant of our chemotype with a methoxy group in the 7-position. Nitration of methyl 3-hydroxy-4-methoxybenzoate (9) followed by triflation and Suzuki-Miyaura coupling afforded methyl 5-(benzo[d][1,3]dioxol-5-yl)-4-methoxy-2-nitrobenzoate (11) in acceptable yields (Scheme 2). Reduction of the nitro group followed by cyclization yielded the required quinazolin-4(3H)-one which was then chlorinated and displaced by selected amines as described above.
Scheme 2.

Reagents and conditions: (i) HNO3, CH3COOH, 0 °C - r.t., 18 h (ii) triflic anhydride, pyridine, DCM, 1 h, 91% (iii) 3,4-(Methylenedioxy)-phenylboronic acid, Pd(PPh3)4, Na2CO3, DME, H2O, 150 °C (μW), 1 h, 55% (iv) Pd/C, H2 (1 atm), EtOH, 18 h, 99% (v) NH2CHO, NH3COOH, 140 °C for 3 h, then r.t. for 18 h, 51% (vi) POCI3, N,N-dimethylaniline, toluene, reflux, 1 h (vii) RNHCH3, iPr2NEt, DMF, 60 °C, 18 h (typical yields: 80–95%).
Utilizing these synthetic procedures we were able to rapidly access numerous analogues of NCGC00010037 (4) to address key SAR questions. To evaluate these agents we utilized a bioluminescent luciferase-based assays capable of visualizing substrate (ATP) depletion or product (ADP) formation.28 Utilizing this assay we ascertained the IC50 values for our agents versus Clk4 and Dyrk1A. To further examine their selectivity, we submitted selected agents to a commercial provider of kinase assays (Reaction Biology Corp) for profiling versus Clk1-4, Dyrk1A and Dyrk1B within a 33P radiolabeled kinase assay. The data from both assay formats was highly consistent, however for uniformity we will report only the values generated by Reaction Biology Corp (refer to PubChem AIDs 1969, 1970, 1983, 2705, 2710, 488872, 488883, and 488887).
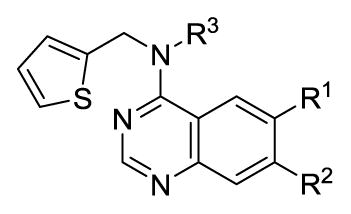
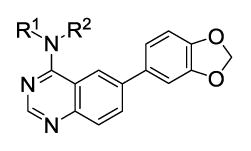
Our previous structure activity evaluations highlighted the utility of a benzo[d][1,3]dioxole moiety at the 6-position of the quinazoline ring. To further our examination at this position we synthesized several derivatives maintaining a common thiophen-2-ylmethanamine substituent in the 4-position while varying the aryl substituent at the 6-position. The results are shown in Table 1 and highlight the activity of the benzo[d][1,3]dioxole and related substitutions [including 2,3-dihydrobenzo[b][1,4]dioxine (29)]. Phenyl ring substitutions were less active as where furan derivatives 16 and 17. Interestingly, furan analogues with substitutions at the 5-position were found to possess activity at Clk4 including the ethyl carboxylate (30) and carboxylic acid (31). Switching the benzo[d][1,3]dioxole from the 6- to the 7-position of the quinazoline ring resulted in a complete loss of activity (analogue 33) as did the incorporation of the methoxy group at the 7-position (analogues 14 [IC50 >10000 for Clk4 and Dyrk1a] and 15).
Table 1.
