

Abstract
New tricyclic HIV-1 integrase (IN) inhibitors were prepared that combined structural features of bicyclic pyrimidinones with recently disclosed 4,5-dihydroxy-1H-isoindole-1,3(2H)-diones. This combination resulted in the introduction of a nitrogen into the aryl ring and the addition of a fused third ring to our previously described inhibitors. The resulting analogues showed low micromolar inhibitory potency in in vitro HIV-1 integrase assays, with good selectivity for strand transfer relative to 3′-processing.
Keywords: HIV-1 integrase, inhibit, Raltegravir, cycloaddition
After more than two decades of intensive research, approximately 30 drugs have been approved by the FDA for the treatment of HIV/AIDS. Included among these is Merck’s Isentress™ (MK-0518 or Raltegravir, 1, Figure 1), which in 2007 became the first marketed drug targeting HIV-1 integrase (IN).1 Raltegravir is one of a number of compounds that inhibit IN by selectively blocking the strand transfer (ST) reaction as opposed to the 3′-processing reaction (3′-P).2 More recently, bicyclic pyrimidinones such as 2 have been reported, which represent an evolution of the N-methylpyrimidinone motif found in Raltegravir.3 We have previously described 4,5-dihydroxy-1H-isoindole-1,3(2H)-diones (3) as structurally simple IN inhibitors that exhibit good potency and ST selectivity in the presence of cofactor Mg2+.4–6 It should be noted that the 1-carbonyl bridge in 3 serves to constrain three critical oxygen atoms (indicated in 3 by “*”) into a coplanar arrangement designed to enhance chelation with the two catalytic divalent metal ions (Figure 1).7
Figure 1.

Structures of HIV-1 integrase inhibitors discussed in the text.
The therapeutic utility of compounds such as 3 is potentially limited by cytotoxicity arising from the embedded catechol functionality. We hypothesized that introduction of a nitrogen substituent into the aryl ring could remove the catechol nature of these inhibitors, but that the resulting derivatives would retain other key features. Based on this rationale, we designed tricyclic hydroxy-pyrrolopyridine-triones (4). These analogues represent a new structural class of IN inhibitors that combine features of 2 with 3 (Figure 1). We report here the highly efficient preparation of these compounds by cycloaddition chemistries and the biological and biochemical evaluation of the compounds.
The efficient synthesis of the key 7-hydroxy-1H-pyrrolo[3,4-c]pyridine-1,3,6(2H,5H)-trione skeleton of 4 (ring numbering is indicated in Figure 1) was achieved by application of Padwa’s “Pummerer cyclization deprotonation cycloaddition” cascades of imidosulfoxides (Scheme 1).8, 9 Conversion of amides 5 to imidosulfoxides 7 via sodium periodate oxidation of α-thioimides 6 (obtained by acylation with freshly prepared 2-ethylthioacetyl chloride in benzene) and reaction with acetic anhydride, led to the in situ formation of un-isolated intermediates (8 and 9). These were subjected to 1,3-dipolar cycloaddition with suitable alkenes (10) to form oxide-bridged products (11). Further treatment with Lewis acids, such as BF3·OEt2, provided an efficient synthesis of 2(1H)-pyridones 4.8, 9
Scheme 1.

Synthesis of substituted 3-hydroxy-2(1H)-pyridones (4) using a “Pummerer cyclization deprotonation cycloaddition” cascade of imidosulfoxides (7).
Synthesis of the analogue 4a, containing a 5-membered “A” ring proved to be problematic using the above approach. Therefore, an alternate route was used that employed the [3+2] cycloaddition of phenylsulfonyl-substituted mesoionic oxazolium ylide (isomünchnone) dipole intermediates (Scheme 2).10, 11 Treatment of α-phenylsulfony-containing imide 12 with p-acetamidobenzenesulfonyl azide (13)12 and triethylamine in acetonitrile, provided the diazoimide 14. Reaction of 14 with a catalytic amount of rhodium(II) acetate (benzene at 80° C) resulted in formation of the corresponding isomünchnone dipole (15), which was trapped by [3+2] cycloaddition with N-(3′-chloro-4′-fluoro-benzyl)maleimide 10-1, to yield the oxide-bridged product (16). Refluxing 16 with BF3·OEt2 in CH2Cl2, resulted in ring opening to form the 7-hydroxy-1H-pyrrolo[3,4-c]pyridine-1,3,6(2H,5H)-trione (4a-1, Scheme 2).