IC50 (nM) values of selected active quinazolines (4, 15–33)a
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Clk4 | Dyrk1A | Compound | R1 | R2 | R3 | Clk4 | Dyrk1A |
| 4 | 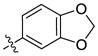 |
H | H | 39 | 62 | 24 |  |
H | H | 3981 | >10000 |
| 15b |  |
OMe | CH3 | >10000 | >10000 | 25 | 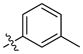 |
H | H | 7943 | >10000 |
| 16 | 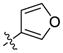 |
H | H | 249 | 1262 | 26 | 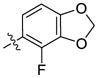 |
H | H | 128 | 164 |
| 17 | 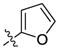 |
H | H | 1090 | 3480 | 27 | 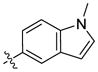 |
H | H | >10000 | 4517 |
| 18 | 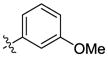 |
H | H | 1051 | 5697 | 28 | 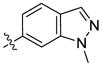 |
H | H | 1642 | >10000 |
| 19 | 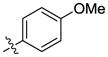 |
H | H | 331 | 3152 | 29 | 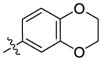 |
H | H | 126 | 180 |
| 20 |  |
H | H | 3675 | >10000 | 30 | 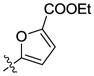 |
H | H | 88 | 1437 |
| 21 |  |
H | H | >10000 | >10000 | 31 | 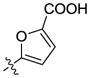 |
H | H | 199 | >10000 |
| 22 | 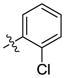 |
H | H | 7943 | 9012 | 32 | 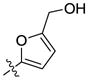 |
H | H | 241 | 4657 |
| 23 | 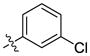 |
H | H | >10000 | >10000 | 33 | H | 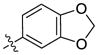 |
H | >10000 | >10000 |
IC50 values determined by Reaction Biology (www.reactionbiology.com). Compounds were tested in 10-dose IC50 mode with 3-fold serial dilution starting at 50 μM. Reactions were carried out at 10 μM ATP.
IC50 values determined using KinaseGlo (Promega) assay.
We next considered alternate substitutions at the 4-position of the quinazoline ring system while maintaining the benzo[d][1,3]dioxole moiety at the 6-position. These agents were profiled versus all Clk isoforms and Dyrk1A and Dyrk1B (Table 2). The results confirmed that these agents possessed selectivity for Clk1 and Clk4 over Clk2 and Clk3. Five-membered heterocycles were found to have the best activity as larger ring systems began to lose activity (analogue 55, for example). The role of adding (and varying) an alkyl group to the 4-position amine was also explored (direct comparisons include analogues 4, 34 and 35; 39 and 40; 45 (NCGC00185963, ML197) and 46 (NCGC00185981, ML195); 47 and 48; 50 and 51). In general, the addition of an alkyl group led to increased activity but at least one related pair had the opposite affect (analogues 50 and 51). Several agents had pan-activity versus Clk1, Clk4, Dyrk1A and Dyrk1B including 34, 46 and 49. Other agents were found to have specified selectivity’s for these targets. For instance, the profile of 38 revealed that this analogue had modest selectivity for Clk1, Clk4 and Dyrk1A while analogue 54 (NCGC00229610, ML196) possesses selectivity for Clk4 and Dyrk1A over the other isoforms. Analogue 50 possess a modest selectivity for Clk4 over all other isoforms.
Table 2.
IC50 (nM) values of selected active quinazolines (4, 34–58)
 | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | Clk1 | Clk2 | Clk3 | Clk4 | Dyrk1A | Dyrk1B | Compound | R1 | R2 | Clk1 | Clk2 | Clk3 | Clk4 | Dyrk1A | Dyrk1B |
| 4 | H | 59 | 1902 | 6936 | 39 | 62 | 697 | 46 | CH3 | 20 | 186 | 1924 | 11 | 14 | 25 | ||
| 34 | CH3 | 11 | 173 | 4949 | 12 | 31 | 73 | 47 | H | 176 | 414 | 3527 | 50 | 70 | 446 | ||
| 35 | CH2CH3 | 14 | 447 | 6172 | 14 | 38 | 231 | 48 | CH3 | 28 | 589 | 4655 | 20 | 51 | 180 | ||
| 36 | H | 888 | >10000 | >10000 | 117 | 1158 | >10000 | 49 | CH3 | 128 | 195 | 3019 | 37 | 26 | 48 | ||
| 37 | CH3 | 89 | 962 | >10000 | 29 | 260 | 1781 | 50 | H | 191 | 3594 | >10000 | 34 | 155 | 1125 | ||
| 38 | H | 69 | 2163 | 6555 | 35 | 35 | 506 | 51 | CH3 | 480 | 3331 | >10000 | 120 | 922 | 4036 | ||
| 39 | H | 192 | 301 | 2556 | 79 | 84 | 534 | 52 | H | 931 | 2219 | >10000 | 161 | 541 | 2271 | ||
| 40 | CH3 | 22 | 692 | 8744 | 36 | 98 | 452 | 53 | CH3 | 98 | 3118 | >10000 | 45 | 120 | 1441 | ||
| 41 | H | 130 | 801 | 2017 | 42 | 74 | 130 | 54 | H | 522 | 1055 | 3642 | 141 | 93 | 734 | ||
| 42 | H | 86 | 2725 | 6450 | 47 | 57 | 431 | 55 | H | 598 | 882 | 5461 | 257 | 135 | 856 | ||
| 43 | H | 111 | 877 | 4650 | 38 | 126 | 938 | 56 | H | 90 | 1810 | 3294 | 29 | 30 | 458 | ||
| 44 | H | 39 | 651 | >10000 | 15 | 93 | 846 | 57 | H | 227 | 486 | 1320 | 90 | 76 | 313 | ||
| 45 | H | 96 | 1327 | 7448 | 40 | 206 | 1510 | 58 | H | 173 | 584 | 435 | 70 | 17 | 83 | ||
IC50 values determined by Reaction Biology (www.reactionbiology.com). Compounds were tested in 10-dose IC50 mode with 3-fold serial dilution starting at 50 μM. Reactions were carried out at 10 μM ATP.