Scheme 2.

Synthesis of substituted 3-hydroxy-2(1H)-pyridone 4a-1 via an isomünchnone-based [3+2] cycloaddition approach.
The tricyclic hydroxy-pyrrolopyridine-triones (4) were designed to combine features of Merck’s bicyclic pyrimidinones (2)3 with those of our previously reported isoindolediones (3, Figure 1).4–6 A triad of oxygens (Figure 1 indicated in 3 by “*”) has been hypothesized to chelate Mg2+ ions involved with IN catalysis, while the halogenated N-benzyl group has been proposed to interact with the penultimate cytosine as the 3′-end of the viral DNA in the IN·DNA complex.13, 14 As reported herein, modification of the known inhibitors 2 by introduction of the 3-carbonyl (giving a third “C”-ring) was done to facilitate this metal chelation by constraining the carbonyl group at the 1-position of 4 into co-planar arrangement with the oxygen atoms at the 6- and 7-positions of the “B”-ring of 4. A similar approach has been shown to be highly effective in the design of 34–6 and in related IN inhibitors.7
Because for the parent compounds 2, inhibitory potency has been shown to vary with ring size, we generated and tested analogues with “A”-rings ranging in size from 5 to 8 members.3 Using 4-fluoro-3-chloro substituents on the N-benzyl group, the resulting tricyclic analogues were shown to be highly selective for the ST step relative to the 3′-P reactions (Table 1). Inhibitory potencies increased slightly with ring size, such that analogue 4d-1, having an 8-membered A-ring was twofold more potent than 4a-1, having a 5-membered ring A-ring.
Table 1.
Integrase Inhibitory Potencies of Tricyclic Hydroxy-pyrrolopyridine Triones.a
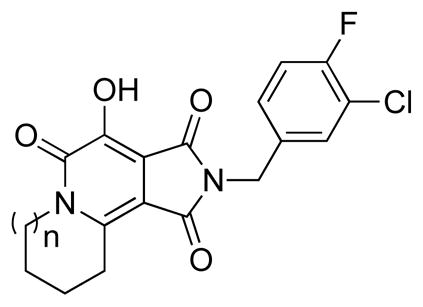 | |||
|---|---|---|---|
| No. | n | 3′-Processing IC50 (μM) | Strand transfer IC50 (μM) |
| 4a-1 | 0 | > 111 | 10.9 ± 1.4 |
| 4b-1 | 1 | > 111 | 9.1 ± 1.1 |
| 4c-1 | 2 | > 111 | 6.0 ± 0.7 |
| 4d-1 | 3 | > 111 | 5.4 ± 0.8 |
Data was obtained from in vitro IN assays as described in reference 4.
For the parent compounds 34–6 and numerous other ST-selective IN inhibitors,7 potency is highly dependent on the nature of the N-benzyl group: In general, benzyl rings with halogen substituents are favored. Using the 7-membered A-ring-containing 4c as a platform, amides containing halogenated (4c-1 through 4c-4) as well as non-halogenated (4c-5 and 4c-6) groups were prepared and compared with the corresponding 4,5-dihydroxy-1H-isoindole-1,3(2H)-diones (3-1 through 3-6). Dramatic substituent-dependent differences were observed (Table 2). In the new pyridone series deletion of halogen functionality resulted in either complete (4c-5, IC50 >111 μM) or near-complete loss (4c-6, IC50 = 90 μM) of ST inhibitory potency. Notably, for the parent catechol-containing compounds, ST inhibitory activity was maintained after deletion of halogen functionality (3-5, IC50 = 0.4 μM and 3-6, IC50 = 0.7 μM, Table 2). However, 3′-P inhibitory activity tended to be reduced after halogen removal (3-5, IC50 = 72 μM and 3-6, IC50 = 150 μM, Table 2). All halogen-containing members of the series 4c exhibited low micromolar ST inhibitory potency and good selectivity relative to 3′-P inhibition.
Table 2.
Effects of N-arylmethyl substituents on integrase inhibitory potencies of hydroxy-pyrrolopyridine triones (4c) as compared with 4,5-dihydroxy-1H-isoindole-1,3(2H)-diones (3).a
 | ||||||
|---|---|---|---|---|---|---|
| No. | R | IC50 (μM)
|
No. | IC50 (μM)
|
||
| 3′-Processing | Strand Transfer | 3′-Processing | Strand Transfer | |||
| 4c-1 |
|
> 111 | 6.0 ± 0.7 | 3-1 | 14 ± 3 | 0.17 ± 0.06 |
| 4c-2 |
|
> 111 | 10.4 ± 1.0 | 3-2 | 8 ± 3 | 5.3 ± 1.5 |
| 4c-3 |
|
> 111 | 6.01 | 3-3 | 27 ± 1 | 0.14 ± 0.03 |
| 4c-4 |

|
> 111 | 9.2 ± 1.4 | 3-4 | 25 ± 16 | 1.7 ± 0.8 |
| 4c-5 |
|
> 111 | > 111 | 3-5 | 72 ± 23 | 0.39 ± 0.21 |
| 4c-6 |
|
> 111 | 90.5 ± 4.7 | 3-6 | 150 ± 36 | 0.72 ± 0.25 |
Data was obtained from in vitro IN assays as described in reference 4.
Divalent metal cofactors are essential both for the proper function of IN and inhibitory potencies of ST inhibitors.15 The recent co-crystal structures of ST inhibitors bound to the prototype foamy virus (PFV) IN complexed to 3′-processed DNA substrate have confirmed the critical roles of two catalytic divalent ions in the binding of these inhibitors.13, 14, 16, 17 Although Mg2+ is believed to be preferred by IN in vivo, assays either Mg2+ or Mn2+ can be used in vitro. Activities of some ST inhibitors differ in assays with the two metals.4, 18 In order to investigate the metal dependency of the newly synthesized 7-hydroxy-1H-pyrrolo[3,4-c]pyridine-1,3,6(2H,5H)-triones (Table 1), we performed assays using either Mg2+ or Mn2+. For Raltegravir (1), inhibitory potencies against both 3′-P and ST reactions were relatively similar when Mn2+ or Mg2+ was used. In contrast, 4b-1 showed an increase in both 3′-P and ST inhibitory potencies in the presence of Mn2+ (Table 3). This indicated a difference in the interaction of 4b-1 and the metal cofactors compared to Raltegravir.
Table 3.
Metal-dependency of Integrase Inhibitory Potencies In Vitro.a
| No. | Metal Cofactor | IC50 (μM)
|
|
|---|---|---|---|
| 3′-P | ST | ||
| 1 | Mg2+ | > 4.5 | 0.067 |
| Mn2+ | > 4.5 | 0.074 | |
| 4b-1 | Mg2+ | > 111 | 9.1 ± 1.1 |
| Mn2+ | 28 ± 2 | 1.2 ± 0.3 | |
Data was obtained from in vitro IN assays using the indicated metal cofactors as described in the Supporting Information.
Resistance to Raltegravir (1) that arises in HIV-1 infected patients frequently involves the following integrase mutations: G140S/Q148H, Y143R and N155H.19 In order to test the susceptibility of the 7-hydroxy-1H-pyrrolo[3,4-c]pyridine-1,3,6(2H,5H)-triones to these mutations, IN was assayed in vitro using pre-cleaved (3′-processed) DNA with Mg2+ as the metal cofactor. Inhibitory potencies were determined for a panel of enzymes that included the wild-type (WT) IN and the three key mutant forms indicated above (Table 4). Although none of the new compounds was as potent against the wild-type IN as Raltegravir, the inhibitory potency of 4b-1 was less susceptible to the Raltegravir resistant mutants. Raltegravir was 105-fold less potent against the G140S/Q148H mutant as compared to the WT enzyme; however, inhibitor 4b-1 showed only a 6-fold loss of potency. Inhibitor 4b-1 was also more than 10-fold less sensitive than Raltegravir to the effects of the Y143R mutation and more than 2-fold less sensitive to the effects of the N155H mutation (Table 4). Interpretation of this data should be done with the understanding that the overall potency of 4b-1 is suboptimal.
Table 4.
Integrase Inhibitory Potencies In Vitro Using Wild-Type (WT) and Mutant Enzymes.a
| No. | ST IC50 (μM, WT) | Mutantsb |
||
|---|---|---|---|---|
| G140S/Q148H | Y143R | N155 H | ||
| 1 | 0.067 | 105 x | 26 x | 7 x |
| 4b-1 | 9.1 ± 1.1 | 6 x | 2 x | 3 x |
Data was obtained from in vitro IN assays using Mg2+ cofactor as described in the Supporting Information.