Hoping to further define selective analogues we next examined several compounds with specific parings of the optimal amines at the 4-position and various substituted furans at the 6-position. The results in Table 3 highlight the fact that, in general, these derivatives possess higher affinity for Clk4 as compared to Clk1. At least three analogues were found that were highly specific for Clk4 over all other Clk and Dyrk1 isoforms (analogues 61, 63 and 65). It should be noted that while these activities were consistent with in-house results (where assays were run in triplicate with good SEM values – see PubChem AIDs) the usefulness of these compounds as tools requires confirmation in cell-based assays.
Table 3.
IC50 (nM) values of selected active quinazolines (16, 59–65)
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Clk1 | Clk2 | Clk3 | Clk4 | Dyrk1A | Dyrk1B |
| 16 | H | 915 | 714 | >10000 | 249 | 1262 | 405 | ||
| 59 | CH3 |  |
899 | 1209 | >10000 | 269 | 1136 | 304 | |
| 60 | CH3 |  |
206 | 889 | >10000 | 81 | 557 | 352 | |
| 61 | H | 798 | 8510 | >10000 | 88 | 1437 | 4269 | ||
| 62 | H | 6164 | 8480 | >10000 | 1666 | 594 | 333 | ||
| 63 | H | 1522 | 1648 | >10000 | 136 | >10000 | 4420 | ||
| 64 | H | 2083 | 6181 | >10000 | 229 | 3629 | 7990 | ||
| 65 | H | 1561 | 1361 | >10000 | 104 | >10000 | 4927 | ||
IC50 values determined by Reaction Biology (www.reactionbiology.com). Compounds were tested in 10-dose IC50 mode with 3-fold serial dilution starting at 50 μM. Reactions were carried out at 10 μM ATP.
While understanding the activity for these agents versus the related Clk and Dyrk1 isoforms is a key element to defining novel probes of these targets, we also endeavored to understand their selectivity in the context of the entire kinome. As previously discussed, quinazoline-based kinase inhibitors are well known to possess a wide range of activity and it remained critical to show that these agents were not promiscuous across numerous targets. Our previous report included a profile of analogue 4 across a commercial kinase panel (DiscoveRx). We also included TG003 (1) in this profile and the results demonstrated that both agents were remarkably selective (4 was shown to possess activity [Kd values less than 300 nM] versus Clk1, Clk4, Dyrk1A and EGFR while 1 possessed activity versus Clk1, Clk2, Clk4, Csnk1d, Csnk1e, Dyrk1A, Dyrk1B, Pim1, Pim3, and Ysk4).15 Often, when new analogues are examined (even highly structurally related compounds) new activities are formed. To assure that these analogues still possessed the high selectivity for the Clk and Dyrk1 class of kinases we selected four representative inhibitors (45, 46, 54, 63) to examine in the same kinome-wide assay (possessing 442 kinases at the time of the profile). The kinome scan records activity as a percentage of kinase bound to an immobilized ligand in the presence and absence of each compound. In accordance with our previous work, activities beyond a selected threshold were submitted for Kd determination. The resulting Kd values further validated the selectivity of 45 and 54 for the Clk and Dyrk classes of kinases (Figure 3). Compound 46, although slightly less selective, is highly active against the desired targets as well as undesired kinases, Mek5 (Kd = 47 nM), a potential prostate cancer target41, and the kinase encoded by PIK3C2G (PI3K family)(Kd = 40 nM), which is involved in the pathophysiology of diabetes.42 The results for 63 suggested that this agent is somewhat promiscuous across several kinases and not acceptable as a probe of Clk and Dyrk1 activity (and highlights the utility of these profiles).
Figure 3.

Dendrogram representation of the human kinome demonstrating kinase selectivity of reported inhibitors over a panel of 442 kinases. Activity for 45: Clk1 = 50 nM, Clk2 = 380 nM, Clk4 = 43 nM, Dyrk1A = 82 nM, PIP5K2C protein = 280 nM. Activity for 46: Clk1 = 18 nM, Clk2 = 59 nM, Clk4 = 5 nM, Dyrk1A = 13 nM, Dyrk1B = 300 nM, Dyrk2 = 480 nM, Erk8 = 430 nM, Mek5 = 47 nM, PIK3C2B protein = 340 nM, PIK3C2G protein = 40 nM, PIK3CG protein = 370 nM, PIP5K2C protein = 360 nM, Ysk4 = 190 nM. Activity for 54: Clk1 = 72 nM, Clk2 = 320 nM, Clk4 = 30 nM, Dyrk1A = 27 nM, PIK3C2B protein = 410 nM, PIK4CB protein = 430 nM, PIP5K2C protein = 310 nM. Data from DiscoveRx (http://kinomescan.com).
Our previous report included a docking study of 4 within a homology model of Clk4. This model highlighted a potential H-bond between an amide NH within the ATP binding pocket and the quinazoline core. In this study, we hoped to utilize these models to better understand the interaction mode and selectivity profiles of the lead compounds within the Clk and Dyrk subfamilies. In order to generate useful models (particular of the Dyrk1 family) we performed multiple protein sequence alignments to derive homology models for Clk4 and Dyrk1B for which there are no published X-ray structures.15 The homology model of Clk4 was developed by utilizing the X-ray structure of Clk1 as the template (86% sequence identity at the catalytic domain), while the Dyrk1B homology model was built based upon the highly homologous Dyrk1A (77% sequence identity) using MOE molecular modeling software (Figure 4A).43 Several of our lead compounds were then docked into the ATP binding domains of these Clk and Dyrk1 models to achieve an optimal binding pose using FRED (OpenEye Scientific Software suite)(Figure 4B).44 The resulting docking poses were considered in the context of the experimentally determined IC50 and Kd values. In agreement with our previous docking results, the quinazoline core adopted a common pose within the ATP binding pocket forming previously validated hydrogen bonds with the hinge region (Figure 4B highlights the docking of 46 with Clk4). As previously discussed, when an alkyl group was added to the 4-position amine (either a methyl or ethyl) activity generally improved. Our model rationalizes this result due to a small hydrophobic pocket (as indicated by a white line) in which the alkyl group is oriented which would likely increase specified van der Waals interactions and lock the inhibitor in a preferred conformation (Figure 4C). Interestingly, the SAR surrounding the amine side-chain suggests that several variations are well tolerated. This model suggests that the primary role of this moiety is space-filling rather than interacting with specific protein residues via H-bonding or electrostatic interactions.
Figure 4.

(A) Ribbon representation of the catalytic clefts in the Clk1 crystal structure (green; PDB 1Z57), Clk4 homology model (cyan), Dyrk1a crystal structure (orange, PDB 2VX3) and Dyrk1b homology model (purple). The ligand shown is the co-crystal ligand in Dyrk1A. (B) Docking model of 46 in the Clk4 catalytic cleft. The binding pocket is depicted by molecular surface and the hydrogen bonds are labeled as yellow dotted lines. (C) The close view of the small hydrophobic pocket (as indicated by a white line) in which the methyl group is sitting in the docking model of 46 within the Clk4 catalytic cleft. (D) Docking model of 63 in the Clk4 catalytic cleft superimposed with Clk1, Dyrk1a and Dyrk1b. The hydrogen bond from the hydroxyl group to Asp248 is labeled as a yellow dotted line. Clk4 kinase domain is a homology model derived from the X-ray structure of Clk1 (PDB code: 1Z57). This figure was prepared with the program VIDA (OpenEye Scientific Software).