Fold-loss of potency relative to WT enzyme for ST.
To complement the in vitro data, we developed HIV-1 vectors that replicated the resistant IN mutants G140S/Q148H, Y143R and N155H. These mutant vectors were challenged with Raltegravir (1) and 4b-1 in cultured cells. One of the design rationales for introducing a nitrogen into the parent 4,5-dihydroxy-1H-isoindole-1,3(2H)-diones (3) was to remove catechol functionality and thereby reduce cytotocity. As shown in Table 5, the cytotoxicity of 4b-1 (CC50 = 291 μM) was 30-fold lower than the previously reported value of the related catechol-containing analogue 3-1 (CC50 = 9.5 μM),5 In addition, consistent with the in vitro data obtained for the Raltegravir-resistant IN mutants (Table 4), Raltegravir showed a greater than 400-fold loss of antiviral efficacy with the G140S/Q148H vector as compared to the wild-type IN vector (Table 5). In contrast to Raltegravir, inhibitor 4b-1 displayed only a 2-fold loss of potency when challenged with the G140S/Q148H mutant vector, thereby making it 200-times less sensitive to the effects of this mutation. Similarly, consistent with the in vitro data in Table 4, analogue 4b-1 showed a 10-fold lower loss of potency compared to Raltegravir, with vectors that individually carry the Y143R (53-fold loss for Raltegravir and 4-fold loss for 4b-1) and N155H mutations (35-fold loss for Raltegravir and 3-fold loss for 4b-1).
Table 5.
Antiviral Potencies in Cells Infected with HIV-1 Containing Wild-Type or Mutant Integrase Enzymes.a
| No. | CC50 (μM) | EC50 (μM, WT) | Mutantsb |
||
|---|---|---|---|---|---|
| G140S/Q148H | Y143R | N155H | |||
| 1 | N/A | 0.004 ± 0.002 | 425 x | 53 x | 35 x |
| 4b-1 | 291 ± 51 | 61 ± 9 | 2 x | 4 x | 3 x |
Data was obtained as indicated in the Supporting Information.
Fold-loss of potency relative to virus containing wild-type (WT) enzyme.
The tricyclic hydroxy-pyrrolopyridine triones (4) combine features of bicyclic pyrimidinones (2) with those of our previously reported isoindolediones (3). Introduction of the 2(1H)-pyridone moiety into the previously 1H-isoindole-1,3(2H)-dione-based analogues significantly increases the requirement for halogen substituent(s) within the key benzylamide pharmacophore. The tricyclic hydroxy-pyrrolopyridine trione inhibitors (4) maintain good selectivity for ST reactions relative to the 3′-P reactions and exhibit reduced cytotoxicity relative to the catechol-containing compounds (3). In both in vitro and in vivo assays the new analogue 4b-1 was less sensitive than Raltegravir to resistance incurred by G140S/Q148H, Y143R and N155H mutations in integrase. The enhanced activity toward the Y143R mutation relative to Raltegravir is consistent with recent findings based on co-crystal structures of the homologous PFV integrase, which show that π–π stacking interactions occur between Y212 (corresponding to Y143 in the HIV-1 IN) and the oxadiazole ring of bound Raltegravir. 13, 14, 16, 17 The new compound 4b-1, does not contain equivalent functionality and 4b-1 would not be expected to be as susceptible (in terms of a fold increase in IC50 value) as Raltegravir to the effects of mutations at Y143.
Compound 4b-1 is also less susceptible (in terms of the increase in IC50 value) to the effects of the G140S/Q148H double mutant. In this case, the new PFV IN structural data does not provide as clear an answer to the question of why the two compounds differ in their relative susceptibility to the two mutants. However, the tricyclic hydroxy-pyrrolopyridine triones (4) showed differential activity in the presence of Mn2+, which Raltegravir (1) does not. This suggests the possibility that there are subtle differences in the way(s) in which the two compounds interact with metals at the active site. It is possible that the G140S/Q148H double mutant changes the active site in a way that alters the interactions of Raltegravir with metals, and because 4b-1 interacts differently with metals, it is less susceptible.