A primary hope was that these models might yield insight into defining more selective inhibitors of the Clk and Dyrk1 targets. One insight surrounds Clk4, which is nearly identical to Clk1 at the catalytic domain, but shows a key divergence in residue Asp248. Models docking 63 at Clk4 and Clk1 suggest that this molecule’s hydroxyl group is within an appropriate proximity to Asp248 (~3.2 Å) to form a hydrogen bond (Figure 4D). This potential interaction would help rationalize the potency for 63 versus Clk4 (136 nM) as compared to Clk1 (1522 nM). In the Dyrk1A crystal structure and the Dyrk1B homology model, the Asp residue at the same position shifted more than 6 Å away from the hydroxyl group in analogue 63 due to a backbone conformational change. This distance is too far for a viable hydrogen bond and offers a basis for the potency decrease versus Dyrk1A and Dyrk1B (>10,000 nM and 4420 nM, respectively). This interaction also highlights a potential route to furthering the SAR of this chemotype.
As it was our goal to define probe compounds for these targets that would have use within cell-based studies it was important for us to examine the water solubility, stability and membrane permeability of selected analogues. To accomplish this, we submitted selected analogues (45, 46 and 54) to commercial providers of kinetic aqueous solubility data (Analiza, Inc.) and Caco-2 permeability data (Cyprotex PLC). We further examined the stability of these agents in aqueous buffered environment for 48 hours (results are based upon retention of UV/Vis signal and mass assessment within a standard HPLC gradient). The outcome of these studies is shown in Table 4. Compounds 45 and 46 were found to possess only modest aqueous solubility while 54 was highly soluble. The Caco-2 profile suggested that each agent was capable of passive membrane permeability and had favorable efflux ratios. Finally, each agent was highly stable in an aqueous environment for up to 48 hours.
Table 4.
Aqueous solubility and stability and Caco-2 permeability data for selected agents.
| Analogue | Kinetic Solubiltya (μM) | Papp(A–B)b (10−6 cm/s) | Papp(B–A)b (10−6 cm/s) | 48 hr Aqueous Stabilityc (DPBS buffer, pH 7.4) |
|---|---|---|---|---|
| 45 | 4.3 | 25.5 | 12.7 | > 95% |
| 46 | 6.6 | 18.9 | 8.9 | > 95% |
| 54 | 143.3 | 35.4 | 23.1 | > 95% |
Solubility measurements done at Analiza, Inc. (http://analiza.com).
Caco-2 permeability was measured at Cyprotex (www.cyprotex.com/adme).
Aqueous stability measured over 48 h in pH 7.4 DPBS buffer.
In conclusion, the expansion of our previous efforts surrounding specific 6-arylquinazolin-4-amines as potent Clk and Dyrk1 inhibitors has yielded several agents with impressive potency and selectivity. Selected inhibitors possess activity versus Clk1, Clk4 and Dyrk1A below 100 nM. A broad kinome scan has confirmed that these compounds are highly selective for these targets and molecular docking studies highly suggest that they bind at the kinase hinge region. This work also defined the first reported inhibitor of Dyrk1B. These agents provide useful tool compounds to probe the role of these targets in pre-mRNA splicing and, in the case of Dyrk1B, specified roles in cancer.
Supplementary Material
Scheme 1.

Reagents and conditions: (i) aryl-boronic acid, Pd(PPh3)4, Na2CO3, DME, H2O,150 °C (μW), 1 h (typical yields: 40–50%) (ii) POCI3, N,N-dimethylaniline, toluene, reflux, 1 h (iii) R,R′NH, iPr2NEt, DMF, rt -to- 60 °C, 2h (typical yields: 80–95%).
Acknowledgments
We greatly appreciate the assistance of William Leister and Thomas Daniel for compound analysis and purification and Chris LeClair, Paul Shinn and Danielle VanLeer for compound management. This research was supported by the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research and the Intramural Research Program of the National Human Genome Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Prasad J, Manley JL. Mol Cell Bio. 2003;23:4139–4149. doi: 10.1128/MCB.23.12.4139-4149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black DL. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 3.Wang Z, Burge CB. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Faustino NA, Cooper TA. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 5.He C, Zhou F, Zuo Z, Cheng H, Zhou R. PLoS ONE. 2009;4:e4732. doi: 10.1371/journal.pone.0004732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matlin AJ, Clark F, Smith CWJ. Nature. 2005;6:386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- 7.Jiang K, Patel NA, Watson JE, Apostolatos H, Kleiman E, Hanson O, Hagiwara M, Cooper DR. Endocrinology. 2009;150:2087–2097. doi: 10.1210/en.2008-0818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwertz H, Tolley ND, Foulks JM, Denis MM, Risenmay BW, Buerke M, Tilley RE, Rondina MT, Harris EM, Kraiss LW, Mackman N, Zimmerman GA, Weyrich AS. J Exp Med. 2006;203:2433–2440. doi: 10.1084/jem.20061302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muraki M, Ohkawara B, Hosoya T, Onogi H, Koizumi J, Koizumi T, Sumi K, Yomoda J-i, Murray MV, Kimura H, Furuichi K, Shibuya H, Krainer AR, Suzuki M, Hagiwara M. J Biol Chem. 2004;279:24246–24254. doi: 10.1074/jbc.M314298200. [DOI] [PubMed] [Google Scholar]
- 10.Yomoda J-i, Muraki M, Kataoka N, Hosoya T, Suzuki M, Hagiwara M, Kimura H. Genes Cells. 2008;13:233–244. doi: 10.1111/j.1365-2443.2008.01163.x. [DOI] [PubMed] [Google Scholar]
- 11.Hartmann AM, Rujescu D, Giannakouros T, Nikolakaki E, Goedert M, Mandelkow EM, Gao QS, Andreadis A, Stamm S. Mol Cell Neuro. 2001;18:80–90. doi: 10.1006/mcne.2001.1000. [DOI] [PubMed] [Google Scholar]
- 12.Perni RB, Bemis J, Nunes JJ, Szczepankiewicz BG. WO 2009/085226 A2. Inhibitors of the CDC2-like Kinases and Methods of Use Therof. 2009 July 9;
- 13.Hekimi S, McBride K, Hihi AK, Kianjcka I, Wang Y, Hayes SL, Guimond M-P, Sevigny G, Dumas D, Smith J. 60/832937. New Quinoline Derivatives. 2006 July 25;
- 14.Fedorov O, Huber K, Eisenreich A, Filippakopoulos P, King O, Bullock AN, Szklarczyk D, Jensen LJ, Fabbro D, Trappe J, Rauch U, Bracher F, Knapp S. Chem Biol. 201(18):67–76. doi: 10.1016/j.chembiol.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mott BT, Tanega C, Shen M, Maloney DJ, Shinn P, Leister W, Marugan JJ, Inglese J, Austin CP, Mistelli T, Auld DS, Thomas CJ. Bioorg Med Chem Lett. 2009;19:6700–6705. doi: 10.1016/j.bmcl.2009.09.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Becker W, Weber Y, Wetzel K, Eirmbter K, Tejedor FJ, Joost HG. J Biol Chem. 1998;273:25893–25902. doi: 10.1074/jbc.273.40.25893. [DOI] [PubMed] [Google Scholar]
- 17.Alvarez M, Estivill X, Luna Sdl. J Cell Sci. 2003;116:3099–3107. doi: 10.1242/jcs.00618. [DOI] [PubMed] [Google Scholar]
- 18.Graaf Kd, Czajkowska H, Rottmann S, Packman LC, Lilischkis R, Luscher B, Becker W. BMC Biochem. 2006:7. doi: 10.1186/1471-2091-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park J, Song WJ, Chung KC. Cell Mol Life Sci. 2009;66:3235–3240. doi: 10.1007/s00018-009-0123-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida K. Biochem Pharm. 2008;76:1389–1394. doi: 10.1016/j.bcp.2008.05.021. [DOI] [PubMed] [Google Scholar]