The range of inhibitory potencies shown by our title inhibitors is less than we expected based on the structures of other highly potent ST inhibitors7 and recent co-crystal data of inhibitors bound to the homologous PFV integrase. 13, 14, 16, 17 In spite of their modest (low micromolar) potencies, these new compounds, which should be viewed as a new structural class, are promising. We are particularly interested in understanding why the new compounds retain activity against the G140S/Q148H double mutant. If this property could be incorporated into more potent derivatives, they would have real potential for the treatment of drug-resistant viruses.
Supplementary Material
Acknowledgments
This work was supported in part by the Intramural Research Program of the NIH, Center for Cancer Research, NCI-Frederick and the National Cancer Institute, National Institutes of Health and the Joint Science and Technology Office of the Department of Defense. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Electronic supplementary information (ESI) available: Biological and synthetic experimental procedures and analytical data for synthetic products are reported.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Gonzalez Paz O, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M. J Med Chem. 2008;51:5843. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 2.Marchand C, Maddali K, Metifiot M, Pommier Y. Curr Top Med Chem. 2009;9:1016. doi: 10.2174/156802609789630910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muraglia E, Kinzel O, Gardelli C, Crescenzi B, Donghi M, Ferrara M, Nizi E, Orvieto F, Pescatore G, Laufer R, Gonzalez-Paz O, Di Marco A, Fiore F, Monteagudo E, Fonsi M, Felock PJ, Rowley M, Summa V. J Med Chem. 2008;51:861. doi: 10.1021/jm701164t. [DOI] [PubMed] [Google Scholar]
- 4.Zhao XZ, Semenova EA, Vu BC, Maddali K, Marchand C, Hughes SH, Pommier Y, Burke TR., Jr J Med Chem. 2008;51:251. doi: 10.1021/jm070715d. [DOI] [PubMed] [Google Scholar]
- 5.Zhao XZ, Maddali K, Vu BC, Marchand C, Hughes Stephen H, Pommier Y, Burke Terrence R., Jr Bioorg Med Chem Lett. 2009;19:2714. doi: 10.1016/j.bmcl.2009.03.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao XZ, Maddali K, Marchand C, Pommier Y, Burke TR., Jr Bioorg Med Chem. 2009;17:5318. doi: 10.1016/j.bmc.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liao C, Marchand C, Burke TR, Jr, Pommier Y, Nicklaus MC. Future Med Chem. 2010;2:1107. doi: 10.4155/fmc.10.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Padwa A, Heidelbaugh TM, Kuethe JT. J Org Chem. 1999;64:2038. doi: 10.1021/jo982315r. [DOI] [PubMed] [Google Scholar]
- 9.Padwa A, Heidelbaugh TM, Kuethe JT. J Org Chem. 2000;65:2368. doi: 10.1021/jo9915729. [DOI] [PubMed] [Google Scholar]
- 10.Sheehan SM, Padwa A. J Org Chem. 1997;62:438. doi: 10.1021/jo961690l. [DOI] [PubMed] [Google Scholar]
- 11.Padwa A, Sheehan SM, Straub CS. J Org Chem. 1999;64:8648. [Google Scholar]
- 12.Baum JS, Shook DA, Davies HML, Smith HD. Synth Commun. 1987;17:1709. [Google Scholar]
- 13.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Nature. 2010;464:232. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krishnan L, Li X, Naraharisetty HL, Hare S, Cherepanov P, Engelman A. Proc Natl Acad Sci U S A. 2010;107:15910. doi: 10.1073/pnas.1002346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grobler Jay A, Stillmock K, Hu B, Witmer M, Felock P, Espeseth Amy S, Wolfe A, Egbertson M, Bourgeois M, Melamed J, Wai John S, Young S, Vacca J, Hazuda Daria J. Proc Natl Acad Sci U S A. 2002;99:6661. doi: 10.1073/pnas.092056199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hare S, Vos AM, Clayton RF, Thuring JW, Cummings MD, Cherepanov P. Proc Natl Acad Sci USA. 2010;107:20057. doi: 10.1073/pnas.1010246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cherepanov P. EMBO Reports. 2010;11:328. doi: 10.1038/embor.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marchand C, Johnson AA, Karki RG, Pais GCG, Zhang X, Cowansage K, Patel TA, Nicklaus MC, Burke TR, Jr, Pommier Y. Mol Pharmacol. 2003;64:600. doi: 10.1124/mol.64.3.600. [DOI] [PubMed] [Google Scholar]
- 19.Croxtall JD, Scott LJ. Drugs. 2010;70:631. doi: 10.2165/11204590-000000000-00000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.