- 21.Kim ND, Yoon J, Kim JH, Lee JT, Chon YS, Hwang MK, Hac I, Songc WJ. Bioorg Med Chem Lett. 2006;16:3772–3776. doi: 10.1016/j.bmcl.2006.04.042. [DOI] [PubMed] [Google Scholar]
- 22.Seifert A, Allan LA, Clarke PR. FEBS Journal. 2008;275:6268–6280. doi: 10.1111/j.1742-4658.2008.06751.x. [DOI] [PubMed] [Google Scholar]
- 23.Guimera J, Casas C, Estivill X, Pritchard M. Genomics. 1999;57 doi: 10.1006/geno.1999.5775. [DOI] [PubMed] [Google Scholar]
- 24.Tejedor FJ, Hammerle B. FEBS Journal. 2011:278. doi: 10.1111/j.1742-4658.2010.07954.x. [DOI] [PubMed] [Google Scholar]
- 25.Wegiel J, Gong CX, Hwang YW. FEBS Journal. 2011;278:236–245. doi: 10.1111/j.1742-4658.2010.07955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toiber D, Azkona G, Ben-Ari S, Toran N, Soreq H, Dierssen M. Neurobiol Dis. 2010;40:348–359. doi: 10.1016/j.nbd.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 27.Guo X, Williams JG, Schug TT, Li X. J Biol Chem. 2010;285:13223–13232. doi: 10.1074/jbc.M110.102574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yabut O, Domogauer J, D’Arcangelo G. J Neuro. 2010;30:4004–4014. doi: 10.1523/JNEUROSCI.4711-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, Gao X, Neilson JR, Chen L, Heit JJ, Kim SK, Yamasaki N, Miyakawa T, Francke U, Graef IA, Crabtree GR. Nature. 2006;441:595–600. doi: 10.1038/nature04678. [DOI] [PubMed] [Google Scholar]
- 30.Mercer SE, Friedman E. Cell Biochem Biophys. 2006;45:303–315. doi: 10.1385/CBB:45:3:303. [DOI] [PubMed] [Google Scholar]
- 31.Lee K, Deng X, Friedman E. Cancer Res. 2000;60:3631–3637. [PubMed] [Google Scholar]
- 32.Friedman E. Cancers. 2010;2:1492–1512. doi: 10.3390/cancers2031492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin K, Park S, Ewton DZ, Friedman E. Cancer Res. 2007;67:7247–7255. doi: 10.1158/0008-5472.CAN-06-4099. [DOI] [PubMed] [Google Scholar]
- 34.Friedman E. J Cell Bio. 2007;102:274–279. doi: 10.1002/jcb.21451. [DOI] [PubMed] [Google Scholar]
- 35.Jin K, Ewton DZ, Park S, Hu J, Friedman E. J Biol Chem. 209;284:22916–22925. doi: 10.1074/jbc.M109.035519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao J, Zheng Z, Rawal B, Schell MJ, Bepler G, Haura EB. Cancer Biology & Therapy. 2009;8:1671–1679. doi: 10.4161/cbt.8.17.9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JSC, Alessi DR, Cohen P. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gockler N, Jofre G, Papadopoulos C, Soppa U, Tejedor FJ, Becker W. FEBS Journal. 2009;276:6324–6337. doi: 10.1111/j.1742-4658.2009.07346.x. [DOI] [PubMed] [Google Scholar]
- 39.Becker W, Sippl W. FEBS Journal. 2011;278:246–256. doi: 10.1111/j.1742-4658.2010.07956.x. [DOI] [PubMed] [Google Scholar]
- 40.www.tarceva.com
- 41.Mehta PB, Jenkins BL, McCarthy L, Thilak L, Robson CN, Neal DE, Leung HY. Oncogene. 2003;22:1381–1389. doi: 10.1038/sj.onc.1206154. [DOI] [PubMed] [Google Scholar]
- 42.Daimon Makoto, Sato Hidenori, Oizumi Toshihide, Toriyama Sayumi, Saito Takafumi, Karasawa Shigeru, Jimbu Yumi, Wada Kiriko, Kameda Wataru, Susa Shinji, Yamaguchi Hiroshi, Emi Mitsuru, Muramatsu Masaaki, Kubota Isao, Kawata Sumio, Kato T. Biochem Biophys Res Commun. 2008;365:466–471. doi: 10.1016/j.bbrc.2007.10.180. [DOI] [PubMed] [Google Scholar]
- 43.MOE Molecular Operating EnVironment. 2008.10. Chemical Computing Group Inc; Montreal, Canada: 2008. [Google Scholar]
- 44.FRED. OpenEye Scientific Software, Inc; Santa Fe, NM: 2010. http://www.eyesopen.com/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.